

Abstract
The TRIB1 locus has been linked to both cardiovascular disease and hepatic steatosis. Recent efforts have revealed TRIB1 to be a major regulator of liver function, largely, but not exclusively, via CEBPA degradation. We recently uncovered a functional interaction between TRIB1 and HNF4A, another key regulator of hepatic function, whose molecular underpinnings remained to be clarified. Here we have extended these findings. In hepatoma models, HNF4A levels were found to depend on TRIB1, independently of its impact on CEBPA. Using a reporter assay model, MTTP reporter activity, which depends on HNF4A, positively correlated with TRIB1 levels. Confocal microscopy demonstrated partial colocalization of TRIB1 and HNF4A. Using overexpressed proteins we demonstrate that TRIB1 and HNF4A can form complexes in vivo. Mapping of the interaction interfaces identified two distinct regions within TRIB1 which associated with the N-terminal region of HNF4A. Lastly, the TRIB1-HNF4A interaction resisted competition with a CEPBA-derived peptide, suggesting different binding modalities. Together these findings establish that TRIB1 is required for HNF4A function. This regulatory axis represents a novel CEBPA-independent aspect of TRIB1 function predicted to play an important role in liver physiology.
Introduction
The TRIBBLES proteins form a family of 3 mammalian proteins (TRIB1, 2 and 3) sharing ~30% overall identity, characterized by the presence of a relatively well-conserved core kinase-like domain and divergent N and C-termini. Unlike bona fide kinases, the TRIBBLES exhibit no detectable kinase activity (with the exception of TRIB2) as a result of mutations at key catalytic residues1, 2. Rather, they act as molecular adaptors by regulating other kinases and/or targets thereof. Over the years numerous roles, pathological and physiological roles have been ascribed to the TRIBBLES3, 4. For TRIB1 specifically, research has focused on leukemia and more recently, lipid and lipoprotein metabolism. Genome-wide Association Studies (GWAS) have identified a locus proximal (~30 kb) to TRIB1 that associates with increased plasma triglycerides and a predisposition for cardiovascular disease (CAD)5. Importantly, TRIB1 has also been linked to hepatic steatosis6. In a previous work we observed an inverse correlation between the top CAD risk single nucleotide polymorphism (SNP) and TRIB1 expression levels in whole blood on the one hand and circulating lipids on the other hand, suggesting that TRIB1 may play a role in reducing hepatic triglyceride synthesis and secretion in humans7.
Whole animal models have uncovered roles for TRIB1 in both lipid and glucose metabolism8. Of the numerous proximal targets of TRIB1 identified over the years, there is a consensus on the ability of TRIB1 to promote CEBPA degradation9. Recently Bauer et al. have demonstrated a close functional relationship between these proteins in mouse liver10. More specifically, liver-specific TRIB1 deficiency could be partially rescued by CEBPA knock-out hinting that a major function of TRIB1 in the liver is to regulate CEBPA. Importantly, while circulating lipid levels could be rescued by CEBPA knock-out, hepatic lipid accumulation (steatosis) could not, indicating that TRIB1 has roles transcending CEBPA regulation.
We recently identified a functional interaction between HNF4A and TRIB1 11. TRIB1 suppression resulted in impaired HNF4A function inferred from reduced HNF4A, HNF1B and increased SNAI1 transcripts in primary hepatocytes. In HepG2 cells, a widely used hepatic cell model, HNF4A protein levels were reduced as a result of TRIB1 suppression while HNF4A suppression increased TRIB1 transcript abundance. HNF4A is a highly conserved member (NR2A1) of the nuclear receptor family and is unique among the nuclear receptor superfamily in its ability to bind DNA exclusively as a homodimer and activate transcription in the absence of exogenous ligand12. HNF4A plays a pivotal metabolic role by regulating the expression of liver and intestinal genes13, 14. HNF4A is essential for TG, cholesterol homeostasis and bile acid metabolism and helps regulate the expression of several key lipoprotein regulators including APOC3 and MTTP 15–19. In addition, loss of HNF4A perturbs the function of key regulators of the mesenchymal-to-epithelial transition (EMT) and is associated with the development of hepatic steatosis and hepatocellular carcinoma20, 21. Interestingly HNF4A and CEBPA co-localize extensively on chromatin and loss of Hnf4a reduces the ability of Cebpa to bind DNA and vice versa22. In this work the interplay between TRIB1 and HNF4A is explored and a general requirement for the TRIBBLES in sustaining HNF4A protein levels is demonstrated. In addition a protein-protein interaction between HNF4A and TRIB1 is described and mapped.
Results
TRIB1 regulates HNF4A in HuH-7 hepatoma cells
In our previous work we observed that TRIB1 suppression led to reduced HNF4A expression in both HepG2 cells and human primary hepatocytes11. Interestingly while TRIB1 suppression is associated with reduced HNF4A transcript levels in primary hepatocytes, no such change is obvious in HepG2 (Suppl Fig. 1), suggesting that TRIB1 may utilize transcriptional and non-transcriptional mechanisms to regulate HNF4A. To examine how prevalent this relationship was, we examined the impact of TRIB1 silencing in another widely studied human hepatoma cell line, HuH-7 cells where TRIB1 suppression led to reduced HNF4A protein (Fig. 1A). This change was associated with a 26% reduction in HNF4A transcript (74 ± 18% of control (n = 6, p = 0.02). Thus HuH-7 cells, in contrast to HepG2 cells, seem to have retained some capacity to sustain HNF4A transcript levels via TRIB1. Yet as HNF4A protein levels in HuH-7 and HepG2 cells exhibit similar and pronounced (~50% reduction and11) sensitivities to TRIB1 silencing, this suggests that transcriptional impacts may not single-handedly account for lower HNF4A protein expression in HuH-7 cells.
Figure 1.
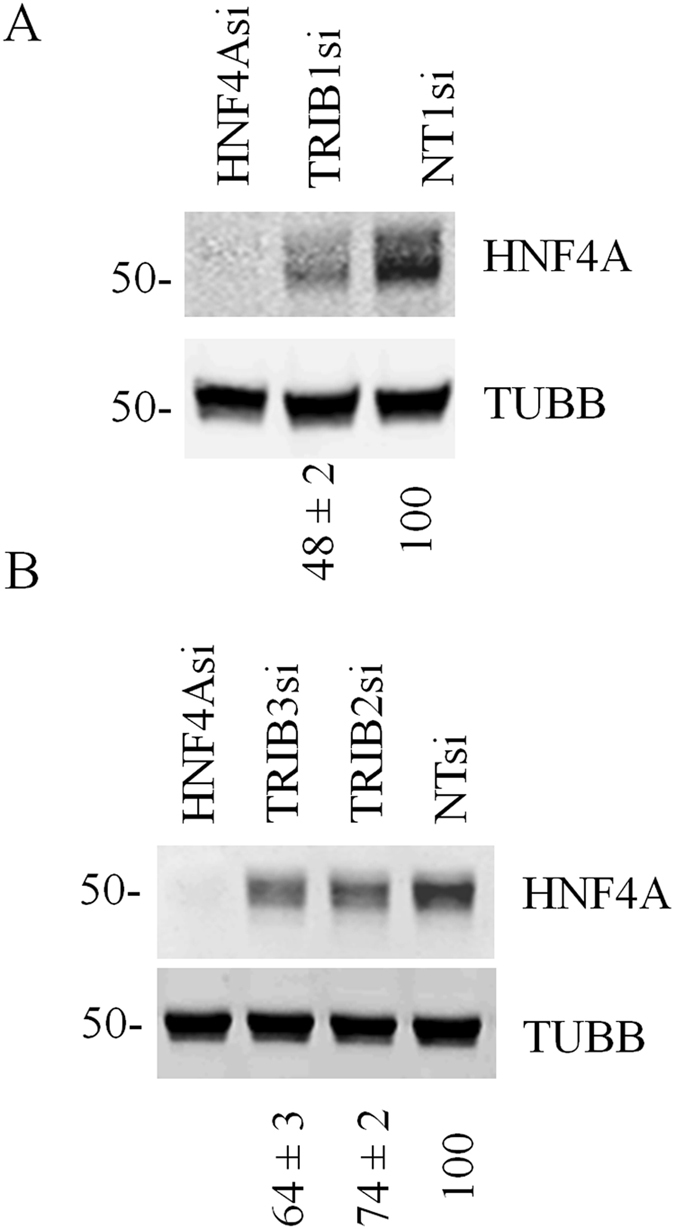
HNF4A expression depends on all the TRIBBLES, with a greater contribution of TRIB1. (A,B) HuH-7 cells were treated for 72 h with the indicated siRNAs and analyzed for protein content by Western blotting. Quantifications of HNF4A (relative to TUBB and then normalized to the NT value) are shown under the blot (means of 3 biological replicates ± S.D). Changes in HNF4A transcript were tested for statistical significance using one-way ANOVA followed by between group comparisons using Tukey’s post-hoc test. TRIB1 vs TRIB2/TRIB3: p < 0.01; TRIB1/2/3 vs NT: p < 0.001.
