

Abstract
Advanced colorectal cancer (CRC) remains a critical health care challenge worldwide. Various TGF-β superfamily members are important in colorectal cancer metastasis, but their signaling effects and predictive value have only been assessed in isolation. Here, we examine cross-regulation and combined functions of the two most prominent TGF-β superfamily members activin and TGF-β in advanced colorectal cancer. In two clinical cohorts we observed by immune-based assay that combined serum and tissue activin and TGF-β ligand levels predicts outcome in CRC patients and is superior to single ligand assessment. While TGF-β growth suppression is independent of activin, TGF-β treatment leads to increased activin secretion in colon cancer cells and TGF-β induced cellular migration is dependent on activin, indicating pathway cross-regulation and functional interaction in vitro. mRNA expression of activin and TGF-β pathway members were queried in silico using the TCGA data set. Coordinated ligand and receptor expression is common in solid tumors for activin and TGF-β pathway members. In conclusion, activin and TGF-β are strongly connected signaling pathways that are important in advanced CRC. Assessing activin and TGF-β signaling as a unit yields important insights applicable to future diagnostic and therapeutic interventions.
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide resulting in approximately 50,000 deaths per year in the US alone1. While the overall incidence of CRC is decreasing likely due to increased screening resulting in prevention or early detection2, the prognosis of late stage disease remains poor. Risk stratification for metastatic disease continues to be challenging. In addition there is no consensus on management for suspected early metastatic disease and use of adjuvant or post-surgery chemotherapy in stage 2 CRC remains controversial3.
Given the relatively low recurrence rates in stage 2 CRCs4, the risk benefit ratio does not favor treating all patients for suspected metastasis. However, there is a small but important subpopulation of stage 2 CRC patients that later present with metastases and likely would benefit from early systemic treatment5. Once CRC has metastasized distally, palliative and supportive approaches are the standard of care6 with a five year survival rate as low as 10%1. Therefore, both new treatment approaches and biomarkers for risk stratification for metastatic disease are needed in CRC.
TGF-β superfamily signaling is established as a crucial signaling family in CRC7, with a dominant growth suppressive function at early stages of tumorigenesis, yet parallel pro-metastatic effects at later time points8. The most involved superfamily members in sporadic CRC are TGF-β and activin, each having several ligand isoforms. In this study, we chose to focus on the most prominent isoforms of activin and TGF-β in CRC, activin A and TGFB1, respectively as they are the two cancer dominant isoforms.
After ligand binding to respective type II receptors, both signal through subsequent dimerization with and phosphorylation of their specific type I receptors9 (also called activin receptor like kinases or ALKs) to activate shared canonical and distinct non-canonical pathways10.
In the canonical pathway phosphorylation of SMAD 2 and/or 3, is followed by dimerization with SMAD411 and SMAD2/3-SMAD4 complex translocation to the nucleus, where a substantial number of genes are either transcriptionally activated or repressed12. In contrast, a number of CRC oncogenic pathways, including but not limited to PI3K/AKT13, 14, MAPK/ERK13, 15, WNT16, and notch17, can be activated by TGF-β superfamily signaling independent of SMADs and are thus called non-canonical pathways.
We have previously reported that while activin and TGF-β share canonical signaling, in CRC they diverge in their non-canonical pathways with activin primarily signaling through PI3K/AKT while TGF-β activates MAP/ERK13. Numerous ligand isoforms, ligand promiscuity of some type I and type II receptors, and substantial crosstalk between canonical and non-canonical pathways contribute to a complex yet poorly understood system which allows context-specific and dynamic functions of TGF-β superfamily signaling10.
This is emphasized by the fact that despite a number of available compounds for TGF-β pathway inhibition18 and promising data in animal models19–21, to date no clinical benefit of targeting TGF-β signaling has been reported in solid tumors22. In vivo data showed that disruption of epithelial TGF-β receptor signaling may lead to a more invasive and aggressive CRC phenotype through unknown mechanisms23, 24 likely involving stromal signaling. Recent data from our group implies that TGF-β inhibition through neutralization of TGF-β ligand may be detrimental in a subset of CRCs25.
Given activin and TGF-β’s overlap in their canonical pathway and divergence in their downstream oncogenic signaling as well as the frequent co-occurrence of mutations in their respective receptors26–28, we here focus on understanding the mechanisms of activin and TGF-β crosstalk in metastatic signaling in CRC.
Results
An elevated combined activin/TGF-β ligand expression score is predictive of a worse prognosis in patients with CRC
Because of the overlap of the canonical and divergence of non-canonical activin and TGF-β functions, we hypothesized that both pathways need to be assessed simultaneously to comprehensively predict TGF-β superfamily effects. To test our hypothesis, we determined activin and TGF-β serum ligand levels in a subset of 120 stage II patients (60 with and 60 without recurrence) from the Quasar trial29 and correlated both single ligands and a combined ligand score with clinical outcome.
