

ABSTRACT
The transcription factor NRF2 (nuclear factor [erythroid-derived 2]-like 2) plays crucial roles in the defense mechanisms against oxidative stress and mediates anti-inflammatory actions under various pathological conditions. Recent studies showed that the dysfunction of regulatory T cells (Tregs) is directly linked to the initiation and progression of various autoimmune diseases. To determine the Treg-independent impact of NRF2 activation on autoimmune inflammation, we examined scurfy (Sf) mice, which are deficient in Tregs and succumb to severe multiorgan inflammation by 4 weeks of age. We found that systemic activation of NRF2 by Keap1 (Kelch-like ECH-associated protein 1) knockdown ameliorated tissue inflammation and lethality in Sf mice. Activated T cells and their cytokine production were accordingly decreased by Keap1 knockdown. In contrast, NRF2 activation through cell lineage-specific Keap1 disruption (i.e., in T cells, myeloid cells, and dendritic cells) achieved only partial or no improvement in the inflammatory status of Sf mice. Our results indicate that systemic activation of NRF2 suppresses effector T cell activities independently of Tregs and that NRF2 activation in multiple cell lineages appears to be required for sufficient anti-inflammatory effects. This study emphasizes the possible therapeutic application of NRF2 inducers in autoimmune diseases that are accompanied by Treg dysfunction.
KEYWORDS: KEAP1, NRF2, scurfy mice, inflammation, autoimmune diseases, scurfy mouse, autoimmunity
INTRODUCTION
The Kelch-like ECH-associated protein 1–nuclear factor (erythroid-derived 2)-like 2 (KEAP1-NRF2) system plays important roles in the antioxidant response and contributes to cytoprotection from various redox disturbances (1, 2). Under normal conditions, the transcription factor NRF2 is negatively regulated by KEAP1 via polyubiquitination and subsequent degradation. When cells are exposed to oxidative stress, NRF2 escapes from KEAP1-mediated degradation, translocates into the nucleus, and enhances the expression of various genes encoding antioxidant proteins and detoxifying enzymes.
In addition to this antioxidant function, we recently showed that NRF2 exerts anti-inflammatory effects by decreasing the mRNA levels of proinflammatory cytokine genes, including Il6 and Il1b (3). Indeed, the critical contributions of NRF2 to reduced inflammation have been demonstrated in mouse models of various pathological conditions, such as elastase-induced emphysema (4), cecal ligation and puncture-induced sepsis (5), dextran sulfate sodium-induced colitis (6), allergen-driven airway inflammation (7), and dystrophin-deficient muscular dystrophy (8).
NRF2 has also been shown to mitigate autoimmune-mediated inflammation. NRF2 deficiency exacerbates rheumatoid arthritis (RA) (9) and systemic lupus erythematosus (SLE) (10, 11), while NRF2 activation ameliorates experimental autoimmune encephalomyelitis (3). These studies exploited experimentally induced autoimmunity in mice, which does not completely mimic the pathogenesis of autoimmune diseases in humans. At present, decreased numbers and/or functional impairments of regulatory T cells (Tregs), which enable the aberrant activation of autoreactive T cells (12–15), have been shown to contribute to autoimmune conditions in human patients, such as those with RA, SLE, primary Sjögren's syndrome, and multiple sclerosis (MS) (16–18). Under autoimmune-mediated inflammatory conditions, Tregs are suppressed due to the high levels of proinflammatory cytokines that are produced by other immune cells and/or tissue cells. To correct autoimmunity by reactivating Tregs and subsequently restoring self-tolerance, the activities of the cells that produce proinflammatory cytokines need to be controlled (17–19). Thus, in addition to the direct modulation of Tregs to increase their beneficial activities, appropriate control of inflammatory cells other than Tregs is important for the improvement of autoimmune diseases.
A recent study demonstrated that T cell-specific activation of NRF2 increases the number of Tregs (20), which suggests that NRF2 inhibits the inflammatory response by potentiating Treg-mediated immune suppression. However, it remains unknown whether NRF2 has any inhibitory effects on autoimmune-mediated inflammation in a Treg-independent manner.
