

Abstract
Commercially available DOTAP is a racemic mixture of two enantiomers. The adjuvanticity of each isomer was examined using a peptide/lipid complex as a therapeutic vaccine in an established murine cervical cancer model. This simple vaccine consists of a cationic lipid (DOTAP) and a major histocompatibility complex (MHC) class I–restricted epitope of the Human Papillomavirus (HPV) 16 protein E7. Dose-dependent tumor regression experiments have been completed for racemic DOTAP/E7, (R)-DOTAP/E7 and (S)-DOTAP/E7. Tumor-bearing mice treated with (R)-DOTAP/E7 complexes have shown tumor regression in a dose-dependent manner comparable to those mice treated with a racemic DOTAP with E7 peptide. These data are supported by IFN-γ production by CD8+ splenocytes, in vivo cytotoxic T-lymphocytes (CTL) response, CD8+ tumor-infiltrating lymphocytes (TIL), and IFN-γ production by CD8+ TIL in (R)-DOTAP/E7-vaccinated mice. When (S)-DOTAP/E7 is delivered, tumor progression is delayed. While IFN-γ production is absent from CD8+ splenocytes in mice vaccinated with (S)-DOTAP/E7, IFN-γ production by CD8+ TIL is present, supporting our hypothesis that (S)-DOTAP has limited activity. Activation of bone marrow-derived dendritic cells by the enantiomeric formulations has also been evaluated, as well as cytokine production and toxicity with no considerable differences between the groups. The results show the DOTAP enantiomers act differently as adjuvants in vivo, with (R)-DOTAP being more effective at stimulating a CD8+ anti-tumor response.
Keywords: Therapeutic vaccine, Cervical cancer, Enantiomers, Cationic lipid
Introduction
Therapeutic cancer vaccines are safe treatments that can create a potent immune response against transformed host cells. Vaccines and immunotherapy avoid the toxicity of harsh chemotherapy that includes side effects such as weight loss, hair loss, nausea, and leukopenia. Instead of targeting all rapidly dividing cells, as is the case of many small molecule chemotherapeutic drugs, vaccines can train the immune system how to specifically target cancer cells only. Creating an efficacious cell-mediated response is crucial for anti-tumor effects [1]. Many different types of antigens have been used in cancer vaccines, ranging from whole tumor cell lysate to MHC class I–restricted peptides specifically recognizable by CTLs [2–4]. Only the use of peptide antigens allows for ease of manufacturing, as well as precise control over the purity and specificity of the vaccine. However, peptide antigens need supplementary treatments to increase immunogenicity.
There is very little variety in the adjuvants that are clinically available. The US Food and Drug Administration (FDA) has only approved two adjuvants for use in humans, both aluminum hydroxide based [5]. The first, Alum (aluminum hydroxide), has been approved since the 1920s. The second, AS04 (3-O-desacyl-4′-monophosphoryl lipid A adsorbed on to aluminum as hydroxide salt), approved in the United States in 2009, is only used in one vaccine that is currently on the market (Cervarix) [6]. The scarcity of available adjuvants, particularly in the United States, demonstrates a need for new safe adjuvants allowing more options in formulation and titrating of the desired immune response via immunotherapy.
Development and subsequent approval of new adjuvants face several barriers. Encouraged by the FDA, creating treatments that are chirally pure is a trend growing in popularity across pharmaceutics, with the increased ease of separation of enantiomers after synthesis [7, 8]. However, chiral drugs can have different efficacies in vivo. It is known that stereoisomers of small molecule drugs can have widely varied effects, with one being efficacious and the other showing no therapy, or worse, toxicity [9]. Biology provides an asymmetric environment, containing chiral components at its most basic molecular level, l-amino acids and d-carbohydrates. It is no surprise that highly purified antigens show stereospecificity in all types of immune reactions [10, 11]. However, it has never been shown that the stereospecificity of an adjuvant plays a vital role in its efficacy in producing an immune response.
Our basis for investigating this line of work starts with a therapeutic vaccine we have previously developed for a murine cervical cancer model. This nanoparticle complex consists of three molecules, racemic DOTAP lipid and E7 peptide, an H-2Db-restricted 9-mer from the HPV 16 E7 oncogene, able to cause regression in pre-existing tumors [12]. The MHC class I-restriction of the peptide promotes a cell-mediated response rather than a humoral response against the growing tumor. We have shown that the racemic DOTAP lipid is able to activate bone marrow-derived dendritic cells (BMDC) in vitro to elicit a cytokine response via the ERK pathway [13].