We found that ligand serum levels of activin and TGF-β significantly correlated with each other (r = 0.435, p < 0.05). Neither serum activin nor TGF-β alone significantly correlated with time to recurrence. However, a high combined activin/TGF-β score (see methods) was predictive of decreased recurrence-free survival compared to patients with low or intermediate activin/TGF-β serum levels (median recurrence free survival 415 days versus 715 days, p < 0.05, Fig. 1A), supporting our hypothesis of a combined activin/TGF-β system with pro-metastatic action.
Figure 1.
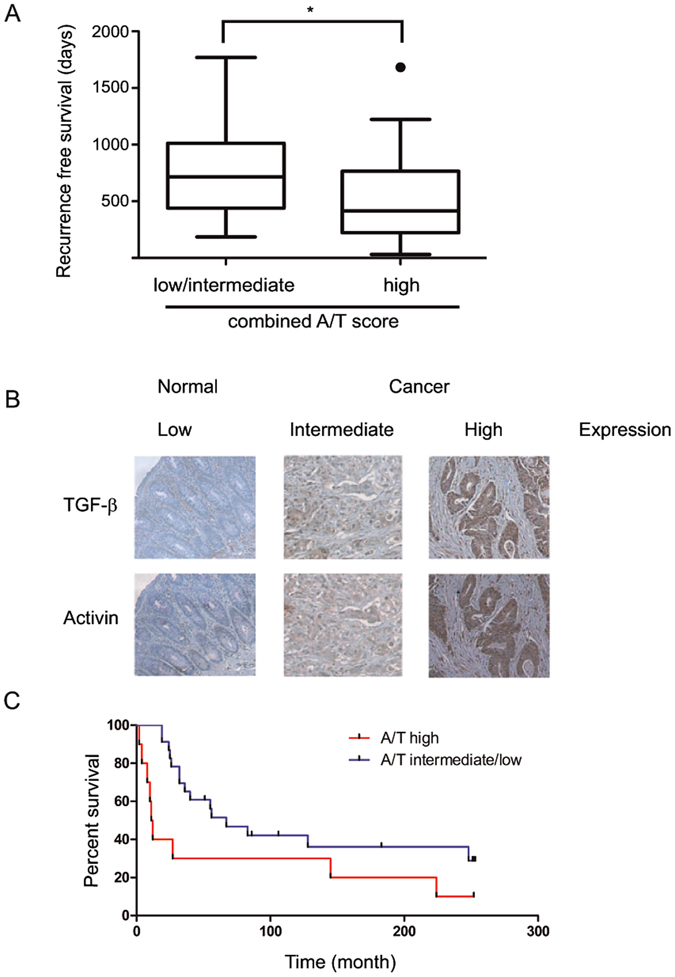
Combined activin and TGF-β ligand scores are strongly predictive of a worse prognosis in colorectal cancer. (A) Serum ligand levels are predictive of shorter recurrence-free survival. Sixty stage 2 CRC patients with recurrence were stratified by activin and TGF-b serum ligand expression and with median recurrence-free survival shown as box plots. The combined activin/ TGF-β score (A/T) is defined in the Materials and Methods. Low/intermediate is defined as a score of 4 or less and high is defined as a score of 5 or higher. (B) Tumoral activin and TGF-β expression are strongly correlated. Representative sections of immunohistochemical staining of TGF-β or activin ligand in a CRC TMA with examples of low, intermediate and high staining. Matched colon cancer sections of adjacent slides are shown to demonstrate correlation of activin and TGF-β expression. (C) Tumoral activin and TGF-β ligand are predictive of shorter overall survival. Kaplan Meier curve of patient survival in either activin/ TGF-β high (A/T) patients or A/T low as defined in the Materials and Methods.
To control for potential non-tumoral factors that may influence activin and TGF-β serum levels, we next determined tumor expression levels of the ligands. For this, we stained adjacent slides of a commercial TMA with 40 CRC, 10 CRC metastasis and 10 control samples30 for activin and TGF-β and correlated expression with each other as well as outcome (example staining, Fig. 1B). Activin and TGF-β tissue ligand expression scores correlated with each other (r = 0.604, p < 0.01). When we correlated activin and TGF-β expression with survival, activin and TGF-β combined, but not activin or TGF-β tissue ligand expression alone, was associated with statistically significant shorter survival (median overall survival 11.5 months versus 67 months, p < 0.05, Fig. 1C). Importantly, this correlation was not solely due to higher expression levels at higher stages (mean combined score stage III-IV 2.19 versus stage I-II 2.23 p = n.s.), indicating that combined tumor activin and TGF-β ligand expression provides additional information to the TNM staging classification31 regarding advanced disease.
TGF-β leads to enhanced epithelial and stromal activin secretion and resulting migration is dependent on activin
Given the strong correlation and predictive power of activin and TGF-β ligand expression in our human cohorts, we next explored the impact of TGF-β on activin secretion. As the tumor microenvironment appears essential to TGF-β superfamily signaling, we also assessed the contribution of fibroblasts to activin ligand secretion using a co-culture approach.