To determine whether NRF2 activation exerts Treg-independent suppression of autoimmune-mediated inflammation, we used scurfy (Sf) mice, which possess a missense mutation in the Foxp3 gene on the X chromosome (21). Because the development and maintenance of Tregs largely depend on the transcription factor FOXP3 (22, 23), Sf mice are almost completely deficient in functional Tregs and thereby develop severe multiorgan inflammation with hyperactivation of autoreactive effector T cells, which results in lethality by 4 weeks of age (24). Thus, we used Sf mice to investigate the Treg-independent suppressive effects of NRF2 on the activation status of inflammatory cells, especially effector T cells.
We found that systemic activation of NRF2 induced by Keap1 knockdown mitigated tissue inflammation and improved the survival of Sf mice. NRF2 also suppressed the activation of effector T cells and reduced their cytokine production. Almost comparable but modest outcomes were observed following the pharmacological activation of NRF2 in Sf mice by administration of an NRF2 inducer, CDDO-Im {oleanolic triterpenoid 1-[2-cyano-3,12-dioxooleane-1,9(11)-dien-28-oyl] imidazole}. In contrast, NRF2 activation by Keap1 disruption in a cell lineage-specific manner, including disruptions in T cells, myeloid cells, and dendritic cells, induced only partial or no improvement in the inflammatory status of Sf mice. These results indicate that systemic activation of NRF2 suppresses the autoimmune response in a Treg-independent manner and suggest that coordination of multiple cell lineages is essential for the NRF2-mediated anti-inflammatory effects in autoimmune diseases with Treg dysfunction.
RESULTS
Systemic NRF2 activation by Keap1 knockdown alleviates multiple-organ inflammation and improves the survival rate of Sf mice.
To investigate the effects of NRF2 activation on the inflammatory milieu resulting from autoimmunity, we genetically activated NRF2 by reducing Keap1 expression in Sf (Foxp3sf/Y) mice. Foxp3sf/X female mice were crossed with male mice possessing knockout (Keap1−) (25) and knockdown-floxed (Keap1FA) (26, 27) alleles of Keap1 to generate Sf::Keap1FA/− (Foxp3sf/Y::Keap1FA/−) mice. We confirmed the decreased expression of Keap1 in representative tissues of mice with the Keap1FA/− background, which was accompanied by increased expression of Nqo1, one of the typical NRF2 target genes, and elevated accumulation of NRF2 protein (Fig. 1). These results indicated successful activation of NRF2 in Sf::Keap1FA/− mice.
FIG 1.

NRF2 activation in representative tissues of Sf mice in the Keap1 knockdown background. (A) Gene expression of Keap1 and an NRF2 target gene, Nqo1, in WT (n = 4), Keap1FA/− (n = 4), Sf (n = 4), and Sf::Keap1FA/− (n = 4) mice. The expression levels were normalized to Gapdh expression, and the expression level in WT mice was set to 1. The values represent the means and SD. (B) Immunoblot analysis of the NRF2 protein in WT, Keap1FA/−, Sf, and Sf::Keap1FA/− mice. α-Tubulin was used as a loading control. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
We first compared the severities of systemic inflammation in Sf::Keap1FA/− mice and Sf mice at the age of 15 to 19 days, which is generally before Sf mice succumb to the disease. The anemia and thrombocytopenia typically found in Sf mice were almost completely improved in Sf::Keap1FA/− mice (Table 1), and the inflammatory skin lesions found on the pinnae and tails of Sf mice were also improved in Sf::Keap1FA/− mice (Fig. 2A). Histological analysis further showed that the infiltration of immune cells was decreased in the skin, liver, and lungs in Sf::Keap1FA/− mice (Fig. 2B). Accordingly, Sf::Keap1FA/− mice exhibited an improved survival rate compared to that of Sf mice (Fig. 2C). However, approximately half of the Sf::Keap1FA/− mice died by 25 days of age, which is very similar to the rate for Sf mice, despite the apparent inhibition of the inflammatory response. This early-phase lethality in Sf::Keap1FA/− mice was likely caused by feeding difficulties due to hyperkeratotic lesions within the upper digestive tract, which is an issue inherent to Keap1FA/− mice (27). A recent report suggested that severe KEAP1 insufficiency causes other complex abnormalities (28), which might have led to the early lethality of Sf::Keap1FA/− mice. Sf::Keap1FA/− mice were consistently smaller than wild-type (WT) and Sf mice at the same age (Fig. 2D and E). It should be noted that all Sf::Keap1FA/− mice that avoided the early onset of lethality survived thereafter.