In this study, we investigate the impact of chirality on the efficacy of DOTAP lipid as an adjuvant. We have evaluated (R)-DOTAP and (S)-DOTAP for its interaction with E7 peptide in formulation properties, measuring peptide encapsulation, particle size, and charge. Lipid alone was used to test the interaction and ability to stimulate BMDC, inducing activation through CD86 up-regulation and CCL2 production. Lipid toxicity was also compared when treating BMDC. In vivo, (R)- and (S)-DOTAP formulations were compared in many ways, testing for effect on tumor regression, and further elucidating the affected cellular mechanisms that cause regression.
Materials and methods
Materials
Racemic DOTAP (99.9% purity), (R)-DOTAP (100% purity), and (S)-DOTAP (99.9% purity) were obtained from Merck KGaA (Darmstadt, Germany). (R)-DOTAP stock used had a specific optical rotation [α]25D of −1.77°, while (S)-DOTAP stock used had a specific optical rotation [α]25D of +1.99°. H-2Db-restricted peptides E7 (H-RAHYNIVTF-OH, amino acids 49–57 from the E7 protein of HPV16) and NP (H-ASNENMETM-OH, amino acids 366–374 from the influenza virus strain A/PR/8/34 nucleoprotein) were obtained from the University of Pittsburgh Peptide Synthesis Facility (Pittsburgh, PA).
Tumor cell culture
TC-1 cells are lung endothelial cells isolated from a C57BL/6 mouse that were transfected with HPV 16 oncogenes E6 and E7 and activated human H-ras and are used as a model for cervical cancer [14]. TC-1 cells were obtained from American Type Culture Collection (Manassas, VA) and maintained in RPMI 1640 media supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 100 U/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen).
Preparation and evaluation of vaccine formulations
DOTAP/E7, (R)-DOTAP/E7, and (S)-DOTAP/E7 complexes were made as described previously with few modifications [12]. Briefly, lipids dissolved in chloroform were dried in glass vials under a stream of nitrogen and stored under vacuum in a desiccator overnight. Lipid/peptide complexes were formed by thin film hydration, using a solution of E7 peptide in molecular grade water (Cellgro, Manassas, VA) and subsequent vortexing. After 2 h at room temperature, the suspensions were extruded through two 100 nm polycarbonate membranes (Fisher Healthcare, Houston, TX) to a size of roughly 120 nm. Particle size and zeta potential were measured with a Malvern Zetasizer Nano ZS in water (Malvern, Worcestershire United Kingdom). E7 encapsulation was determined by Microcon® centrifugal filtrate device (Millipore, Bedford, MA). Unbound peptide was measured by Micro BCA™ Protein Assay Kit (Rockford, IL). Amount encapsulated calculated as (100–% unbound peptide).
Bone marrow-derived dendritic cells (BMDC)
Methods to culture bone marrow-derived dendritic cells have been described previously [13]. Briefly, bone marrow from the femurs and tibias of 6-week-old C57BL/6 mice was isolated in serum-free RPMI 1640 media. Red blood cells were lysed using ACK Lysing Buffer (Invitrogen) and cells were plated for 2 h in serum-free RPMI 1640 to remove suspension cells (lymphocyte precursors). Remaining adherent cells were cultured in RPMI 1640 media containing 10% fetal bovine serum, non-essential amino acids (Invitrogen), antibiotic/antimycotic (Sigma–Aldrich, St. Louis, MO) with 1,000 U/ml granulocyte–macrophage colony-stimulating factor (GM-CSF), and 1,000 U/ml interleukin-4 (IL-4) (R&D Systems, Minneapolis, MN) for 6 days changing the media every 2 days. BMDCs were used in experiments on day 6.
Interaction of formulations with BMDC in vitro
To measure CD86 expression, a sign of activation, BMDC were treated with different concentrations of liposomes (free of peptide) for 18 h. Cells were stained with anti-CD86 (GL1) and anti-CD11c (HL3) antibodies (BD Biosciences, San Jose, CA) and analyzed using a FACS Canto II flow cytometer (BD Biosciences). One hundred ng/ml LPS served as a control. Isotype controls were used. To measure toxicity, BMDC were treated with different concentrations of liposomes (free of peptide) for 18 h, stained with propidium iodide (PI) (Sigma–Aldrich), and evaluated using flow cytometry. To measure production of chemokine C–C motif ligand 2 (CCL2) in a dose-dependent manner, BMDC were treated with varying doses of (R)-DOTAP and (S)-DOTAP liposomes (free of peptide). After 24 h, cell supernatants were collected and analyzed by ELISA (BD Biosciences).