Stromal CCD18 and epithelial CRC FET cells were either grown separately, or in co-culture using a trans-well approach and activin ligand expression analyzed following TGF-β stimulation. After TGF-β treatment, activin secretion was increased both in SMAD4 wild-type FET and SMAD4 null SW480 colon cancer cells (Fig. 2A). Interestingly, levels of activin secretion both at baseline and after TGF-β treatment were substantially higher either in stromal CCD18 cells alone or in co-cultures of stromal with epithelial cells, indicating that the stroma is a significant source of secreted activin (Fig. 2B).
Figure 2.
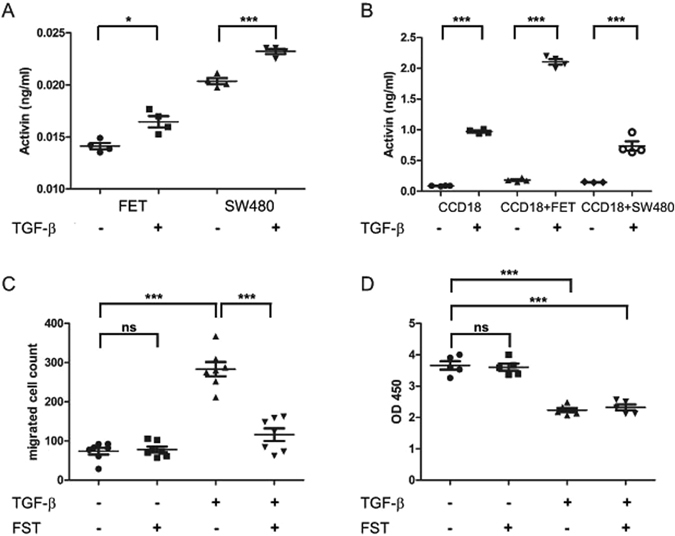
TGF-β’s pro-metastatic function is activin dependent while its anti-proliferative function is independent of activin. TGF-β increases activin secretion in both epithelial (A) and stromal (B) cells. (A) Activin ligand from respective CRC cell culture supernatants as measured by ELISA with and without treatment with 10 ng/ml of TGF-β. (B) Activin ligand from CRC cells with or without co-culture with CCD18 stromal cells. Cells were treated with either vehicle control (PBS) or 10 ng/ml TGF-β. (C) TGF-β pro-migratory effects are activin dependent. Transwell migration assay of FET colon cancer cells treated with either the activin inhibitor follistatin (FST, 100 ng/ml) or TGF-β (10 ng/ml) or both as indicated. Cells were imaged and counted as described in Materials and Methods. D) TGF-β induced anti-proliferative effects are activin independent. Metabolic activity as an approximation of cell proliferation of FET CRC cells treated with either follistatin (FST, 100 ng/ml) or TGF-β (10 ng/ml) or both as indicated was measured by wst-8-based cell counting assay and reported as OD450. All experiments shown were reproduced at least twice with at least a total n = 5.
As TGF-β showed a substantial impact on activin secretion, we next explored its effects on pro-metastatic functions. For this, we examined cell motility measured by transwell migration. As reported previously13, 32 and confirmed in Supplementary Figure S1, both activin and TGF-β individually increase the number of FET colon cancer cells which migrate through transwells. To delineate how much of the TGF-β migratory effect is dependent on activin, here we inhibited activin signaling using its specific antagonist follistatin (FST), which does not bind TGF-β but neutralizes activin with high affinity by acting as a ligand trap33, 34. We found that the pro-migratory effect of TGF-β was abrogated in the presence of follistatin, indicating that the TGF-β induced pro-metastatic function is dependent on activin signaling. Consistent with this, follistatin alone did not change migration (Fig. 2C). Similar, albeit more modest effects were observed in SMAD4 mutated SW480 colon cancer cells (Supplementary Figure S2).
To investigate the impact of activin inhibition on the anti-proliferative actions of TGF-β, we next measured metabolic activity following TGF-β treatment in the presence and absence of follistatin. Interestingly, in the presence or absence of TGF-β, follistatin had no impact on proliferation in TGF-β superfamily signaling wild-type FET colon cancer cells (Fig. 2D), confirming that TGF-β induced activin pathway activation selectively affects colon cancer cell migration and not TGF-β- induced growth suppression.
Activin and TGF-β pathway member expression correlate on receptor and ligand level in patients with CRC
To further test our hypothesis that activin and TGF-β expression and signaling are closely connected in colorectal cancer and to further support data obtained in our clinical cohorts, we performed an in silico analysis using The Cancer Genome Atlas (TCGA) colorectal dataset35. This is a publicly available and NIH curated in silico data base providing high quality mRNA-seq data from solid tumor tissues compared to paired normal tissue.