TABLE 1.
Peripheral blood analysis of Sf::Keap1FA/− micea
| Parameter | Valueb for: |
P value |
|||
|---|---|---|---|---|---|
| WT mice | Sf mice | Sf::Keap1FA/− mice | WT vs Sf mice | Sf vs Sf::Keap1FA/− mice | |
| WBC count (102/μl) | 60 ± 18 | 61 ± 25 | 48 ± 16 | NS | NS |
| RBC count (104/μl) | 513 ± 117 | 374 ± 60 | 495 ± 102 | <0.005 | <0.005 |
| Hb concn (g/dl) | 7.2 ± 1.5 | 5.4 ± 1.0 | 7.0 ± 1.3 | <0.006 | <0.007 |
| Hct (%) | 24.2 ± 5.9 | 18.7 ± 2.8 | 26.3 ± 5.8 | <0.05 | <0.007 |
| MCV (fl) | 47.1 ± 2.4 | 50.2 ± 1.1 | 50.4 ± 1.5 | <0.02 | NS |
| MCH level (pg) | 14.2 ± 1.3 | 14.5 ± 1.5 | 14.3 ± 0.8 | NS | NS |
| MCHC (g/dl) | 30.1 ± 2.1 | 28.9 ± 2.6 | 28.3 ± 1.7 | NS | NS |
| PLT count (104/μl) | 108 ± 31 | 82 ± 17 | 143 ± 42 | <0.04 | <0.004 |
WBC, white blood cell; RBC, red blood cell; Hb, hemoglobin; Hct, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; PLT, platelet; NS, not significant.
Values are presented as the means ± SD for WT (n = 7), Sf (n = 11), and Sf::Keap1FA/− (n = 8) mice.
FIG 2.

Systemic NRF2 activation by Keap1 knockdown alleviates tissue inflammation in Sf mice. (A) Macroscopic observation of the pinnae and tails of WT, Sf, and Sf::Keap1FA/− mice. (B) HE staining of the skin (pinnae), livers, and lungs of WT, Sf, and Sf::Keap1FA/− mice. Arrows indicate leukocyte infiltration. (C) Survival curves for WT (n = 30), Sf (n = 26), and Sf::Keap1FA/− (n = 17) mice. (D) Macroscopic observation of WT, Sf, and Sf::Keap1FA/− mice. (E) Body weight measurements of WT (n = 10), Sf (n = 10), and Sf::Keap1FA/− (n = 10) mice at 14 days of age. The values represent the means and SD. *, P < 0.05; ***, P < 0.0005.
To further confirm the anti-inflammatory effect of NRF2 in Sf mice, we used additional Keap1 knockdown mice, i.e., Keap1FA/+ and Keap1FA/FA mice, which have higher Keap1 expression than that of Keap1FA/− mice (Fig. 3A and B) and do not suffer from feeding difficulties or other abnormalities. All Sf::Keap1FA/+ and Sf::Keap1FA/FA mice exhibited increased survival rates compared to that for the Sf mice (Fig. 3C). Importantly, Sf, Sf::Keap1FA/+, and Sf::Keap1FA/FA mice showed a decreasing order of Keap1 expression and an increasing order of NRF2 protein and Nqo1 mRNA levels (Fig. 3A and B). In good agreement with the graded NRF2 activity, Sf, Sf::Keap1FA/+, and Sf::Keap1FA/FA mice tended to show an increasing order of survival, implying a correlation between NRF2 activation and improved survival, although the difference between Sf::Keap1FA/+ and Sf::Keap1FA/FA mice did not reach statistical significance (Fig. 3C). Collectively, these results suggest that systemic activation of NRF2 by Keap1 knockdown effectively alleviates the chronic inflammation caused by autoimmunity in Sf mice.