Mice and immunizations
Six-week-old female C57BL/6 mice were used in all studies. On day 0, 105 TC-1 cells were injected subcutaneously into the hair-trimmed abdomen. On day 6, formulations (100 μl) were subcutaneously injected into the contralateral side of the abdomen in a 5% dextrose solution. In the initial tumor growth delay experiment, 100 nmol of lipid and 20 μg of E7 peptide were used. After this initial experiment, an optimal dose was determined by delivering lipid in concentrations ranging from 3 to 600 nmol, with a constant dose of 20 nmol of peptide. In all future in vivo experiments, lipid formulations contained 300 nmol lipid and 20 nmol of E7 peptide. Unencapsulated peptide was not removed. Tumors were measured every 2–3 days with calipers, calculating area as (length) x (width). Humane sacrifice of mice was performed after tumors reached 20 mm in one dimension. All animal protocols were approved by the University of North Carolina at Chapel Hill’s Institutional Animal Care and Use Committee.
Interferon gamma (IFN-γ) production by CD8+ T cells
Mice were injected with 105 TC-1 cells on day 0. On day 6, mice were vaccinated with either (R)-DOTAP/E7 or (S)-DOTAP/E7 with lipid and peptide concentration of 300 and 20 nmol, respectively. On day 14, mice were sacrificed, spleens sterilely removed, and processed to a single cell suspension by crushing the spleen through a 70 μm filter (BD Biosciences). After removal, cells were incubated in RPMI 1640 media supplemented with NEAA and antibiotic/antimycotic, 1 μl/ml GolgiStop™ (BD Biosciences), and 5 μM E7 or NP peptide for 6 h at 37°C. Cells were removed, washed with staining buffer (BD Pharmingen, San Diego, CA), and stained with anti-CD8 antibody (53-6.7). Cells were then treated with Cytofix/Cytoperm™ kit, stained intracellularly for IFN-γ (XMG1.2), washed, and analyzed by flow cytometry. Percents represent IFN-γ+ CD8+ T cells out of the total CD8+ T-cell population.
In vivo cytotoxic T-lymphocyte assay
The in vivo CTL assay has been described previously and was performed here with slight modifications [15]. Briefly, mice were injected with 105 TC-1 cells on day 0. On day 6, mice were vaccinated with either (R)-DOTAP/E7 or (S)-DOTAP/E7 with lipid and peptide concentration of 300 nmol and 20 nmol, respectively. Eight days later, naïve mice were sacrificed and splenocytes removed. Splenocytes were pulsed with either 10 μM E7 or NP peptide for 1–2 h in complete media at 37°C. Cells were stained with carboxyfluorescein succinimidyl ester (CFSE) (Sigma–Aldrich, St. Louis, MO), with NP peptide-pulsed and E7 peptide-pulsed cells stained with 0.4 and 4 μM, respectively, in serum-free media for 15 min. Cells were then washed with complete media and counted. Equal numbers of CFSEhigh (E7-pulsed cells) and CFSElow (NP-pulsed cells) were mixed together and stained with 8 μM PKH-26 (Sigma–Aldrich) according to manufacturer’s instructions. Vaccinated mice were injected with 107 labeled cells and in vivo killing of the targets was allowed for 16–20 h. After that time, spleens from treated mice were removed and made into a single cell suspension, red blood cells lysed, washed, and resuspended in phosphate-buffered saline (Sigma–Aldrich). The cells were analyzed by flow cytometry, first gating for the lymphocyte population, then for the PKH-26-positive cells, to determine the amount of specific lysis of the E7-labeled cells. The following equation from [15] shows this, with NP and E7 representing the number of cells pulsed with E7 or NP peptide present after in vivo killing.
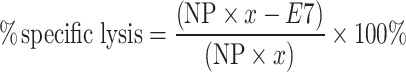 |
.
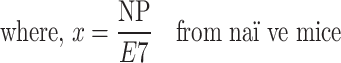 |
.
Tumor-infiltrating lymphocyte analysis
TC-1 tumors from mice vaccinated on day 6 were removed on day 14 in a sterile environment. Tumors were minced and incubated in RPMI 1640 containing 1 mg/ml collagenase type 1 (Worthington, Lakewood, NJ) for 30 min at 37°C. Tumors were put through a 70-μm strainer and formed a single cell suspension. Cells were blocked with Fc block (anti CD16/CD32 (2.4G2)) for 15 min, then stained with anti-CD8 (53-6.7) and anti-CD4 (RM4–5), and analyzed by flow cytometry.