We queried the connection of activin and TGF-β by assessing pathway member correlation in mRNA expression. Indeed, the expression of a substantial number of activin and TGF-β members correlated on the mRNA level (Table 1), most notably the activin ligand measurable subunit INHBA with the TGF-β ligand (TGFB1) (Fig. 3). In order to put the magnitude of the correlation into context, we performed a permutation test and correlated 100,000 random gene pairs (Supplementary Figure S3) revealing the correlation between activin and TGF-β ligand as in the top 1% of most strongly correlated genes in sporadic CRC (Table 1).
Table 1.
Strong correlation of mRNA expression of activin and TGF-β pathway members.
| Activin pathway | TGF-β pathway | rho | p-value | FDR |
|---|---|---|---|---|
| INHBA | TGFB3 | 0.666491996 | <1E-22 | <1E-22 |
| INHBA | TGFB1 | 0.615817318 | <1E-22 | <1E-22 |
| ACVR1 | TGFBR1 | 0.601597339 | <1E-22 | <1E-22 |
| ACVR2A | TGFBR1 | 0.574281616 | <1E-22 | <1E-22 |
| ACVR1 | TGFB2 | 0.50233367 | <1E-22 | <1E-22 |
| INHBA | TGFB2 | 0.488423405 | <1E-22 | <1E-22 |
| ACVR1 | TGFB3 | 0.416297434 | 1.20E-12 | 7.78E-12 |
| ACVR1 | TGFBR2 | 0.345959021 | 6.60E-09 | 3.53E-08 |
| INHBA | TGFBR1 | 0.335048949 | 2.04E-08 | 1.03E-07 |
| ACVR2B | TGFBR2 | 0.3134617 | 1.68E-07 | 6.95E-07 |
| ACVR2A | TGFBR2 | 0.300971359 | 5.30E-07 | 1.93E-06 |
| ACVR1B | TGFBR2 | 0.294959388 | 9.05E-07 | 3.17E-06 |
| ACVR2A | TGFB2 | 0.293090745 | 1.07E-06 | 3.59E-06 |
Activin and TGF-β ligands, receptor and downstream SMAD molecules were interrogated for correlation using Pearson’s correlation coefficient (rho). The p value of the correlation and the false discovery rate (FDR) are included.
Figure 3.
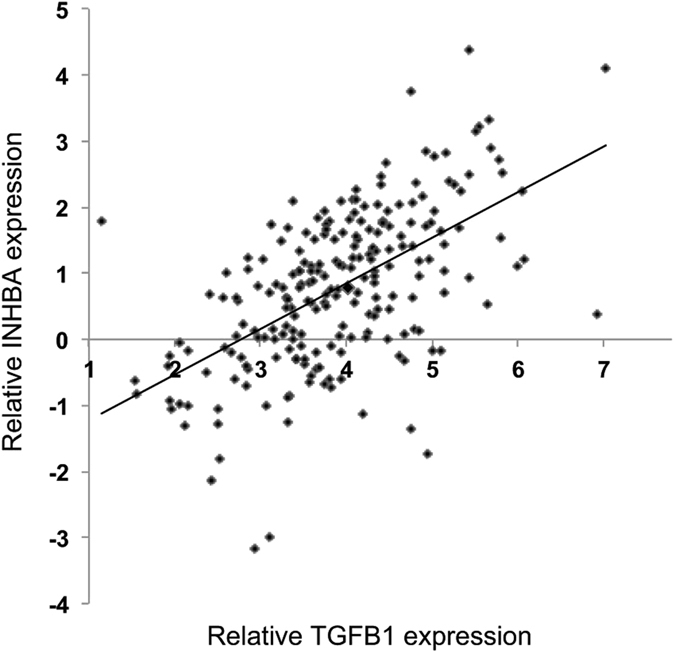
Activin and TGF-β ligand mRNA expression strongly correlate. Relative mRNA expression in the TCGA data set of the Activin A ligand subunit (INHBA) and TGF-β ligand (TGFB1) is shown after global normalization. Each data point represents an individual tumor (r = 0.6158).
These findings support the notion that activin and TGF-β pathways are closely connected in CRC. In addition, we found substantial in silico correlations through analysis in cohorts of other adenocarcinomas (esophageal adenocarcinoma TCGA provisional r = 0.58, p < 0.001, gastric adenocarcinoma TCGA36 r = 0.47, p < 0.01, lung adenocarcinoma TCGA37 r = 0.33, p < 0.05.) hinting towards a cross regulated TGF-β superfamily signaling network in additional solid tumors.
In summary, we here present in vivo, in vitro and in silico data all strongly supporting a close and functionally important connection between the activin and TGF-β pathways in advanced CRC.
Discussion
Mortality from metastatic CRC remains high as prediction and treatment of metastasis are still ineffective. In addition, there is a growing number of patients under 40 years of age who present with advanced disease and did not meet age or risk requirements for screening2.