FIG 3.

Graded expression of Keap1 and NRF2 activation in Sf, Sf::Keap1FA/+, Sf::Keap1FA/FA, and Sf::Keap1FA/− mice. The mice were analyzed 17 to 20 days after birth. (A) Gene expression of Keap1 and Nqo1 in Sf (n = 3), Sf::Keap1FA/+ (n = 3), Sf::Keap1FA/FA (n = 3), and Sf::Keap1FA/− (n = 3) mice. The expression levels were normalized to Gapdh expression, and the expression level in Sf mice was set to 1. The values represent the means and SD. (B) Immunoblot analysis of KEAP1 and NRF2 in Sf, Sf::Keap1FA/+, Sf::Keap1FA/FA, and Sf::Keap1FA/− mice. α-Tubulin was used as a loading control. (C) Survival curves for Sf::Keap1FA/+ (n = 22) and Sf::Keap1FA/FA (n = 26) mice. The data presented in Fig. 2C (survival of WT, Sf, and Sf::Keap1FA/− mice) are shown for comparison. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
Systemic NRF2 activation by Keap1 knockdown suppresses the activation of effector T cells and their cytokine production.
Abnormal activation and proliferation of CD4+ effector T cells have been shown to be major pathogenic factors in Sf mice (29). We next examined the effect of NRF2 activation on the status of effector T cells and their cytokine expression levels. As an initial analysis, the spleen/body weight and lymph node (LN)/body weight ratios were calculated to evaluate the proliferation of leukocytes (Fig. 4A and B). The spleen/body weight and LN/body weight ratios were markedly elevated for Sf mice compared to those for the WT mice. Although the spleen/body weight ratios were not different between the groups, the LN/body weight ratio was significantly lower for Sf::Keap1FA/− mice than for Sf mice, implying that NRF2 activation is effective at suppressing lymphocyte proliferation in LNs.
FIG 4.

Systemic NRF2 activation inhibits the activation of T cells in Sf mice. (A) Ratios of the weights of the spleens and LNs (bilateral axillary, brachial, and inguinal LNs) to the whole-body weights of WT (n = 10), Sf (n = 10), and Sf::Keap1FA/− (n = 10) mice at 17 days of age. The values represent the means and SD. (B) Macroscopic observation of the spleens and LNs of WT, Sf, and Sf::Keap1FA/− mice. (C) Frequencies of T cell populations in the LNs of WT (n = 5), Sf (n = 4), and Sf::Keap1F/− (n = 8) mice. Percentages of CD4 and CD8 single-positive cells in LN cells (left) and percentages of CD25+, CD44+, and CD69+ cells in CD4 single-positive cells (right) are shown. The values indicate the means and SD. (D) Representative histogram data for CD4 single-positive LN cells from WT, Sf, and Sf::Keap1FA/− mice at 15 to 19 days of age, with percentages of CD25+, CD44+, and CD69+ cells. NS, not significant; *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
We then examined the activation status of CD4+ CD8− and CD4− CD8+ T cells (here referred to as CD4+ and CD8+ T cells, respectively) in LNs. The frequencies of CD4+ and CD8+ T cells were not different between Sf and Sf::Keap1FA/− mice (Fig. 4C, left panel). Among CD4+ T cells, activated populations of CD25+, CD44+, and CD69+ cells were markedly increased in Sf mice compared to those in the WT mice, which is consistent with previous findings (29, 30). Moreover, this aberrant increase was mitigated in Sf::Keap1FA/− mice (Fig. 4C, right panel, and D), suggesting that NRF2 activation inhibits the activation of CD4+ T cells.