Alternately, lymphocytes were isolated from the tumors. Tumors were prepared as described above to a single cell suspension. The cells were washed and resuspended in complete media. Tumor-infiltrating lymphocytes (TIL) were separated from the tumor using Ficoll-Paque™ PLUS (GE Healthcare, Uppsala, Sweden), a density gradient method. These tumor-infiltrating lymphocytes were plated at a concentration of 2 × 106 and stimulated with 5 μM E7 or NP peptide for 6 h at 37°C. The TIL were incubated with Fc Block, antibodies for CD8, stained intracellularly for IFN-γ, and analyzed by flow cytometry.
Statistical analysis
Statistical analysis was completed by using a two-tailed Student’s t test. Data were statistically significant if the P value was less than 0.05.
Results
Tumor regression by therapeutic vaccination
One injection of racemic DOTAP/E7 (100 nmol/20 μg) 6 days after TC-1 tumor inoculation caused regression of preformed tumors (Fig. 1a), statistically different than untreated mice (P < 0.0001). (R)-DOTAP/E7 complexes showed the same response, indicating that (R)-DOTAP is the active enantiomer in racemic DOTAP (P = 0.63). (S)-DOTAP/E7 showed a partial response, statistically significant from racemic DOTAP/E7- and (R)-DOTAP/E7-treated mice (P < 0.05). The (S)-DOTAP/E7-treated mice also had tumor growth that was statistically significant from the untreated control group and the control mice treated with E7 alone (P < 0.01).
Fig. 1.

TC-1 tumor growth inhibition by vaccine delivery in vivo. a TC-1 tumor growth inhibition in mice by lipid/E7 complexes. Six-week-old female C57BL/6 mice were inoculated with 10,000 TC-1 cells s.c. in the abdomen on day zero. On day 6, treatments were s.c. injected into the opposite side of the abdomen. Lipid formulations delivered contained 100 nmol lipid and 20 μg of E7 peptide. Sample size and statistics are as follows: n = 5–6, *: P < 0.05, **: P < 0.01, ***: P < 0.0001. b Dose-dependent antitumor activity of racemic DOTAP/E7, (R)-DOTAP/E7, or (S)-DOTAP/E7 complexes. Six-week-old female C57BL/6 mice were injected with 10,000 TC-1 cells s.c. as in previous studies. On day 6, treatments were s.c. injected into the opposite side of the abdomen. The dose of lipid was varied while the dose of E7 peptide was kept constant at 20 nmol. n = 5–6 per dose
(R)-DOTAP/E7 complexes also showed a dose-dependent response comparable to that of racemic DOTAP/E7 when evaluating tumor size at day 23 (Fig. 1b). (S)-DOTAP/E7 never achieved an optimal dose, even at very high concentrations of lipid (600 nmol). The optimal dose assessed from these studies was 300 nmol of lipid, not only because it had the greatest response in terms of tumor regression (for (R)-DOTAP/E7 and racemic DOTAP/E7-treated mice), but it also had the greatest difference between (R)- and (S)-DOTAP/E7-treated groups.
Formulation characterization
As shown in Table 1, the average size of (R)-DOTAP/E7 complexes was 126.8 ± 5.1 nm and (S)-DOTAP/E7 complexes were of comparable size at 125.6 ± 7.8 nm (P = 0.69). Zeta potentials of the (R)- and (S)-DOTAP/E7 complexes also did not vary significantly with values of 54.3 ± 4.0 mV and 55.8 ± 1.9 mV, respectively (P = 0.37). Encapsulation efficiency of E7 peptide with (R)-DOTAP and (S)-DOTAP also did not differ significantly, with values of 31.3 ± 16.6% and 32.1 ± 8.9% at a dose of 300 nmol lipid and 20 nmol E7 peptide (P = 0.87).
Table 1.