TGF-β, initially categorized as a tumor suppressor due to its growth suppressive properties, promotes metastatic CRC at later stages. TGF-β inhibitors are in early phase clinical trials, although results have been mixed22. We here present data suggesting that treatment options focusing on TGF-β alone may not be effective without considering the role of its superfamily member, activin. Activin and TGF-β signaling pathways have been deemed redundant due to shared downstream growth suppressive canonical signaling via SMAD48. Our previously published studies support pro-metastatic actions of activin mechanistically distinct from TGF-β via separate effector mutagenic pathways13, 32. Here, we illustrate how activin and TGF-β are complexly intertwined and need to be interpreted as a unit to better target and predict metastatic CRC.
Here we show that simultaneously considering activin and TGF-β serum ligand levels is superior to assessing either ligand alone in identifying patients with advanced CRC and assessment of serum ligand levels is feasible as a clinical assay to guide treatment options. While baseline levels of activin appear to be low in the general populations (unpublished observations), clinical decisions based on elevated ligand serum levels have limitations in regards to measuring TGF-β superfamily signaling activation in CRC. Circulating levels of TGF-β and activin may be affected by other conditions, especially inflammatory conditions38. We determined here that a combined activin and TGF-β ligand expression score in the tumor was strongly predictive for shorter overall survival in our cohort, however, currently, there are no studies which simultaneously measures tumor and systemic ligand levels to determine if serum ligand levels reflect tumor ligand levels. Therefore, we plan to measure serum and tumor ligand expression in a prospective cohort including all CRC stages to fully evaluate their correlation and respective individual or combined predictive power. While we did not consider confounding factors such as lifestyle or mutational background in this initial study, it is possible and perhaps likely that our findings are influenced by confounders. Analyses of these clinical nuances will be important in effectively translating our observations into the clinical realm.
Follistatin, a naturally occurring activin antagonist which we utilized in our in vitro studies, was utilized by others to inhibit activin signaling and improve outcomes in in vivo models of inflammation39, 40. While follistatin is well tolerated, long term follistatin treatment for either in vivo models of CRC or patient care is challenging due to both its short half-life41 and considerable expense with repeat administration. Other approaches for inhibiting activin signaling are needed.
Our observation that activin inhibition through follistatin did not affect TGF-β-induced growth suppression suggests that activin inhibition may specifically inhibit pro-oncogenic TGF-β functions while preserving its growth suppressive functions. While the inhibition of activin signaling is understudied in comparison to the inhibition of TGF-β signaling, activin inhibition may have further clinical benefits including reversing both cancer associated cachexia42, 43 and anemia44. Additionally, activin inhibition has exhibited a favorable safety profile in a phase II study of cancer associated anemia45, which was unfortunately terminated early due to poor patient enrollment.
The co-occurrence of mutations in activin and TGF-β receptors we observed in silico strongly supports a functional connection between these pathways. This connection suggests substantial mutational pressure on the cancer cell to acquire a mutation in a TGF-β receptor if a mutation is present in an activin receptor, and vice versa. In our in silico analysis, mutations in receptors of both pathways were mutually exclusive with SMAD mutations, an established mechanism to escape TGF-β superfamily induced growth suppression46. This leads us to conclude that it is very likely that dual receptor mutations are an alternative route through which CRC can obviate the early anti-oncogenic effects of growth suppression by activin and TGF-β.
To our knowledge, this is the first report of a functional connection between activin and TGF-β signaling in CRC. In a mouse model of tumor cachexia, activin was shown to induce muscle wasting and inhibition of activin signaling reduced cachexia42. Others have reported activin to be downstream of TGF-β and necessary for its pro-metastatic function in breast adenocarcinoma cell lines47. These in vitro data and the data reported here suggest that assessing activin and TGF-β as a unit may be translatable to other solid tumors.
In summary, our study provides novel insight into the cross-regulation of TGF-β superfamily signaling in CRC and supports the assessment of activin signaling for both risk stratification and potential treatment of advanced CRC.
Materials/Methods
Reagents
Activin A and TGFβ1 were reconstituted in PBS plus 4 mM HCl according to the manufacturer’s instruction (both R&D, Minneapolis, MN). Follistatin 288 (R&D) was reconstituted in PBS.
Cohorts
Serum samples from a subset of patients from the published Quasar colorectal cancer cohort29 were kindly provided by Dr. David Kerr as de-identified and coded. All samples were collected following informed consent and ethics approval for the study was given by the West Midlands Multi Research Ethics Committee at each collection site29. All methods were performed in accordance with the relevant guidelines and regulations of the local research ethics committee. The serum samples were drawn after surgery but prior to randomization into a chemotherapy arm versus a placebo arm. These samples consisted of serum samples from 120 stage II colon cancer patients. 60 patients without disease recurrence were matched by study arm, age, gender and BMI to 60 patients with disease recurrence. A commercial tumor micro-array of 40 CRC tumors, 10 metastasis and 10 control samples30 was employed to assay tumor activin and TGF-β ligand expression. The epidemiologic data of this cohort are listed in Table 2.