This observation was verified by assessing the number of cytokine-positive CD4+ effector T cells, which differentiate from naive CD4+ T cells when they are activated by T cell receptor stimulation. Consistent with the results of a previous study (31), Sf mice exhibited dramatic increases in Th1-, Th2-, and Th17-type effector CD4+ T cells, which are marked by the production of interferon gamma (IFN-γ), interleukin-4 (IL-4), and IL-17A, respectively (Fig. 5). This increase in cytokine-producing T cells was mitigated in Sf::Keap1FA/− mice. In particular, Sf::Keap1FA/− mice exhibited a dramatic decrease in the frequency of IFN-γ+ CD4+ T cells, by more than 80%, compared to that in Sf mice, and IFN-γ+ CD8+ T cells were also similarly decreased in Sf::Keap1FA/− mice. These results suggest that systemic NRF2 activation suppresses the activation of effector T cells and their cytokine production in autoimmune diseases induced by Treg deficiency.
FIG 5.

Systemic NRF2 activation inhibits cytokine production of T cells in Sf mice. (A) Frequencies of effector T cell subsets in the LNs of WT (for IL-10+ cells, n = 4; for others, n = 7), Sf (for IL-10+ cells, n = 3; for others, n = 6), and Sf::Keap1F/− (for IL-10+ cells, n = 4; for others, n = 8) mice. Percentages of the IFN-γ+, IL-4+, IL-17A+, and IL-10+ fractions of CD4 single-positive cells and the percentage of the IFN-γ+ fraction of CD8 single-positive cells are shown. The values represent the means and SD. (B) Representative contour plot data for the IFN-γ+, IL-4+, IL-17A+, and IL-10+ fractions of CD4 single-positive LN cells and the IFN-γ+ fraction of CD8 single-positive LN cells, with the percentages of cytokine-positive fractions shown. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
Previous studies showed that there are Foxp3-independent T cell populations, such as type 1 regulatory T (Tr1) cells, which suppress autoimmunity through secretion of high levels of IL-10 and transforming growth factor beta (TGF-β) (32, 33). Because IL-10-producing T cells do not rely on Foxp3 expression for their differentiation and function (34, 35), these cells may contribute to the NRF2-mediated suppression of autoimmune responses in Sf mice, which are deficient in Foxp3. To examine this possibility, we measured the frequency of IL-10+ CD4+ T cells. However, it was not increased but rather decreased in Sf::Keap1FA/− mice compared to that in Sf mice (Fig. 5). Thus, the anti-inflammatory effect of NRF2 activation in Sf mice is not due to the expansion of IL-10-producing suppressive T cells but to the inhibition of effector T cells, particularly those producing IFN-γ.
IFN-γ production is dramatically suppressed by systemic NRF2 activation.
The inflammatory symptoms in Sf mice are highly dependent on the types of cytokines secreted into the serum; for instance, IFN-γ is abundantly secreted in Sf mice, and its suppression results in prolonged survival and delayed inflammation in the pinnae, skin, lungs, and liver (36). To investigate the effects of NRF2 activation on systemic cytokine production, we assessed serum cytokine levels. In Sf mice, the serum concentrations of proinflammatory cytokines, such as IFN-γ, tumor necrosis factor alpha (TNF-α), and IL-6, were elevated, while the levels of IFN-γ and TNF-α were decreased to nearly basal levels in Sf::Keap1FA/− mice (Fig. 6A). However, there were no significant changes in the serum concentrations of IL-4 and IL-17A between Sf and Sf::Keap1FA/− mice. We also attempted to measure the concentration of IL-10, but it was not detectable (data not shown). These results suggest that the reductions of the serum levels of IFN-γ and TNF-α are closely related to the suppression of autoimmune inflammation by NRF2.
FIG 6.