Physical properties of the vaccine formulations
| Formulation | Particle size (nm)* | Zeta potential (mV)** | Encapsulation efficiency (%)*** |
|---|---|---|---|
| R-DOTAP/E7 | 126.8 ± 5.1 | 54.3 ± 4.0 | 31.3 ± 16.6 |
| S-DOTAP/E7 | 125.6 ± 7.8 | 55.8 ± 1.9 | 32.1 ± 8.9 |
Lipid concentration used in these studies was 6 mM, equivalent to 300 nmol of lipid per mouse
* P = 0.69, n = 8–10
** P = 0.37, n = 8–10
*** P = 0.87, n = 13–14
Evaluation of (R)-DOTAP and (S)-DOTAP in vitro
BMDC treated with (R)-DOTAP and (S)-DOTAP for 18 h both showed similar dose-dependent effects. Activation of BMDC was increased with increasing doses of both (R)- and (S)-DOTAP (Fig. 2a). (S)-DOTAP increased CD86 expression more at lower doses (45 μM) and the peak of (R)-DOTAP-induced CD86 expression came at higher doses (150 μM); however, the difference was not significant. Expression of chemokine CCL2 was increased in (R)-DOTAP-treated BMDC at doses of 45, 75, and 100 μM compared to (S)-DOTAP; however, the difference was not significant (Fig. 2b). After an 18 h treatment with either lipid, toxicity to BMDC was similar shown by the increased signal from PI (Fig. 2c). Dose-dependent effects were observed with greater toxicity at higher doses, showing an increase in toxicity in 75 μM up through the highest dose tested. All toxicity levels were much higher than the LPS-treated BMDC. We believe the BMDC model is not sufficient to explain the profound difference in (R)- and (S)-DOTAP activity, thus moved to whole-body experiments to examine the immune response in the complete mouse.
Fig. 2.

Chiral lipid interaction with BMDC. BMDC were treated with (R)-DOTAP or (S)-DOTAP liposomes (free of peptide) for 18 h and a dose-dependent activation of CD86 was evaluated (n = 3), as well as b dose-dependent CCL2 secretion after 24 h treatment (n = 3). c BMDCs were also evaluated for toxicity by PI staining of cells after 18 h treatment (one representative experiment shown, repeated in triplicate)
IFN-γ production by CD8+ T cells from vaccinated mice
When comparing naïve, tumor-bearing, (R)-DOTAP/E7-treated tumor-bearing mice, and (S)-DOTAP/E7-treated tumor-bearing mice for IFN-γ production by CD8+ T cells, (R)-DOTAP/E7-treated mice showed an antigen-specific response (Fig. 3a). Splenocytes isolated from (R)-DOTAP/E7-treated tumor-bearing mice that had been pulsed with E7 peptide for 6 h produced IFN-γ at a magnitude of 0.8% of the CD8+ T cells present, while the NP peptide-pulsed control splenocytes produced 0.0%. Splenocytes isolated from naïve mice, tumor-bearing mice, and (S)-DOTAP/E7-treated mice, whether pulsed with NP or E7 peptide, produced 0.0% CD8+ T cells. The difference between IFN-γ-secreting CD8+ cells per 104 CD8+ T cells for the two groups was statistically significant (P < 0.01) (Fig. 3b).
Fig. 3.

IFN-γ production from splenocytes of (R)-DOTAP/E7- or (S)-DOTAP/E7-treated mice. a Splenocytes isolated (on day 14) from tumor-bearing mice treated with (R) or (S)-DOTAP/E7 at 300 nmol/20 nmol (on day 6). Splenocytes were pulsed with E7 or an irrelevant peptide (NP) for 6 h, washed, and stained for CD8 and IFN-γ. The numbers on the contour plots indicate percentage of IFN-γ+ CD8+ T cells out of all CD8+ T cells. One representative experiment is shown. b IFN-γ-secreting CD8+ cells per 104 CD8+ T cells after 6 h pulse with peptide. n = 6–8. **: P < 0.01 c IFN-γ production after 12 h pulse with either NP or E7 peptide. d IFN-γ-secreting CD8+ cells per 104 CD8+ T cells after 12 h pulse with peptide. n = 6-8. *: P < 0.02
As no groups but (R)-DOTAP/E7-treated mice pulsed with E7 produced IFN-γ, we incubated the cells with NP or E7 peptide for 12 h to evaluate if a longer incubation would show IFN-γ production in any other group (Fig. 3c). However, while all the NP peptide-pulsed samples remained at 0.0%, as expected, (R)-DOTAP/E7 samples pulsed with E7 increased to 1.2% IFN-γ-producing cells out of all CD8+ cells. (S)-DOTAP/E7-treated samples increased from 0.0% at 6 h to 0.1% at 12 h. Even with this increase, the difference between the (R)- and (S)-DOTAP/E7 groups was still significant (P < 0.02) for the number of IFN-γ-secreting CD8+ cells per 104 CD8+ T cells (Fig. 3d).