Maintaining HNF4A levels requires the three TRIBBLES, but most prominently TRIB1
We previously noted that TRIB1 suppression in primary hepatocytes and HepG2 cells resulted in higher levels of the other TRIBBLES (TRIB2 and TRIB3) 11, which could indicate that TRIB2 and TRIB3 are functionally linked to HNF4A as well; similarly, TRIB2 and TRIB3 transcripts were increased in HuH-7 cells upon TRIB1 silencing (data not shown). To investigate possible contributions of the other TRIBBLES in controlling HNF4A levels. TRIB2 and TRIB3 mRNA were targeted by their cognate siRNAs. As seen with TRIB1, TRIB2 and TRIB3 silencing also reduced HNF4A protein levels (Fig. 1B), albeit more modestly suggesting that all TRIBBLES contribute to maintain HNF4A steady state levels, with TRIB1 playing a prominent role. While validation at the RNA level confirmed that the corresponding TRIBBLES transcripts were reduced, endogenous TRIBBLES proteins could not be detected in cellular extracts by Western blot, in line with low RNA expression (Cp values of ~25–30) (Suppl Fig. 2).
TRIB1 and TRIB3 are reportedly unstable proteins and thus reduced mRNA levels are expected to result in reduced protein expression23. As instability was assessed using overexpressed proteins as proxies, the fate of the endogenous protein remained unclear however. To address this last point, large scale immunoprecipitations on control and silenced lysates were undertaken; we decided to focus on TRIB1 in view of its greater contribution to HNF4A function. Western blot analysis of a large scale TRIB1 immunoprecipitation confirmed the efficacy of the knock-down at the protein level (Suppl Fig. 2C). This finding, in conjoncution with our earlier findings demonstrating that targeting two distinct regions of the TRIB1 transcript resulted in reduced HNF4 expression in HepG2 cells, clearly establish that TRIB1 is needed for sustaining HNF4A expression.
Impact of TRIB1 on HNF4A requires more than CEBPA
An important question relates to the role of the CEBPA-TRIB1 axis in HNF4A regulation given that CEBPA and HNF4A are functionally intertwined22. While our suppression data suggested that all 3 TRIBBLES contribute to the maintenance of HNF4A, recent studies have demonstrated an essential contribution of Cebpa in mediating Trib1 function in murine liver10. A major role previously ascribed to TRIB1 and 2, but not shared by TRIB3 is to degrade CEBPA via COP1 recruitment9. Thus the contribution of CEBPA to HNF4A expression was tested in HuH-7 cells. CEBPA silencing reduced HNF4A levels indicating that CEBPA is necessary for maximal HFN4A expression (Fig. 2). By contrast, TRIB1 suppression increased CEBPA protein levels, a finding that is directionally inconsistent with the reduced HNF4A entailed by suppressing TRIB1. Thus changes in CEBPA are insufficient to account for the impact of TRIB1 silencing on HNF4A.
Figure 2.
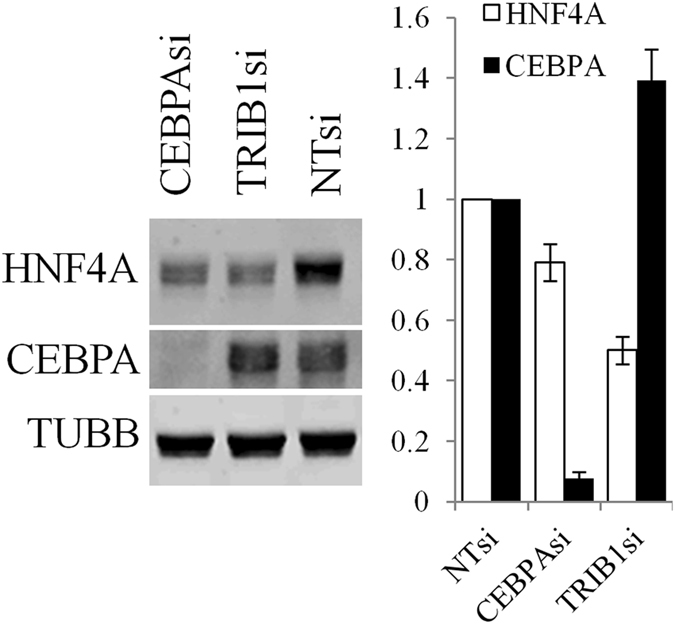
Interplay between TRIB1, CEPBA and HNF4A in HuH-7 cells. HuH-7 cells were silenced with the indicated siRNA for 48 h prior to Western blotting using the indicated antibodies. Representative blot is shown. Quantification of 3 Western blots is shown on the right; bars represent the means of 3 independent biological replicates ± S.D. All changes are statistically significant (p < 0.05) from NTsi.
TRIB1 silencing affects HNF4A function
Next, the impact of TRIB1 suppression on HNF4A function was examined using a MTTP promoter reporter assay previously demonstrated to depend on HNF4A for maximal activation17. In HuH-7 cells HNF4A silencing reduced MTTP promoter (MTTPp) activity to about 10–15% of its maximal value (Fig. 3A), consistent with its strong dependence on HNF4A. Surprisingly a mutated construct that is deficient for HNF4A binding (MTTP4Am), which reduced MTTPp activity by ~35% relative to the Wt construct in naïve cells (data not shown), was similarly sensitive to HNF4A suppression (Fig. 3A). Thus, in HuH-7 cells at least, depletion of HNF4A seems to affect MTTP promoter activity via two distinct mechanisms: proximally, through loss of direct binding of HNF4A (i.e. about 35% of the total signal), as well as distally, possibly through a reprogramming of the cell transcriptional machinery. Be that as it may TRIB1 suppression decreased promoter activities of both constructs by ~30%, a change overall consistent with the HNF4A protein reduction observed. Thus, TRIB1 silencing decreases HNF4 protein level and HNF4A-dependent promoter activity.
Figure 3.