Production of IFN-γ is dramatically reduced in Sf mice in the Keap1 knockdown background. (A) Serum cytokine levels in WT (n = 10), Keap1FA/− (n = 9), Sf (n = 10), and Sf::Keap1FA/− (n = 10) mice. The horizontal bars represent the means. (B) Gene expression of Ifng in the LNs and spleens of WT (n = 4), Keap1FA/− (n = 4), Sf (n = 4), and Sf::Keap1FA/− (n = 4) mice. The expression levels were normalized to Gapdh expression, and the expression level in WT mice was set to 1. The values represent the means and SD. NS, not significant; *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
Because most of the serum IFN-γ is thought to be derived from T cells and natural killer cells (37), we quantified the mRNA levels of Ifng in lymphoid tissues, i.e., the LNs and spleen, to determine whether the reduced production of IFN-γ in Sf::Keap1FA/− mice was caused by a decrease in its mRNA. The Ifng mRNA level was significantly higher in Sf mice than in WT mice, whereas Sf::Keap1FA/− mice showed an Ifng mRNA level almost comparable to that in WT mice, suggesting that NRF2 activation decreases Ifng mRNA in the LNs and spleen (Fig. 6B).
T cell-specific loss of Keap1 attenuates IFN-γ production in T cells and partially improves inflammation.
To determine whether NRF2 activation exerts anti-inflammatory effects in a T cell-autonomous manner, we conducted T cell-specific Keap1 disruption in an Sf background (Sf::Keap1FB/FB::Lck-Cre [Sf::Keap1-TKO]) and compared the resulting animals to Sf::Control mice (Sf::Keap1FB/FB) by utilizing another Keap1 floxed allele, Keap1FB, that expresses the Keap1 gene in an amount comparable to that of the wild-type allele (38). We confirmed that the Keap1 gene was deleted in approximately 70% of T cells, judging from the reduction of Keap1 expression, while the NRF2 target gene Nqo1 was upregulated in the T cells of Sf::Keap1-TKO mice (Fig. 7A). From macroscopic observations, the skin inflammation in the pinnae and tails of Sf::Keap1-TKO mice appeared to be ameliorated (Fig. 7B). Indeed, histological analysis of pinna skin sections showed that the thickening and cellular infiltration observed in Sf::Control mice were reduced in Sf::Keap1-TKO mice (Fig. 7C). However, the heavy infiltration of inflammatory cells in the liver and lungs, as well as anemia and thrombocytopenia, was not improved in Sf::Keap1-TKO mice (Fig. 7C; see Table S1 in the supplemental material).
FIG 7.

T cell-specific Keap1 deletion alleviates skin inflammation. (A) Gene expression of Keap1 and Nqo1 in T cells (CD3e+) and the rest of the cells (CD3e−) from the spleens of control (n = 3) and Sf::Keap1-TKO (n = 3) mice. The expression levels were normalized to Gapdh expression, and the expression level in control mice was set to 1. The values represent the means and SD. (B) Macroscopic observation of the pinnae and tails of control, Sf::Control, and Sf::Keap1-TKO mice. (C) HE staining of the skin (pinnae), livers, and lungs of control, Sf::Control, and Sf::Keap1-TKO mice. Arrows indicate leukocyte infiltration. NS, not significant; *, P < 0.05.
Analysis of LN cells showed that the frequencies of CD4+ and CD8+ T cells were decreased in the Sf::Keap1-TKO mice compared to those in the Sf::Control mice (Fig. 8A, left panel). The frequencies of CD4+ T cells expressing activation markers were not different between Sf::Control and Sf::Keap1-TKO mice, although the frequency of CD25+ CD4+ cells was marginally decreased in the latter group (Fig. 8A, right panel, and B), suggesting that the activation of CD4+ T cells is not suppressed by T cell-specific Keap1 deficiency.
FIG 8.