In vivo cytotoxic T-lymphocyte assay
As expected, tumor-bearing mice injected with NP (CFSElow)- and E7 (CFSEhigh)-pulsed cells showed no specific killing of either population (Fig. 4a) with the two peaks being of equal height and indeed containing similar numbers of cells. Shown in Fig. 4b, (R)-DOTAP/E7-vaccinated mice either with or without a tumor showed similar levels of specific killing of E7-labeled targets that was statistically significant compared to tumor-bearing mice (P < 0.001). (S)-DOTAP/E7-treated mice, even without a tumor, still were statistically different than either (R)-DOTAP/E7-treated group (P < 0.05). Tumor-bearing mice treated with (S)-DOTAP/E7 had specific killing of E7-labeled targets that was not different from tumor-bearing mice that had not been vaccinated (P = 0.20). These results led us to conclude the tumor responds to how the formulation is affecting the immune response, thus we examined the tumor.
Fig. 4.

In vivo cytotoxic T-lymphocyte (CTL) assay. a Targets pulsed with E7 or an irrelevant peptide (NP) were stained with high (E7) or low (NP) concentrations of CFSE, injected into treated mice and in vivo killing was allowed for 16-20 h, then spleens removed and analyzed by flow cytometry. b Percent specific lysis, P < 0.05 between (R)-DOTAP and (S)-DOTAP groups. No statistical difference exists between the two (R)-DOTAP-treated groups, or the (S)-DOTAP tumor-bearing group and tumor-bearing-untreated group. n = 15–18
Tumor-infiltrating lymphocytes and their actions
Whole tumors from mice were broken down to determine the concentration of infiltrating lymphocytes caused by each treatment (Fig. 5a). Tumor-bearing mice that had not been treated with either formulation had 0.7% CD4+ and 0.2% CD8+ cells out of all cells in the solid tumor. (R)-DOTAP/E7-treated mice had much higher levels of CD8+ cells in their tumors compared to (S)-DOTAP/E7-treated mice, 0.9% versus 0.1%, respectively. However, the differences in levels of CD4+ cells in the three groups were less dramatic, 0.5% in (R)-DOTAP/E7-treated mice, compared to 0.2% in (S)-DOTAP/E7-treated mice. (R)-DOTAP/E7-treated mice had greater numbers of CD4+ and CD8+ cells found in the tumor tissue compared to (S)-DOTAP/E7-treated mice (Fig. 5b). The high levels of CD4+ cells in tumor-bearing mice, similar to (R)-DOTAP/E7 treated mice, prompt further study to determine the phenotype of these cells.
Fig. 5.

Tumor-infiltrating lymphocytes. Tumor-infiltrating lymphocytes assayed from tumors of mice treated with (R) or (S)-DOTAP/E7 a CD4+ and CD8+ cells b CD4+ or CD8+ cells per 10,000 cells in the solid tumor. n = 3–4. When comparing (R)-DOTAP to (S)-DOTAP or tumor only, P < 0.01 for CD8+ cells; however, (S)-DOTAP compared to tumor alone, P = 0.57. Looking at the groups with regard to CD4+ cells, all comparisons yield P values greater than 0.3
Lymphocytes isolated from tumors using a density gradient protocol were stimulated with NP or E7 peptides for 6 h and assayed for IFN-γ production (Fig. 6a). All samples showed some IFN-γ activity in their CD8+ cells in both NP- and E7-pulsed treatments. Background levels of IFN-γ production were determined to be the signal from CD8+ NP-pulsed cells. These levels in tumor only, (R)-DOTAP/E7- and (S)-DOTAP/E7-treated mice were 1.2, 10.3, and 3.9%, respectively. CD8+ T cells from (S)-DOTAP/E7 treated mice showed some non-specific activity above control, yet not to the level of cells from (R)-DOTAP/E7-treated mice. However, 32.6% of CD8+ E7-pulsed cells produced IFN-γ when isolated from a (R)-DOTAP/E7-treated mouse. This is compared to 12.0% in an (S)-DOTAP/E7-treated mouse, a significant difference in IFN-γ-secreting CD8 + T cells (Fig. 6b) (P < 0.05). With the E7 pulse, the tumor-bearing mouse had some activity above background, showing some CD8+ T cells had been activated against the tumor by the process of a natural immune response. This experiment showed that although (S)-DOTAP/E7 does not have activity as great as (R)-DOTAP/E7, the treatment does have some ability to recruit E7 reactive lymphocytes to the tumor.
Fig. 6.