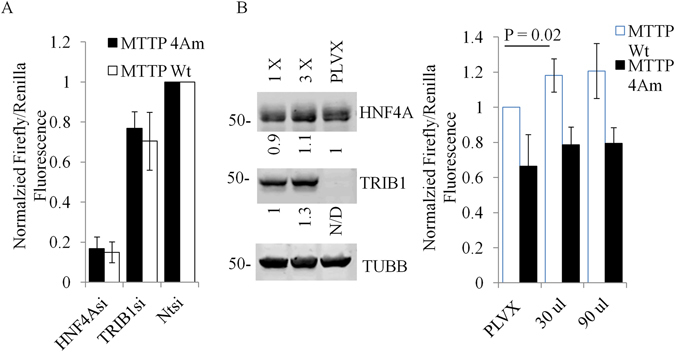
Overexpression of TRIB1 in HuH-7 cells increases MTTP promoter assay activity but does not affect HNF4A protein level. (A), HuH-7 cells were treated for 48 h with indicated siRNAs, washed and transfected with MTTP constructs (and Renilla internal control) for an additional 24 h. Samples were lysed in Passive Lysis Buffer and processed for fluorescence. Results represent the means of 3 experiments (±S.D). Values are normalized to the Non-Target (NT) siRNA-treated samples. (B), A stable pool of HIS-TRIB1 expressing cells was generated by transduction with increasing viral titer (1X, 3X). A control pool was generated using the empty vector (PLVX at 3X titer). Cells transfected with MTTP promoter and renilla internal control constructs for 24 h were lysed and analyzed by Western blotting (left) or luciferase assay (right). Data represent the mean of 5 experiments normalized to PLVX (Wt MTTP) values, ±S.D. Quantifications of the Western blot are relative to TUBB and normalized to either PLVX (HNF4A) or 1X MOI (TRIB1). Statistical significance was assessed using the Student’s 2-tailed t-test.
To assess the impact of TRIB1 overexpression, stable pools of TRIB1 (tagged with a C terminal His tag) were obtained by transducing HuH-7 cells with lentiviral constructs driving TRIB1 expression. Unlike its suppression, introduction of exogenous TRIB1 had no noticeable impact on HNF4A levels (Fig. 3B). When MTTPp activity was measured however, TRIB1 overexpression was associated with a modest but statistically significant increase in Wt promoter activity, in line with increased HNF4A function (Fig. 3B).
TRIB1 co-localizes partially with HNF4A
The experiments above indicated that TRIB1, may regulate HNF4A via multiple pathways. One possible route may involve a direct effect of TRIB1 on HNF4A. Indeed, TRIB1 was previously shown to interact with RAR24, a distantly related nuclear receptor. The localization of both proteins was examined by confocal microscopy, with the expectation that co-localization events represent putative sites of functional convergence and association. Microscopy was performed in several cell types, using either endogenous HNF4A or exogenous HNF4A, in the presence of overexpressed TRIB1; in all cell types examined, endogenous TRIB1 protein was undetectable, defined by a resistance of detectable signal to several TRIB1 siRNA and/or antisense oligonucleotide treatments (data not shown). Both proteins showed qualitatively comparable staining: pan-nuclear signals with some granularity (Fig. 4 and Suppl Figs 3 and 4). Although the signals were largely distinct spatially, consistent with independent functions, some co-localization visualized by orange signals were also observed, indicating that a subset of these proteins have the potential to converge functionally in situ.
Figure 4.
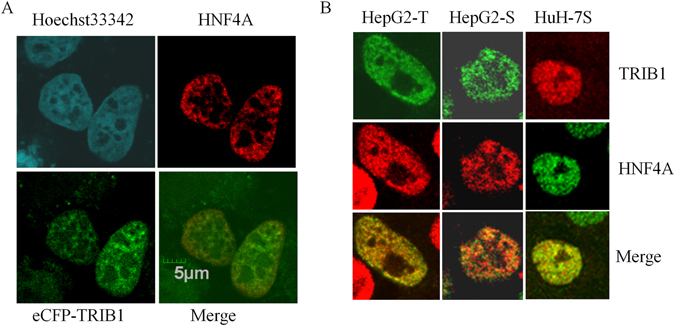
Partial co-localization of TRIB1 and HNF4A in vivo. Confocal microscopy of HNF4A- and TRIB1- expressing cells. (A), HeLa cells co-transfected with plasmids encoding HNF4A and ECFP-TRIB1 for 48 h were fixed, permeabilized, incubated with HNF4A-specific antibodies and visualized by microscopy. (B), Expression in hepatocyte models. Cells were either transfected for 48 h with a plasmid encoding for FLAG-TRIB1 (HepG2-T) or transduced stably with TRIB1 (HepG2-S) or HisTRIB1 (HuH-7S) prior to microscopy. Immunocytochemistry was performed with HNF4A and antibodies specific for either FLAG (HepG2-T) or TRIB1 (HepG2-S, HuH-7S).
TRIB1 forms a complex with HNF4 in vivo
While the microscopy indicated that HNF4A and TRIB1 share some common nuclear territories, confocal resolution is insufficient to examine physical interaction. To assess whether HNF4A/TRIB1 can directly interact in vivo, we employed the BirA biotin ligase system of Roux et al.25 which relies on the ability of a fused BirA to tag vicinal proteins with biotin: presence of biotin in a target reflects its propensity to interact with a BirA fusion bait. TRIB1 tagged at its N-terminus with the BirA moiety (or BirA alone) was co-transfected with a HNF4A expression vector in HEK293T cells, which do not express endogenous HNF4A. Transfection of HNF4A in 293 T cells resulted in the appearance of two major bands at ~48 and 42 kDa, as assessed by Western blot (Fig. 5A). Cells were lysed under denaturing conditions to prevent ex vivo biotinylation and the lysates were fractionated using streptavidin beads under stringent conditions. Western blot analyses revealed that co-transfection with BirA-TRIB1 resulted in the presence of biotinylated HNF4A in the isolate. By comparison, transfection of either control BirA or an empty plasmid resulted in weaker reactive bands. Significantly, unlike seen with BirA-TRIB1 derived lysates, the streptavidin-bound material from either control did not exhibit evident biotinylation of HNF4A as indicated by the absence of matching streptavidin signal. Thus, presence of the TRIB1 moiety permits HNF4A biotinylation, presumably reflecting the formation of TRIB1-HNF4A complexes in situ.
Figure 5.
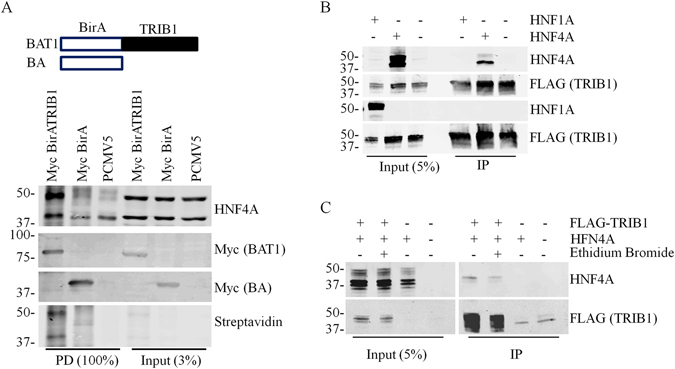
TRIB1 and HNF4A form a complex. (A), Constructs coding for myc-tagged BirA alone or inserted N terminal of TRIB1 were co-transfected with HNF4A expression plasmids in HEK293T for 24 h. Following an additional 24 h growth in media supplemented with 0.1 mM biotin, cells were harvested and lysed under denaturing conditions. Biotinylated targets were then isolated using streptavidin beads and analyzed by Western blotting using cognate antibodies followed by streptavidin coupled to IRDye800CW. (B), Plasmids encoding either HNF4A or HNF1A were transfected alongside FLAG-tagged TRIB1 plasmids in HEK293T cells. Potential complexes were isolated with FLAG-specific immunobeads and analyzed by Western blotting. (C), Interaction is independent of DNA binding. Ethidium bromide (0.2 mg/ml) was included during the immunoprecipitation. Samples were analyzed by Western blotting using the indicated antibodies.