Table 2.
Epidemiologic data of colorectal cancer TMA by Imgenex (San Diego, CA).
| Stage | Stage I: | 2 (5%) |
| Stage II: | 11 (27.5%) | |
| Stage III: | 15 (37.5%) | |
| Stage IV: | 12 (30%) | |
| Metastasis | Yes: | 13 (32.5%) |
| No: | 27 (68.5%) | |
| Gender | Female: | 12 (30%) |
| Male: | 28 (70%) | |
| Age | < 65: | 30 (75%) |
| > 65: | 10 (25%) | |
| Activin/TGF-β staining intensity | High: | 13 (32.5%) |
| Intermediate/low: | 27 (68.5%) | |
| Survival (after 252 months of follow up) | Yes: | 10 (25%) |
| No: | 23 (57.5%) | |
| Lost to follow up: | 7 (17.5%) |
Cohort data from commercial CRC tumor micro-array (TMA) including tumor stage, whether the subject had metastases, gender, age, length of survival, and activin and TGF-β combined staining intensity.
Quantification of activin and TGF-β ligands
Activin and TGF-β from human patient serum samples and cell culture supernatants were measured utilizing activin A and TGFB1 Quantikine© ELISAs (both R&D Systems), respectively, following the manufacturer’s instructions. Cell culture supernatants were analyzed without prior freezing after 5 min centrifugation at 10,000 g, and human serum samples after storage at −80 °C. Activin and TGF-β ligand expression was assigned as score of “high” (3) if expression was higher than the 75th percentile in our cohort, “intermediate” (2) if levels were between the 25th and 75th percentile, and “low” (1) if levels were below the 25th percentile. Compound scores were calculated by adding activin and TGF-β scores. All samples were assessed in duplicates.
Immunohistochemistry
Adjacent slides of a commercial TMA (Imgenex, San Diego, CA) with 40 CRC samples, 10 metastases and 10 adjacent normal samples were stained as previously reported13, 25 with antibodies against the Activin A subunit INHBA (Ansh lab LCC, Webster, TX) and TGFB1 (Abcam, Cambridge, United Kingdom). Slides were reviewed by a pathologist and scored blindly with an additional two individual investigators using a three point scoring system from 0 – weak staining to 2 - strong staining. A compound activin and TGF-β score was calculated by adding both scores.
Cell culture
SW480 colon cancer cells (ATCC, Manassas, VA,) were maintained in DMEM, FET colon cancer cells (gift from Michael Brattain, University of Nebraska, Omaha, NE) and CCD18 fibroblasts (ATCC) cells were maintained in DMEM/F12 50:50 (both Corning Inc., Corning, NY) supplemented with 10% fetal bovine serum and penicillin (100 U/ml) as well as streptomycin (100 µg/ml) (Invitrogen, Carlsbad, CA). Cells were grown at 37 °C in a humidified incubator with 5% CO2. All cells were serum starved for 18 hours prior to treatment to approximate cell cycle synchronization.
Co-culture experiments were performed using trans-well plates (6-well) with 0.4 μm pore inserts. CCD18 fibroblasts were grown to confluency on the inserts and colon cancer epithelial cells (FET or SW480) on the plate surface. Cells were treated with either 10 ng/ml of TGF-β or activin and incubated for an additional 48 hours. Supernatant was collected and assayed for respective ligands by ELISA as described below.
All cell lines were validated for authenticity by 9 STR (short tandem repeat) profiling using CellCheck 9 Plus and tested negative for mycoplasma (both IDEXX, Columbia, MO).
Transwell migration assay
Migration assays were performed as previously described13, 32. Briefly, transwell 12 well plates (8 µm pores, Corning) with fibronectin (Sigma, St. Louis, MO) were seeded with 5 × 105 colon cancer cells per well. Cells were treated with either activin A (10 ng/ml) or TGF-β (10 ng/ml) or follistatin (100 ng/ml) for 30 minutes and TGF-β (10 ng/ml) was added. Cells were then allowed to migrate for 6 hours, stained, and imaged. Images from 5 microscopic fields at the center of each well were counted using ImageJ software48.
Proliferation assay
Cells were seeded in 96 well plates, serum starved for 18 hours and treated with TGF-β at 10 ng/ml, follistatin 288 at 100 ng/ml, or a combination of both. Controls were treated with vehicle (PBS). After 48 hours, the wst-8-based Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Rockville, MD) measuring metabolic activity as an approximation of cell proliferation was used following the manufacturer’s instructions as previously described13.
In silico dataset
The Cancer Genome Atlas (TCGA) CRC dataset was accessed as described in its original publication35 which includes the clinical characteristics of this cohort. TCGA datasets for esophageal adenocarcinoma, gastric adenocarcinoma36, and lung adenocarcinoma37 were accessed via cbioportal.com49, 50.