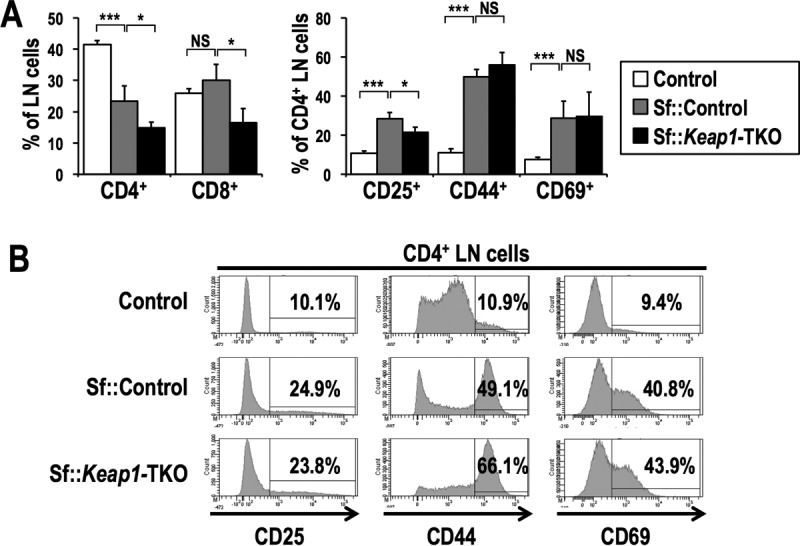
T cell-specific Keap1 deletion does not alter phenotypic activation of T cells in Sf mice. (A) Frequencies of T cell populations in the LNs of control (n = 4), Sf::Control (n = 6), and Sf::Keap1-TKO (n = 3) mice. Percentages of CD4 and CD8 single-positive cells in LN cells (left) and percentages of CD25+, CD44+, and CD69+ cells in CD4 single-positive cells (right) are shown. The values indicate the means and SD. (B) Representative histogram data for CD4 single-positive LN cells from control, Sf::Control, and Sf::Keap1-TKO mice, with percentages of CD25+, CD44+, and CD69+ cells. NS, not significant; *, P < 0.05; ***, P < 0.0005.
Despite the similar phenotypic activation of CD4+ T cells between Sf::Control and Sf::Keap1-TKO mice, the frequencies of CD4+ and CD8+ T cells positive for IFN-γ were significantly decreased in Sf::Keap1-TKO mice, while those of CD4+ T cells positive for IL-4 or IL-17A were unaffected (Fig. 9). These results confirm that the reduction of IFN-γ by NRF2 activation in T cells is a cell-autonomous effect, although the extent of this reduction is not sufficient for the suppression of effector T cell activities, which may explain the limited improvement in inflammation in Sf::Keap1-TKO mice. Thus, NRF2 activation in cell lineages other than T cells is required to recapitulate the anti-inflammatory effects observed in the Keap1 knockdown background.
FIG 9.

T cell-specific Keap1 deletion decreases production of IFN-γ in T cells of Sf mice. (A) Frequencies of effector T cell subsets in the LNs of control (n = 5), Sf::Control (n = 8), and Sf::Keap1-TKO (n = 5) mice. The percentages of the IFN-γ+, IL-4+, and IL-17A+ fractions of CD4 single-positive cells and the percentage of the IFN-γ+ fraction of CD8 single-positive cells are shown. The values represent the means and SD. (B) Representative contour plot data for the IFN-γ+, IL-4+, and IL-17A+ fractions of CD4 single-positive LN cells and the IFN-γ+ fraction of CD8 single-positive LN cells, with the percentages of cytokine-positive fractions shown. NS, not significant; *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
Because NRF2 has been well described as an anti-inflammatory factor in macrophages and dendritic cells (3, 5, 39, 40), we next examined the contribution of NRF2 activation in these antigen-presenting cells to the suppression of autoimmune inflammation. To this end, we conducted myeloid cell-specific and dendritic cell-specific Keap1 disruption in the Sf background and generated Sf::Keap1FB/FB::LysM-Cre (Sf::Keap1-MKO) and Sf::Keap1FB/FB::CD11c-Cre (Sf::Keap1-DKO) mice, respectively. Contrary to our expectations, neither of these Sf mouse strains exhibited any attenuation of inflammation (Fig. 10; Tables S2 and S3). These results indicate that NRF2 activation in macrophages or dendritic cells alone is not sufficient to suppress the autoimmune response in Sf mice. Collectively, NRF2 activation in multiple cell lineages and the coordination of NRF2-driven cellular functions appear to be required for the sufficient anti-inflammatory effects in autoimmune diseases caused by Treg deficiency.