Interaction of TRIB1 with HNF4A is specific and DNA-independent
To obtain additional evidence supporting the formation of complexes in mammalian cells and to gauge the relative strength of the interaction, CoIP experiments were then performed on HEK293T cellular lysates. HEK293T cells were transfected with HNF4A and/or TRIB1 as indicated. HEK293T transiently expressing FLAG-tagged TRIB1 and either HNF4A or HNF1A, a structurally unrelated transcription factor, were subjected to FLAG immunoprecipitation. HNF4A, but not HNF1A, was isolated by the procedure, indicating that TRIB1 and HNF4A can form a specific complex in cellular extracts (Fig. 5B). Moreover the isolation resisted ethidium bromide, an indication that dsDNA binding is not required for complex formation (Fig. 5C).
Interaction of with HNF4A involves multiple TRIB1 interfaces
Next, we explored TRIB1/HNF4A interaction modalities using recombinant TRIB1 protein. GST pull-downs assays performed in HuH-7 and HepG2 cells confirmed the suitability of the method: recombinant TRIB1 could interact with endogenous HNF4A from both cell types (Suppl. Fig. 5). Next, the interaction interface on TRIB1 was investigated using pull-down assays on HEK293T transfected with HNF4A; this approach yielded higher HNF4A levels than could be obtained from either HepG2 or HuH-7 cells thereby rendering the analyses more robust. Contributions of the better characterized interaction epitopes on TRIB1, i.e. the COP1 and the MEK1 binding sites were first evaluated. Mutating either site did not prevent HNF4A association indicating that these regions are not required for binding to HNF4A although deleting the MEK1 interaction interface impaired binding (Fig. 6A). Furthermore, a gain of function mutation (L107R)26 had no detectable impact on the interaction.
Figure 6.
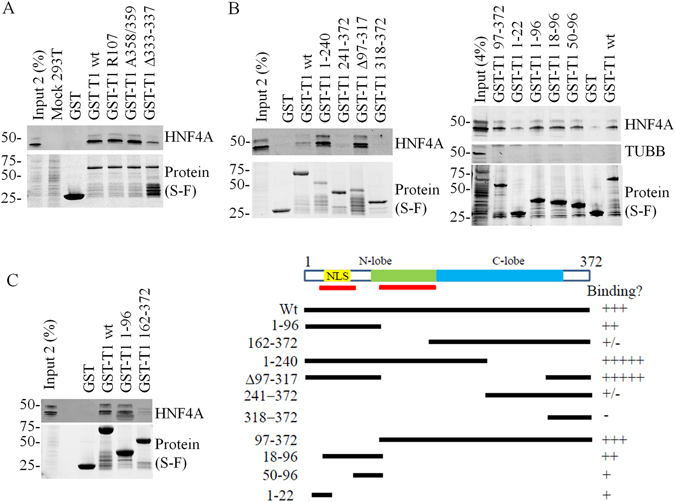
Mapping of the TRIB1 interface associating with HNF4A. Recombinant TRIB1 or fragments thereof, harboring the indicated mutations or deletions were incubated in the presence of HNF4A-expressing HEK293T lysates. A, Interaction between TRIB1 and HNF4A does not require MEK1 or COP1 binding interface, nor is it altered by a gain-of-function mutation. B and C, Multiple TRIB1 epitopes interact with HNF4A in vitro. Bound material was isolated and analyzed by Western blot. Corresponding protein baits were imaged prior to transfer using Stain Free gels. Schema illustrates truncations employed and their relative binding to HNF4A.
The epitope on TRIB1 responsible for its interaction with RAR, a nuclear receptor distantly related to HNF4A, has been mapped to a large central region of TRIB1 comprising the pseudokinase domain24. As this domain is highly conserved among the TRIBBLES, this suggested that other TRIBBLES may also interact with HNF4A. This was tested using TRIB1, TRIB2 and TRIB3 (Suppl Fig. 6). Interestingly all 3 TRIBBLES were found to interact with HNF4A although TRIB1 and TRIB3 displayed higher binding after correcting for the amount of bait. Unexpectedly, unlike observed previously with RAR, the presence of an intact pseudokinase domain is not essential as a TRIB1 construct missing the core pseudokinase region (Δ97–317) still interacted with HNF4A (Fig. 6B). In addition the C-terminal region does not appear to be involved as a construct spanning AA318–372 did not interact with HNF4A; this was confirmed by GST pull-down where region 1–240 was sufficient for binding.
The interaction picture is however complex. First of all, multiple epitopes are involved as we noted that the C-terminal half (241–372 and 162–372) could weakly interact with HNF4A (Fig. 6B and C). Furthermore the interaction was enhanced by removing the C-terminus (1–240 vs. Wt), suggesting that in the intact protein the C-terminal region of TRIB1 plays a dominant role in reducing net binding, perhaps by masking a high affinity binding site and providing an alternate, weaker binding site. Refining the interaction requirements narrowed down an interaction epitope in the N-terminus but also demonstrated that region 97–372 could interact with HNF4A, confirming that presence of at least two distinct interfaces (Fig. 6B ). Finally, pull-down assays performed on HepG2 nuclear lysates confirmed that a fragment spanning AA1–97 was sufficient for HNF4A binding (Suppl Fig. 7).
Mapping of the HNF4A region interacting with TRIB1
Earlier pull-downs indicated that transfected full-length HNF4A resulted in two distinct bands migrating as ~48 kDa and ~42 kDa that were systematically co-isolated by TRIB1 pull-downs. While the slower form was consistent with its predicted mass, we hypothesized that the second form may stem from an alternative translation initiation event. This hypothesis was based on the observation that 1) the epitope recognized by the antibody is located at the very C-terminus of HNF4A, narrowing down mass differences to the N-terminus and 2) initiation at the next methionine (Met84) is predicted to yield a product of ~42 kDa. Indeed, when HNF4A was expressed as three distinct fragments spanning the entire protein, fused to CFP, only the N-terminus (including part of the hinge region) displayed affinity for TRIB1 (Fig. 7A). Further deletions within the N-terminal fragment revealed a progressive loss of interaction (Fig. 7B). Thus the entire N-terminal region of HNF4A is required to maximize binding.
Figure 7.
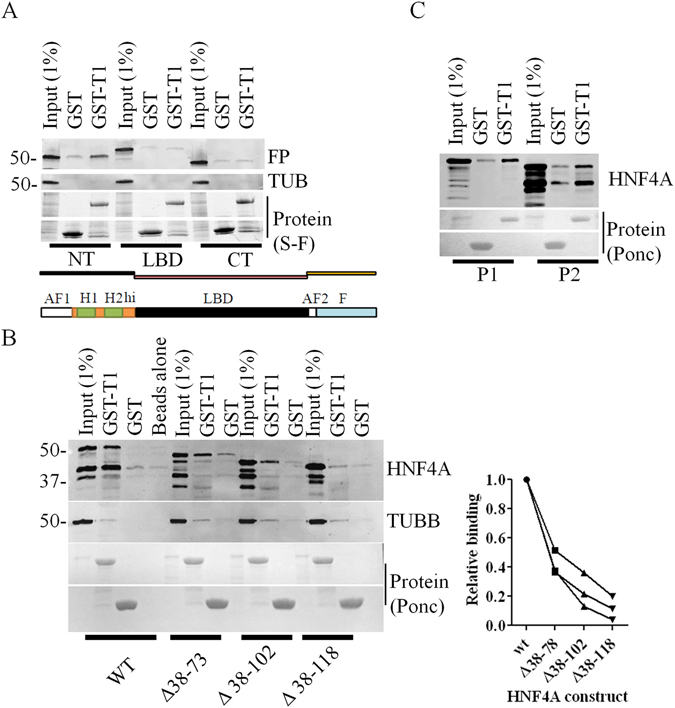
HNF4A interacts with TRIB1 via its N-terminus. Fragments (A) or deletions (B) of the HNF4A P2 variant or the full-length P1 or P2 variants of HNF4A (C) were expressed in HEK293T cells and subjected to pull-downs using either GST or GST-TRIB1 (GST-T1). Results are representative of three independent pull-down assays. In B, quantification of three distinct pull-downs (±S.D) is shown; quantification was performed using the high molecular band of each form and is expressed relative to Wt binding (see Methods). Total protein was resolved on a 4–15% gradient (A) or a 10% SDS-PAGE gels (B,C). Proteins were imaged using either Stain-Free methodology (“S-F”, A) or Ponceau (“Ponc”, B and C).