Statistical methods
Data are shown as mean + SEM if not explicitly stated otherwise. All in vitro experiments were reproduced at least twice. Statistical significance level of α = 0.05 was set before experiments. In silico analysis of the TCGA colorectal dataset was performed by the UIC biostatistical core facility. Mutual exclusivity and co-occurrence were tested using Fisher’s exact test. For correlation of expression, Pearson or Spearman’s correlation were used as applicable. For our permutation analysis, 100,000 random gene pairs from the TCGA colorectal dataset35 were correlated with each other. Two sided t-test with Welch’s correction and one sided ANalysis Of VAriance (ANOVA) with Dunett’s post-test was used for comparison of means in two or more than two groups with one control group respectively. For survival analysis, Log-rank (Mantel-Cox) test was used. All statistical analysis was performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA). All authors had access to the study data and reviewed and approved the final manuscript.
Data Availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
Electronic supplementary material
Acknowledgements
This study was supported by NIH grant R01 CA141057 to BJ and the German Research Foundation stipend STA1458/1-1 to JJS. We thank the UIC bioinformatics core, especially Mark Maienschein-Cline and Zhengdeng Lei for excellent statistical support. This work is dedicated to the late Peter Harwood Curry, Junior.
Author Contributions
Hypothesis was created by J.J.S. and B.J. Study was designed by J.J.S., P.G. and B.J. Experiments were designed by J.J.S. and Ö.Ö. Immunohistochemistry was performed by A.J. and J.J.S., slides were reviewed by G.G. and scored by J.J.S., A.J. and J.B. ELISA experiment were planned and run by J.T. and T.C. Transwell migration assays were performed by J.B. and A.J. Proliferation assays were performed by G.M. Samples from the QUASAR study were managed and provided by D.K. Statistical analyses were performed by J.J.S. and the UIC bio statistical core facility. Manuscript was drafted by J.J.S. and N.K., and written, critically reviewed and edited by J.J.S., Ö.Ö., N.K., D.K., P.G., and B.J.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05907-8
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Lin, J. S. et al. Screening for Colorectal Cancer: A Systematic Review for the U.S. Preventive Services Task Force. (Agency for Healthcare Research and Quality, Rockville (MD), 2016). [PubMed]
- 3.Carrato A. Adjuvant treatment of colorectal cancer. Gastrointest Cancer Res. 2008;2:S42–46. [PMC free article] [PubMed] [Google Scholar]
- 4.Mouradov D, et al. Survival in stage II/III colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am J Gastroenterol. 2013;108:1785–1793. doi: 10.1038/ajg.2013.292. [DOI] [PubMed] [Google Scholar]
- 5.Primrose JN, et al. Effect of 3 to 5 years of scheduled CEA and CT follow-up to detect recurrence of colorectal cancer: the FACS randomized clinical trial. JAMA. 2014;311:263–270. doi: 10.1001/jama.2013.285718. [DOI] [PubMed] [Google Scholar]
- 6.Edwards MS, Chadda SD, Zhao Z, Barber BL, Sykes DP. A systematic review of treatment guidelines for metastatic colorectal cancer. Colorectal Dis. 2012;14:e31–47. doi: 10.1111/j.1463-1318.2011.02765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellam N, Pasche B. Tgf-beta signaling alterations and colon cancer. Cancer Treat Res. 2010;155:85–103. doi: 10.1007/978-1-4419-6033-7_5. [DOI] [PubMed] [Google Scholar]
- 8.Principe, D. R. et al. TGF-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst106, djt369, doi:10.1093/jnci/djt369 (2014). [DOI] [PMC free article] [PubMed]
- 9.Willis SA, Zimmerman CM, Li LI, Mathews LS. Formation and activation by phosphorylation of activin receptor complexes. Mol Endocrinol. 1996;10:367–379. doi: 10.1210/mend.10.4.8721982. [DOI] [PubMed] [Google Scholar]
- 10.Weiss A, Attisano L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol. 2013;2:47–63. doi: 10.1002/wdev.86. [DOI] [PubMed] [Google Scholar]
- 11.Wrighton KH, Lin X, Feng XH. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009;19:8–20. doi: 10.1038/cr.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bauer J, et al. Activin and TGFbeta use diverging mitogenic signaling in advanced colon cancer. Mol Cancer. 2015;14:182. doi: 10.1186/s12943-015-0456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Zhou F, ten Dijke P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends Biochem Sci. 2013;38:612–620. doi: 10.1016/j.tibs.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Chapnick DA, Warner L, Bernet J, Rao T, Liu X. Partners in crime: the TGFbeta and MAPK pathways in cancer progression. Cell Biosci. 2011;1:42. doi: 10.1186/2045-3701-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piersma B, Bank RA, Boersema M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front Med (Lausanne) 2015;2:59. doi: 10.3389/fmed.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu Y, et al. Differential regulation of transforming growth factor beta signaling pathways by Notch in human endothelial cells. J Biol Chem. 2009;284:19452–19462. doi: 10.1074/jbc.M109.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang YA, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109:1607–1615. doi: 10.1172/JCI200215333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melisi D, et al. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7:829–840. doi: 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calon A, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neuzillet C, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31. doi: 10.1016/j.pharmthera.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Munoz NM, et al. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006;66:9837–9844. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- 24.Biswas S, et al. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004;64:4687–4692. doi: 10.1158/0008-5472.CAN-03-3255. [DOI] [PubMed] [Google Scholar]