IFN-γ production by tumor-infiltrating lymphocytes. TIL were isolated from tumors of mice vaccinated with (R)-DOTAP/E7 or (S)-DOTAP/E7 and pulsed for 6 h with E7 or an irrelevant peptide (NP). a Percentages are IFN-γ+CD8+ cells out of all CD8+ cells. One dot plot is shown for each group that is representative of three experiments. b IFN-γ+-producing cells per 10,000 CD8+ cells, P < 0.05. n = 6–7
Discussion
With the advancements of chiral separation technology, stereospecific drugs are not solely for academic investigation. The pharmaceutical industry has developed an interest in delivering enantiomerically pure formulations, understanding that the chirality of a drug can affect how the molecule acts in the biological milieu.
By simplifying our formulation to one enantiomer of DOTAP and the E7 peptide, we were able to further investigate the immunological action of the DOTAP lipid in formulation; both in vitro and in vivo. Our results demonstrate (R)-DOTAP is the active component in the DOTAP racemic mixture used by our group in previous vaccine studies. This is evident in dose-dependent tumor growth delay studies (Fig. 1).
(R)-DOTAP and (S)-DOTAP have similar formulation characteristics of size, charge, and encapsulation (Table 1). E7 peptide has two positively charged amino acids (arginine and histidine (only partial)), and its interaction with a cationic lipid is unexpected. Previously, our group correlated increased antigen encapsulation with increased vaccine efficiency when studying an acylated version of E7 peptide, using racemic DOTAP [16]. Thus, even in the presence of (S)-DOTAP (in the racemic mixture), a high amount of E7 lipopeptide loading into the liposomes caused regression of TC-1 tumors both when delivered on 6 and 10 days after tumor inoculation. DOTAP has been hypothesized to facilitate endosomal escape through association with anionic lipids (phosphatidylserine) in the interior of the endosome causing destabilization [17]. Thus, as APC endocytose the vaccine, E7 peptide will be released into the cytosol available to be presented by MHC Class I. As the two lipids have similar encapsulation efficiencies (Table 1), no more antigen could be delivered by the (R)-DOTAP than the (S)-DOTAP formulation into the APC.
DOTAP has previously been shown to induce a tunable amount of ROS which was required for anti-tumor efficacy and correlated with activation, CCL2, and toxicity [18]. In our in vitro studies, (R)- and (S)-DOTAP produce similar levels of activation and toxicity (Fig. 2a, c) when dosed to BMDC; therefore, we do not expect that ROS induction is the mechanism with which (R)- and (S)-DOTAP differ. While ROS levels induced could be the same, the difference in antigen loading does not clearly explain the stark differences in cytokine levels produced by the splenocytes and TIL in an (R)-DOTAP/E7-vaccinated mouse (Figs. 3, 6).
We hypothesize the presence of the tumor during vaccine administration amplifies the difference between (R)- and (S)-DOTAP, while the amplification mechanism is not known (Fig. 7). In vitro, the enantiomeric lipids behave similarly to one another in regards to BMDC activation, cytokine production, and toxicity (Fig. 2). However, the difference between the two molecules in eliciting in vivo CTL response (Fig. 4) is most dramatic and indeed statistically significant when a tumor is present. Strong differences can be seen in splenocyte production of IFN-γ in tumor-bearing-vaccinated mice, (Fig. 3) and most obviously in the tumor growth delay data (Fig. 1). The large numbers of TIL that are immunologically active (Fig. 6) support the tumor growth delay data and our hypothesis. The phenotype of the CD4+ cells in the tumors of (R)-DOTAP/E7-treated mice should be investigated as their function could indicate the exact contribution of the tumor.
Fig. 7.

Proposed tumor interaction with DOTAP enantiomers. In vitro (R)- and (S)-DOTAP show little difference in stimulating BMDC, shown by the size of the straight arrows. Due to an unknown interaction between the immune system and the tumor, the presence of a tumor in the in vivo system amplified the difference between (R)- and (S)-DOTAP reflected by the size of the ribbon arrows. The distinction between the two formulations is further amplified via talk back from the tumor to the immune system
There are three requirements for a peptide vaccine to be effective: an antigen specific for the target (tumor), an efficacious adjuvant that can magnify the second signals needed for dendritic cells to produce a strong T-cell response, and a delivery mechanism. (R)-DOTAP/E7 complexes provide all three to cause regression in this cervical cancer model. Future studies will expand the application of this adjuvant to other tumor models and investigate the mechanism of action that lies behind the enantiospecific activity.
Acknowledgments
Angela Yang’s assistance with tissue processing and formatting of figures is greatly appreciated. This research was supported by PDS Biotechnology Corporation and by NIH grant CA129421. EAV is supported by a Pre-Doctoral Fellowship in Pharmaceutics from the PhRMA Foundation.