Several isoforms of HNF4A, which result from alternative splicing/promoter usage and reflect tissue specific functions, have been reported. For our original mapping efforts, the shorter P2 form of HNF4A (corresponding to HNF4α7) was chosen to simplify mapping, under the assumption that, by similarity with RAR, HNF4A interacted with TRIB1 via its activation domains. The two HFN4A isoforms share most of their sequence with the exception of the N-termini. In view of the contribution of the HNF4A N-terminal region for its interaction with TRIB1, its relative affinity for both forms (HNF4α7 and α2) was tested in pull-down assays. When expressed in 293 T cells both forms bound TRIB1 (Fig. 7C).
CEBPA and HNF4 interact with TRIB1 via distinct epitopes
Finally we asked whether TRIB1 displayed exclusive or additive interactions with CEBPA and HNF4A. The evidence of multiple interacting regions on TRIB1 would suggest a possible non-exclusive binding model whereby TRIB1 could interact with both proteins simultaneously. First the ability of TRIB1 to interact with CEBPA was confirmed (Suppl Fig. 8). Interestingly HNF4A and CEBPA exhibited similar binding affinities to TRIB1 as expressed relative to input signal.
To probe the interfaces involved, competition assays employing peptides derived from CEBPA were optimized. This approach took advantage of the recent identification of a region of CEBPA sufficient for binding TRIB127. To overcome solubility limitations, targeted non-essential point mutations were introduced first and the resulting peptide, together with a control peptide containing key mutations at conserved residues, were tested for their ability to prevent TRIB1/CEBPA interaction. Inclusion of the CEBPA-derived peptide, but not the control, reduced co-isolated CEBPA (Fig. 8A). However, when tested for their impact on HNF4A, neither peptide affected HNF4A binding with TRIB1, consistent with the utilization of distinct TRIB1 interfaces (Fig. 8B).
Figure 8.
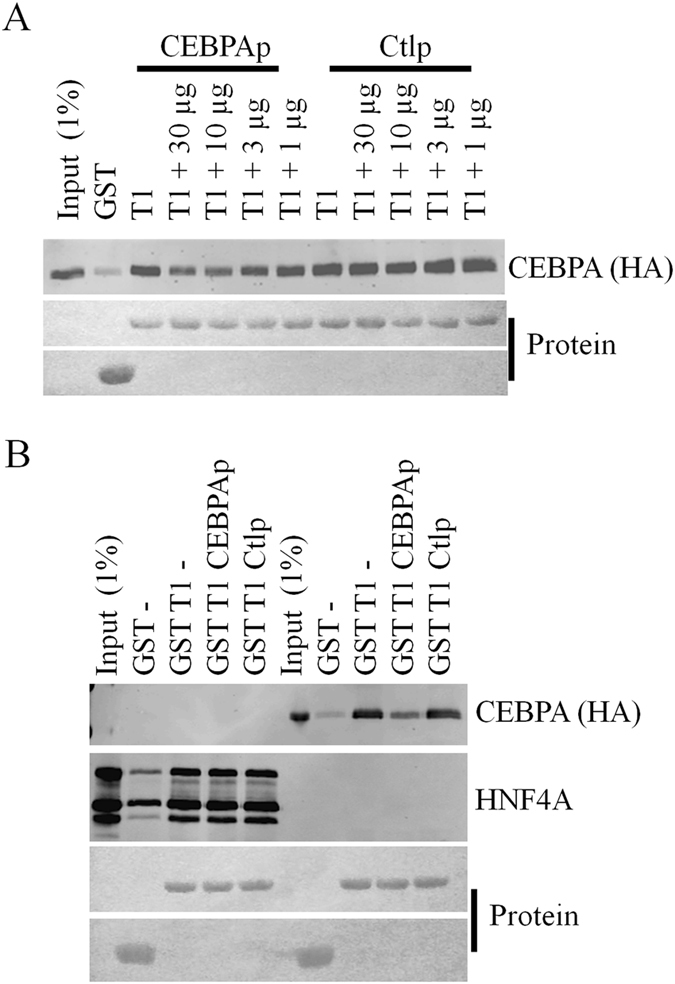
A CEBPA derived peptide interferes with CEBPA binding, but not HNF4A binding, to GST-TRIB1. Pull-down assays of CEBPA-HA and HNF4A. A, lysates of HEK293T transfected with plasmids coding for CEBPA-HA were incubated with GST-TRIB1 or GST in the presence of increasing concentrations of either a peptide matching the CEBPA interface (CEBPAp) or a control (Ctlp), substituted peptide. CEBPA in isolates was quantified by Western blotting and protein input by Ponceau. B, as in A except that lysates expressing either HNF4A or CEBPA were used in each pull-down assay. Where indicated, 10 µg of either peptide were used. Experiments were performed twice with comparable results.
Discussion
Here we have explored the functional relationship between HNF4A and the TRIBBLES, focusing on TRIB1. Our previous work revealed that HNF4A suppression led to TRIB1 upregulation and that TRIB1 suppression using two distinct approaches (siRNA and ASO) targeting different regions of the TRIB1 transcript reduced HNF4A levels and function11. This suggested the presence of a regulatory network involving both proteins. We now demonstrate that TRIB1 suppression and TRIB1 overexpression respectively reduce and increase HNF4A activity. In addition we demonstrate that the other two mammalian TRIBBLES are also required for optimal HNF4A protein expression, although their contributions are more limited in that regard. Although off-target effects remain possible and should be excluded by using additional siRNA targeting distinct regions of TRIBBLES transcripts, the finding that all three TRIBBLES siRNA reduce HNF4A protein expression reinforce the notion that individual TRIBBLES are contributing to maintain optimal HNF4A levels.
In addition to demonstrating a requirement for the TRIBBLES, our results reveal that the three TRIBBLES can physically associate with HNF4A in vitro; further work will be needed to assess whether TRIB2 and TRIB3 can directly interact with HNF4A in vivo. While we demonstrate the presence of an in vivo complex between (overexpressed) TRIB1 and HNF4A, contributions of this complex to HNF4A function remain unclear. Judging from the low abundance of TRIB1 relative to HNF4A, inferred from both qRT-PCR (~60 X difference) and Western blot evidence, the endogenous TRIB1 is predicted to sequester only a small fraction of the HNF4A population at any given time. Thus TRIB1, and perhaps all the TRIBBLES, operate in part by steering subpopulations of HNF4A towards particular transcriptional programs. HNF4A is unique among the nuclear factor superfamily in that coactivator binding, rather than ligand binding (which is necessary but not sufficient) locks its active conformation28. Binding of TRIB1 to its N-terminal, which includes its DNA binding domain and part of the hinge region linking it to the LBD, is predicted to affect its ability to interact with cognate DNA sequences. As TRIB1 overexpression was associated with increased transcriptional output on a reporter system, anchoring of TRIB1 on HNF4A is predicted to augment its activity. Alternatively, cellular changes secondary to TRIB1 overexpression may be invoked. Nonetheless, binding to the N-terminal region of TRIB1 leaves open the possibility for the formation of higher order complexes comprised of HNF4A and other interactors identified over the years, which have been shown to bind elsewhere on TRIB1 (e.g. CEBPA, CEBPB, MEK1, MLXIPL, RARA, RXRA, SAP18). TRIB1 (and the TRIBBLES in general) may thus serve as a transcription modulator that operates by integrating converging transcription programs.