- 25.Principe DR, et al. Loss of TGFbeta signaling promotes colon cancer progression and tumor-associated inflammation. Oncotarget. 2016 doi: 10.18632/oncotarget.9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung B, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126:654–659. doi: 10.1053/j.gastro.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Jung BH, et al. Activin type 2 receptor restoration in MSI-H colon cancer suppresses growth and enhances migration with activin. Gastroenterology. 2007;132:633–644. doi: 10.1053/j.gastro.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biswas S, et al. Mutational inactivation of TGFBR2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor beta resistant cells. Genes Chromosomes Cancer. 2008;47:95–106. doi: 10.1002/gcc.20511. [DOI] [PubMed] [Google Scholar]
- 29.Quasar Collaborative G, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet. 2007;370:2020–2029. doi: 10.1016/S0140-6736(07)61866-2. [DOI] [PubMed] [Google Scholar]
- 30.Sporn JC, Hothorn T, Jung B. BARD1 expression predicts outcome in colon cancer. Clin Cancer Res. 2011;17:5451–5462. doi: 10.1158/1078-0432.CCR-11-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471–1474. doi: 10.1245/s10434-010-0985-4. [DOI] [PubMed] [Google Scholar]
- 32.Bauer J, Sporn JC, Cabral J, Gomez J, Jung B. Effects of activin and TGFbeta on p21 in colon cancer. PLoS One. 2012;7:e39381. doi: 10.1371/journal.pone.0039381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashimoto O, et al. Difference between follistatin isoforms in the inhibition of activin signalling: activin neutralizing activity of follistatin isoforms is dependent on their affinity for activin. Cell Signal. 2000;12:565–571. doi: 10.1016/S0898-6568(00)00099-1. [DOI] [PubMed] [Google Scholar]
- 34.Thompson TB, Lerch TF, Cook RW, Woodruff TK, Jardetzky TS. The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev Cell. 2005;9:535–543. doi: 10.1016/j.devcel.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature487, 330-337, doi:10.1038/nature11252 (2012). [DOI] [PMC free article] [PubMed]
- 36.Cancer Genome Atlas Research, N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature513, 202-209, doi:10.1038/nature13480 (2014). [DOI] [PMC free article] [PubMed]
- 37.Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature511, 543-550, doi:10.1038/nature13385 (2014). [DOI] [PMC free article] [PubMed]
- 38.Hedger MP, Winnall WR, Phillips DJ, de Kretser DM. The regulation and functions of activin and follistatin in inflammation and immunity. Vitam Horm. 2011;85:255–297. doi: 10.1016/B978-0-12-385961-7.00013-5. [DOI] [PubMed] [Google Scholar]
- 39.Dohi T, et al. Therapeutic potential of follistatin for colonic inflammation in mice. Gastroenterology. 2005;128:411–423. doi: 10.1053/j.gastro.2004.11.063. [DOI] [PubMed] [Google Scholar]
- 40.Jones KL, et al. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci USA. 2007;104:16239–16244. doi: 10.1073/pnas.0705971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kogure K, et al. Intravenous administration of follistatin: delivery to the liver and effect on liver regeneration after partial hepatectomy. Hepatology. 1996;24:361–366. doi: 10.1002/hep.510240212. [DOI] [PubMed] [Google Scholar]
- 42.Chen JL, et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 2014;28:1711–1723. doi: 10.1096/fj.13-245894. [DOI] [PubMed] [Google Scholar]
- 43.Zhou X, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 44.Raje N, Vallet S. Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein for the treatment of anemia and bone loss. Curr Opin Mol Ther. 2010;12:586–597. [PubMed] [Google Scholar]
- 45.Raftopoulos H, et al. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: results from two phase 2 studies. Support Care Cancer. 2016;24:1517–1525. doi: 10.1007/s00520-015-2929-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou S, et al. Targeted deletion of Smad4 shows it is required for transforming growth factor beta and activin signaling in colorectal cancer cells. Proc Natl Acad Sci USA. 1998;95:2412–2416. doi: 10.1073/pnas.95.5.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Howley BV, Hussey GS, Link LA, Howe PH. Translational regulation of inhibin betaA by TGFbeta via the RNA-binding protein hnRNP E1 enhances the invasiveness of epithelial-to-mesenchymal transitioned cells. Oncogene. 2016;35:1725–1735. doi: 10.1038/onc.2015.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).