Conflict of interest
Grants from PDS Biotechnology Corporation and the NIH have funded this research. LH is a co-founder and on the scientific board of advisors of PDS Biotechnology Corporation, in addition to financial interests in the company. EAV, WC, and LH are named in a US pending patent on the technology described in this paper.
Abbreviations
- BMDC
Bone marrow-derived dendritic cells
- CCL2
Chemokine C–C motif ligand 2
- CFSE
Carboxyfluorescein succinimidyl ester
- (R)-DOTAP
(R)-1,2-dioleoyl-3-trimethylammonium-propane
- (S)-DOTAP
(S)-1,2-dioleoyl-3-trimethylammonium-propane
- HPV
Human Papillomavirus
- IFN-γ
Interferon gamma
- MHC
Major histocompatibility complex
- ROS
Reactive oxygen species
- TIL
Tumor-infiltrating lymphocytes
References
- 1.Palena C, Schlom J. Vaccines against human carcinomas: strategies to improve antitumor immune responses. J Biomed Biotechnol. 2010;2010:380697. doi: 10.1155/2010/380697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4:328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 3.Hsu FJ, Benike C, Fagnoni F, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 4.Syrengelas AD, Chen TT, Levy R. DNA immunization induces protective immunity against B-cell lymphoma. Nat Med. 1996;2:1038–1041. doi: 10.1038/nm0996-1038. [DOI] [PubMed] [Google Scholar]
- 5.De Gregorio E, Tritto E, Rappuoli R. Alum adjuvanticity: unraveling a century old mystery. Eur J Immunol. 2008;38:2068–2071. doi: 10.1002/eji.200838648. [DOI] [PubMed] [Google Scholar]
- 6.Harper DM, Franco EL, Wheeler C, et al. Efficacy of a bivalent L1 virus-like particle vaccine in prevention of infection with human papillomavirus types 16 and 18 in young women: a randomised controlled trial. Lancet. 2004;364:1757–1765. doi: 10.1016/S0140-6736(04)17398-4. [DOI] [PubMed] [Google Scholar]
- 7.Francotte ER. Enantioselective chromatography as a powerful alternative for the preparation of drug enantiomers. J Chromatogr A. 2001;906:379–397. doi: 10.1016/S0021-9673(00)00951-1. [DOI] [PubMed] [Google Scholar]
- 8.FDA’s policy statement for the development of new stereoisomeric drugs Chirality. 1992;4:338–340. doi: 10.1002/chir.530040513. [DOI] [PubMed] [Google Scholar]
- 9.Simonyi M. On chiral drug action. Med Res Rev. 1984;4:359–413. doi: 10.1002/med.2610040304. [DOI] [PubMed] [Google Scholar]
- 10.Schlossman SF, Herman J, Yaron A. Antigen recognition: in vitro studies on the specificity of the cellular immune response. J Exp Med. 1969;130:1031–1045. doi: 10.1084/jem.130.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbier P, Benezra C. Stereospecificity of allergic contact dermatitis (ACD) induced by two natural enantiomers, (+)- and (−)-frullanolides, in guinea pigs. Naturwissenschaften. 1982;69:296–297. doi: 10.1007/BF00396445. [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Yan W, Huang L. A simple but effective cancer vaccine consisting of an antigen and a cationic lipid. Cancer Immunol Immunother. 2008;57:517–530. doi: 10.1007/s00262-007-0390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan W, Chen W, Huang L. Mechanism of adjuvant activity of cationic liposome: phosphorylation of a MAP kinase, ERK and induction of chemokines. Mol Immunol. 2007;44:3672–3681. doi: 10.1016/j.molimm.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 14.Lin KY, Guarnieri FG, Staveley-O’Carroll KF, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 15.Byers AM, Kemball CC, Moser JM, Lukacher AE. Cutting edge: rapid in vivo CTL activity by polyoma virus-specific effector and memory CD8+ T cells. J Immunol. 2003;171:17–21. doi: 10.4049/jimmunol.171.1.17. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Huang L. Induction of cytotoxic T-lymphocytes and antitumor activity by a liposomal lipopeptide vaccine. Mol Pharm. 2008;5:464–471. doi: 10.1021/mp700126c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y, Szoka FC., Jr Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry. 1996;35:5616–5623. doi: 10.1021/bi9602019. [DOI] [PubMed] [Google Scholar]
- 18.Yan W, Chen W, Huang L. Reactive oxygen species play a central role in the activity of cationic liposome based cancer vaccine. J Control Release. 2008;130:22–28. doi: 10.1016/j.jconrel.2008.05.005. [DOI] [PubMed] [Google Scholar]