HNF4A stands out amongst the TRIB1 interactors given that its expression correlates positively with that of TRIB1. This may reflect in part distinct binding modalities. The observation that binding of HNF4A to TRIB1 resists competition with the CEBPA peptide is consistent with this model. Thus binding by HNF4A does not, unlike CEBPA association, lead to its degradation. Unfortunately, limited additional information can be gleaned from the relatively better characterized CEBPA/TRIB1 interaction picture as it still unclear where CEBPA docks on the TRIB1 surface. Some binding characteristics have been defined however: a requirement for the pseudokinase domain, an intact pseudocatalytic loop as well as the MEK binding interface (but not the more distal COP1 binding region)27, 29. Interestingly we observed a reduction in binding when the MEK binding region was deleted. As the C-terminal tail is not needed for binding, we speculate that the deletion may lead to its inappropriate positioning over the interaction interface located close to the N terminus, which accords with the ability of the C-terminal tail to fold back onto the N-terminus of the protein27.
Our findings are consistent with a model (Suppl Fig. 9) whereby TRIB1 is required to maintain a suitable cellular environment for proper HNF4A expression and function. In the absence of TRIB1 (and TRIBBLES), ensuing changes lead to reduced HNF4A levels. How this is achieved is likely multi-faceted as HNF4A was reduced either in the absence of significant transcript change (HepG2) or its presence (HuH-7 and primary human hepatocytes)11. As TRIB1 overexpression did not measurably increase HNF4A protein level, this further suggests that its impact on HNF4A stability is indirect or requires limiting factors that remain to be identified. Judging from the profound impact of silencing of TRIB1 in various liver models, numerous pathways could be affected. One of these may involve the MEKs which orchestrate a plethora of signaling cascades and which constitute TRIBBLES regulatory targets. The TRIBBLES exhibit a complex regulatory relationship with the MAPKKs30, 31 and their absence would be predicted to influence MAPKK activity, in turn affecting HNF4A. Indeed, activation of MEK1/2 by IL1β has been reported to attenuate HNF4A levels, by reducing transcript level as well as promoting protein degradation32. In addition, HNF4A has been reported to be destabilized by successive SUMOylation and ubiquitination events during hepatocyte maturation33. By interfering with these processes, TRIB1 could be instrumental in helping to sustain higher basal HNF4A levels.
There is now considerable evidence pointing to a convergence of HNF4A and TRIB1 in regulating energy metabolism. Both proteins have been linked to glucose metabolism in the liver. HNF4A is required for gluconeogenesis, a role it performs in conjunction with the PPAR co-activator PPARGC1A (PGC-1alpha)34. Recent work has uncovered a role for TRIB1 in the regulation of circulating glucose levels in the mouse29. Moreover TRIB1 suppression is associated with the widespread downregulation of enzymes involved in glucose metabolism in human primary hepatocytes11. HNF4A and TRIB1 have also been associated with altered lipid traits in humans35, 36. A large GWAS identified a missense mutation in HNF4A that correlates with reduced HDL levels35 and common non-coding variants within the HNF4A gene have been linked to elevated plasma lipids37. TRIB1 polymorphisms located ~30 kb away have been linked to changes in plasma triglycerides, LDL-cholesterol and HDL-cholesterol7, 38–40. A direct contribution of TRIB1 is also supported by animal studies demonstrating that circulating plasma lipids and Trib1 levels are inversely correlated10, 29, 41, 42. This effect may be due in part due to its effect on MTTP encoding the microsomal triglyceride transfer protein, which is under the transcriptional control of HNF4A and is essential for VLDL lipidation and secretion43. Indeed, MTTP expression is proportional to that of TRIB1 44. Mice lacking hepatic Hnf4a show a dramatic reduction in Mttp expression and exhibit severe reductions in circulating cholesterol and triglyceride levels, and hepatic lipid accumulation (hepatosteatosis)18. Significantly, TRIB1-linked SNPs are associated with nonalcoholic fatty liver disease (NAFLD), defined as hepatosteatosis29. This phenotype is also observed in liver-specific Trib1 knockout mice and cannot be phenocopied by CEBPA overexpression, consistent with a role for TRIB1 in orchestrating non-CEBPA related functions10. Here we establish that the TRIBBLES, and TRIB1 in particular are positive regulators of HNF4A, supporting the hypothesis that reduced HNF4A, may in part account for the CEBPA-independent effects of TRIB1 suppression.
Methods
Cell culture and treatments
HepG2 and HEK293T cells were obtained from ATCC (www.atcc.org) and maintained in low or high glucose DMEM (LG-DMEM/HG-DMEM), respectively. HuH-7 is a human differentiated hepatocellular carcinoma (JCRB cell bank). SiRNA treatments were performed for 48–72 h at 20 nM, using 2 ul of RNAiMax per 12 well plate well; siRNAs are listed in the Supplementary Materials section.
Quantitative Real-Time RT-PCR and qPCR arrays
RNA was isolated using a High Pure RNA isolation kit (Roche) and reverse transcribed. Quantitative PCR was performed on a LightCycler480 using the Roche LightCycler480 SybrGreen I Master mix (Roche). Total RNA (0.5–1 μg) was reverse-transcribed with the Transcriptor First Strand cDNA synthesis kit (Roche Diagnostics) using a combination of oligo-dT and random hexamers. Crossing point (Cp) values for each sample were first normalized to PPIA. Oligonucleotides used for Quantitative Real-Time RT-PCR (qRT-PCR) are listed in the Supplementary Materials section.
Expression constructs
Human TRIB1 (372 aa) was subcloned using PCR and restriction digests from Origene’s construct (PCMV6-XL5TRIB1). TRIB1 was cloned in pLVX (HepG2 and HuH-7 stables) or PLVXHIS (in frame with a C-terminal HIS tag), pCMV-Tag (transfection experiments) or pGEX4 T (bacterial expression). TRIB3 was amplified from liver cDNA using Q5 polymerase. The P2 form of HNF4A (α7; NP_001025174)) and HNF1A were amplified from human liver cDNA and cloned in pLVX. HNF4A (α2; NP_000448), TRIB2 and CEBPA constructs were purchased from Bio Basic Inc. The CEBPA coding sequence, corresponding to the p42 form (Uniprot identifier P49715-2) was optimized to reduce its GC content using IDT’s (http://www.idtdna.com/site) proprietary tool. All three constructs contained C-terminal spacers and HA tags. PCR and Mutagenesis were performed using the Q5 high fidelity polymerase as per the supplier’s (New England Biolab) instructions. For the BirA system, the mycBioID plasmid was a gift from Kyle Roux obtained via Addgene (Addgene plasmid 35700). TRIB1 was cloned in C-terminal to the mycBioID tag using conventional molecular biology methods. Integrity of the constructs was confirmed by sequencing of the open reading frames.
Protein interaction studies
Recombinant proteins were isolated 48 h after transfections with either Fugene 6 (Promega) or Lipfectamine 3000 (ThermoFisher) and the appropriate expression constructs. Cells were rinsed in PBS, scraped and lysed for 5 min in ice cold IP buffer (20 mM HEPES, 120 mM NaCl, 0.5% NP40, pH 7.4) containing phosphatase and protease inhibitors cocktails (Roche Life Science). Insoluble debris were cleared by centrifugation (1 min, 15,000 x g) and complexes were snap-frozen to be used as a source of HNF4A for pull-down experiments or used fresh for coIP experiments. For bacterial expression (Rosetta2 strains, Novagen), fusion proteins were cloned in pGEX4T1. Overnight bacterial cultures were diluted 1:100 in 100 ml fresh LB broth (+Ampicillin) and bacteria were grown until OD600 of ~0.7 and induced for 3 h with 0.2 mM isopropyl beta-D-thiogalactoside at 30 °C. Bacteria were recovered in 5 ml of 25 mM Tris-HCl, 0.15 M NaCl, 1% Triton X-100, 0.5 mM EDTA, pH 8 and sonicated in a Bioruptor (Diagenode). Bacterial debris were removed by centrifugation and fusion proteins were bound to GST agarose beads (GE Health Care Life Sciences) and washed in IP buffer. Pull-downs were then performed using 0.2 mg of HEK293T lysates transfected with either HNF4A or CEBPA (snap frozen and stored at −80 °C) or HuH-7 lysed in 0.5 ml of IP buffer and 10 µl of GST bead volume. For pull-down assays, prey binding was first corrected for the amount of bait and then divided by input intensity; binding was then normalized to the wild-type form.
Peptide competition assays were performed using either a CEBPA derived peptide (Biotin-SEICEDENSIDISAYIDPAAFND) with conservative point mutations to facilitate synthesis, or a control peptide harboring damaging mutations (Biotin-GGACEHATSDDASAYADPAAFND) (Novopep, Shanghai, China). Peptides were added concurrently with the prey lysates.
For BioID tagging, 6 well plates of HEK293T were co-transfected with 2 µg per well with HNF4A (1.5 µg per well) and either empty plasmid (0.5 µg), BirA (0.1 µg) or BirATRIB1 (0.5 µg) for 48 h. During the last 24 h of transfection, medium was replaced with fresh medium containing biotin (0.1 mM). Cells were quickly rinsed and recovered in ice cold PBS, resuspended briefly and hot (95 °C) lysis buffer (50 mM Tris, pH 7.4, 200 mM NaCl, 1% SDS, 5 mM EDTA, 1 mM DTT) was added to the samples. Lysates were mixed well and transferred to 95 °C for a further 10 min with occasional mixing. Samples were then diluted 5 X in dilution buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 0.25% TRITON X-100, 5 mM EDTA) and sonicated 2 X 5 min (30 /30 cycles) in a Bioruptor (Diagenode). Insoluble material was removed by centrifugation at 15,000 x g for 10 min and the supernatant was incubated with Streptavidin-coupled magnetic beads (Life Technologies) for 1 h. Samples were washed 4 X in dilution buffer and analyzed by Western blot. Streptavidin (IRdye800CW; LI-COR) diluted at 1:50,000 in PBS was used to detect the biotin-modified protein.
Western blotting, immunocytochemistry and immunoprecipitation
Cellular extracts were obtained by the addition of RIPA buffer (50 mM Tris-HCl, 1% NP40, 0.1% SDS, 0.5% NaDeoxycholate, 0.15 M NaCl, pH 8.0) supplemented with Complete protease inhibitors and PhoStop tablets (Roche) for 5 min to the permeabilized cells on ice. Western blotting was performed on ~30 µg per sample or isolate, as appropriate,that was resolved under reducing and denaturing conditions on TGX 4–15% polyacrylamide gels (Bio-Rad). Where indicated Stain-Free pre-cast gels (Bio-Rad) were used and imaged for total protein content prior to transfer using tryptophan-trihalo fluorescence. Proteins were transferred to nitrocellulose, blocked in PBS/5% milk or Odyssey Blocking buffer (Li-Cor) for 20 min prior to detection. Detection was performed for 16 h using 1:2000 dilutions of the primary antibodies and cognate LI-COR secondary antibodies (1:25,000 dilutions in PBS). All washes were in PBS (4 × 5 min). Imaging was performed on an Odyssey Imager (LI-COR).
For immunocytochemistry, cells seeded on glass coverslips were transfected for 48 h with HNF4A and/or TRIB1 constructs and fixed in 4% PFA for 15 min. Permeabilization was achieved with 0.1% Triton X-100 in PBS for 10 min and followed by a blocking step in 5% bovine serum/PBS (10 min) and incubation for 1 h with antibodies diluted at 1:500 in PBS. Alexa-coupled secondary antibodies were used (Donkey anti-Goat 633 and Donkey anti-Rabbit 546; Life Technologies) at 1:2,000 dilution. Image acquisition was performed using an Olympus confocal microscope equipped with a 100 X oil immersion lens and operated by the FluoView software. Image processing was performed using the FluoView viewer software with subsequent minor adjustments in Microsoft PowerPoint. Antibodies used are listed in the Supplementary Materials section.
Immunoprecipitation of TRIB1 was performed on denatured lysates. HuH-7 cells were treated for 48 h with siRNAs, washed in PBS, harvested by scraping and lysed in 1 ml (per two 10 cm dishes) of PBS/0.1% Triton X-100 supplemented with Complete protease inhibitors (Roche) for 5 min with rotation; using flagTRIB1 as a guide, optimization indicated that this condition was sufficient to recover significant (~10% of total) TRIB1. Samples were then centrifuged and the soluble fractions were recovered, adjusted to 1% SDS and heated at 95 °C for 5 min. Following a dilution in four volumes of 25 mM Tris-HCl, 0.15 M NaCl, 1% Triton X-100, 0.5 mM EDTA, pH 8 to displace the SDS from the protein surface and allow the formation of antigen-antibody complexes45, the lysates were cleared through 0.4 µM PVDF disks. The filtrates were then subjected to immunoprecipitation for 16 h at 4 °C using 10 µl of a rabbit antibody to TRIB1 (N2C3, Genetex) and 20 µl of rinsed protein A/G mix (PureProteome Magnetic Beads, Millipore). Detection was performed using a goat polyclonal antibody to TRIB1 (Genetex).
Statistical analyses
To assess statistical significance of changes relative to a control value (e.g. Non-Target oligonucleotide), unpaired 2-tailed student’s t-tests were performed unless mentioned otherwise. For in-between group comparisons, one way ANOVA was performed, followed by Tukey’s post-hoc test in GraphPad Prism 5.
Electronic supplementary material
Acknowledgements
We would like to thank MM Hussain (State University of New York) for the MTTP promoter constructs. We are indebted to Paulina Lau for her technical support and critical input. This research was supported by the Heart & Stroke Foundation of Canada # T-7268 and the Canadian Institutes of Health Research.
Author Contributions
A.M. and S.S. designed, performed and analyzed experiments; R.M. and S.S. wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05768-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sébastien Soubeyrand, Email: ssoubeyrand@ottawaheart.ca.
Ruth McPherson, Email: rmcpherson@ottawaheart.ca.
References
- 1.Bailey FP, et al. The Tribbles 2 (TRB2) pseudokinase binds to ATP and autophosphorylates in a metal-independent manner. Biochem. J. 2015;467:47–62. doi: 10.1042/BJ20141441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eyers, P. A. et al. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 686–693, doi:10.1016/j.tcb.2016.11.002 (2016). [DOI] [PMC free article] [PubMed]
- 3.Dugast, E., Kiss-Toth, E., Soulillou, J. P., Brouard, S. & Ashton-Chess, J. The Tribbles-1 protein in Humans: roles and functions in health and disease. Curr. Mol. Med (2012). [PubMed]
- 4.Lohan F, Keeshan K. The functionally diverse roles of tribbles. Biochem. J. 2013;41:1096–1100. doi: 10.1042/BST20130105. [DOI] [PubMed] [Google Scholar]
- 5.Nurnberg ST, et al. From Loci to Biology. Circ. Res. 2016;118:586–606. doi: 10.1161/CIRCRESAHA.115.306464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speliotes EK, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits 148. PLoS.Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Douvris A, et al. Functional analysis of the TRIB1 associated locus linked to plasma triglycerides and coronary artery disease. J. Am. Heart Assoc. 2014;3:e000884. doi: 10.1161/JAHA.114.000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stylianou IM, Bauer RC, Reilly MP, Rader DJ. Genetic basis of atherosclerosis: insights from mice and humans. Circ.Res. 2012;110:337–355. doi: 10.1161/CIRCRESAHA.110.230854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dedhia PH, et al. Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood. 2010;116:1321–8. doi: 10.1182/blood-2009-07-229450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer RC, et al. Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPα. J. Clin. Invest. 2015;125:3809–3818. doi: 10.1172/JCI77095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soubeyrand S, Martinuk A, Naing T, Lau P, Mcpherson R. Role of Tribbles Pseudokinase 1 (TRIB1) in human hepatocyte metabolism. Biochim. Biophys. Acta. 2016;1862:223–232. doi: 10.1016/j.bbadis.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Sladek FM. What are nuclear receptor ligands? Mol Cell Endocrinol. 2011;334:3–13. doi: 10.1016/j.mce.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sladek FM, Zhong WM, Lai E, Darnell JE. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990;4:2353–65. doi: 10.1101/gad.4.12b.2353. [DOI] [PubMed] [Google Scholar]
- 14.Babeu JP, Boudreau F. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol. 2014;20:22–30. doi: 10.3748/wjg.v20.i1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin L, Ma H, Ge X, Edwards PA, Zhang Y. Hepatic hepatocyte nuclear factor 4alpha is essential for maintaining triglyceride and cholesterol homeostasis. Arterioscler.Thromb.Vasc.Biol.. 2011;31:328–336. doi: 10.1161/ATVBAHA.110.217828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue Y, Yu AM, Inoue J, Gonzalez FJ. Hepatocyte nuclear factor 4alpha is a central regulator of bile acid conjugation. J Biol Chem. 2004;279:2480–2489. doi: 10.1074/jbc.M311015200. [DOI] [PubMed] [Google Scholar]
- 17.Dai K, Hussain MM. NR2F1 disrupts synergistic activation of the MTTP gene transcription by HNF-4alpha and HNF-1alpha. J.Lipid Res. 2012;53:901–908. doi: 10.1194/jlr.M025130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. 2001;21:1393–1403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheena V, et al. Transcriptional regulation of human microsomal triglyceride transfer protein by hepatocyte nuclear factor-4alpha. J Lipid Res. 2005;46:328–341. doi: 10.1194/jlr.M400371-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Santangelo L, et al. The stable repression of mesenchymal program is required for hepatocyte identity: a novel role for hepatocyte nuclear factor 4alpha. Hepatol. 2011;53:2063–2074. doi: 10.1002/hep.24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonzo JA, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J Biol.Chem. 2012;287:7345–7356. doi: 10.1074/jbc.M111.334599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stefflova K, et al. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell. 2013;154:530–40. doi: 10.1016/j.cell.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soubeyrand S, Martinuk A, Lau P, McPherson R. TRIB1 Is Regulated Post-Transcriptionally by Proteasomal and Non-Proteasomal Pathways. PLoS One. 2016;11:e0152346. doi: 10.1371/journal.pone.0152346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imajo M, Nishida E. Human Tribbles homolog 1 functions as a negative regulator of retinoic acid receptor. Genes Cells. 2010;15:1089–97. doi: 10.1111/j.1365-2443.2010.01445.x. [DOI] [PubMed] [Google Scholar]
- 25.Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J.Cell Biol. 2012;196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yokoyama T, et al. Identification of TRIB1 R107L gain-of-function mutation in human acute megakaryocytic leukemia. Blood. 2012;119:2608–2611. doi: 10.1182/blood-2010-12-324806. [DOI] [PubMed] [Google Scholar]
- 27.Murphy, J. M. et al. Molecular Mechanism of CCAAT-Enhancer Binding Protein Recruitment by the TRIB1 Pseudokinase. Structure 1–11, doi:10.1016/j.str.2015.08.017 (2015). [DOI] [PubMed]
- 28.Duda K, Chi Y-I, Shoelson SE. Structural basis for HNF-4alpha activation by ligand and coactivator binding. J. Biol. Chem. 2004;279:23311–6. doi: 10.1074/jbc.M400864200. [DOI] [PubMed] [Google Scholar]
- 29.Ishizuka Y, et al. TRIB1 down-regulates hepatic lipogenesis and glycogenesis via multiple molecular interactions. J.Mol Endocrinol. 2014;52:145–158. doi: 10.1530/JME-13-0243. [DOI] [PubMed] [Google Scholar]
- 30.Kiss-Toth E, et al. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J.Biol.Chem. 2004;279:42703–42708. doi: 10.1074/jbc.M407732200. [DOI] [PubMed] [Google Scholar]
- 31.Guan H, et al. Competition between members of the tribbles pseudokinase protein family shapes their interactions with mitogen activated protein kinase pathways. Sci. Rep. 2016;6:32667. doi: 10.1038/srep32667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simó R, Barbosa-Desongles A, Hernandez C, Selva DM. IL1β Down-regulation of Sex Hormone-Binding Globulin Production by Decreasing HNF-4α Via MEK-1/2 and JNK MAPK Pathways. Mol. Endocrinol. 2012;26:1917–1927. doi: 10.1210/me.2012-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou W, et al. SUMOylation of HNF4α regulates protein stability and hepatocyte function. J. Cell Sci. 2012;125:3630–5. doi: 10.1242/jcs.102889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon JC, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 35.Kathiresan S, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat.Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weissglas-Volkov D, et al. Common hepatic nuclear factor-4alpha variants are associated with high serum lipid levels and the metabolic syndrome. Diabetes. 2006;55:1970–7. doi: 10.2337/db06-0035. [DOI] [PubMed] [Google Scholar]
- 38.Aung LHH, et al. Association of the TRIB1 tribbles homolog 1 gene rs17321515 A > G polymorphism and serum lipid levels in the Mulao and Han populations. Lipids Health Dis. 2011;10:230. doi: 10.1186/1476-511X-10-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park MH, Kim N, Lee JY, Park HY. Genetic loci associated with lipid concentrations and cardiovascular risk factors in the Korean population. J.Med.Genet. 2011;48:10–15. doi: 10.1136/jmg.2010.081000. [DOI] [PubMed] [Google Scholar]
- 40.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat.Genet. 2008;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burkhardt R, et al. Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J.Clin.Invest. 2010;120:4410–4414. doi: 10.1172/JCI44213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bauer RC, Yenilmez BO, Rader DJ. Tribbles-1: a novel regulator of hepatic lipid metabolism in humans. Biochem. Soc. Trans. 2015;43:1079–1084. doi: 10.1042/BST20150101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chamberlain AJ, et al. Extensive variation between tissues in allele specific expression in an outbred mammal. BMC Genomics. 2015;16:993. doi: 10.1186/s12864-015-2174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makishima S, et al. Sin3A-associated protein, 18 kDa, a novel binding partner of TRIB1, regulates MTTP expression. J. Lipid Res. 2015;56:1145–1152. doi: 10.1194/jlr.M057802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dimitriadis GJ. Effect of detergents on antibody-antigen interaction. Anal. Biochem. 1979;98:445–51. doi: 10.1016/0003-2697(79)90165-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.