FIG 10.

Dendritic cell- or myeloid cell-specific Keap1 deletion does not ameliorate the autoimmune response in Sf mice. (A and C) Macroscopic observations of Sf::Keap1-MKO (A) and Sf::Keap1-DKO (C) mice compared to control and Sf::Control mice. (B and D) Frequencies of T cell populations in the LNs of Sf::Keap1-MKO (n = 5) (B) and Sf::Keap1-DKO (n = 4) (D) mice compared to control (n = 6 for panel B; n = 4 for panel D) and Sf::Control (n = 7 for panel B; n = 4 for panel D) mice. Percentages of CD4 and CD8 single-positive cells in LN cells (left) and percentages of CD25+, CD44+, and CD69+ cells in CD4 single-positive cells (right) are shown. The values indicate the means and SD. NS, not significant; *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
The NRF2 inducer CDDO-Im reduces inflammation in Sf mice.
Finally, to determine whether pharmacological activation of NRF2 suppresses the autoimmune response in Sf mice, we treated WT and Sf mice with four intraperitoneal injections of CDDO-Im (5 μmol/kg of body weight) or vehicle on alternating days starting 7 or 8 days after birth. We examined these mice one day after the final injection (Fig. 11A). CDDO-Im treatment increased the NRF2 protein levels and upregulated the expression of Nqo1 in the spleen, skin, liver, and lungs (Fig. 11B and C). Although the decreases in red blood cell and platelet counts were not significantly different between vehicle-treated and CDDO-Im-treated Sf mice (Table S4), histological analysis revealed that inflammatory cell infiltration in the skin and liver, but not in the lungs, was decreased by CDDO-Im treatment (Fig. 11D). CDDO-Im treatment tended to improve the survival rate of Sf mice, although the effect did not reach statistical significance (Fig. 11E).
FIG 11.

CDDO-Im treatment partially alleviates inflammation in Sf mice. (A) Regimen of CDDO-Im treatment. WT and Sf mice were injected intraperitoneally with vehicle or CDDO-Im every other day starting 7 or 8 days after birth. Mice were analyzed one day after the final treatment. (B) Immunoblot analysis of the NRF2 protein in representative tissues of WT and Sf mice treated with vehicle or CDDO-Im. α-Tubulin was used as a loading control. (C) Gene expression of Nqo1 in representative organs of vehicle-treated WT (n = 4; white bars), CDDO-Im-treated WT (n = 4; light gray bars), vehicle-treated Sf (n = 4; dark gray bars), and CDDO-Im-treated Sf (n = 4; black bars) mice. The expression levels were normalized to Gapdh expression, and the expression level in vehicle-treated WT mice was set to 1. The values represent the means and SD. *, P < 0.05. (D) HE staining of the skin (pinnae), livers, and lungs of vehicle or CDDO-Im-treated Sf mice. Arrows indicate leukocyte infiltration. (E) Survival curves for Sf mice treated with vehicle (n = 5) or CDDO-Im (n = 5). Treatment was continued throughout the observation period.
The analysis of LN cells showed that CDDO-Im treatment did not change the ratios of CD4+ and CD8+ T cells but decreased the frequencies of activated CD4+ T cells in Sf mice (Fig. 12). CDDO-Im treatment also decreased the frequencies of IFN-γ+ CD4+ and IFN-γ+ CD8+ T cells, whereas it did not affect other cytokine-producing cells (Fig. 13A and B). Furthermore, CDDO-Im treatment tended to reduce Ifng mRNA expression in the spleen (Fig. 13C), consistent with the results for Sf::Keap1FA/− mice (Fig. 6B). Collectively, these results indicate that the NRF2 inducer CDDO-Im alleviates inflammation by suppressing autoimmune responses in the context of Treg deficiency.
FIG 12.
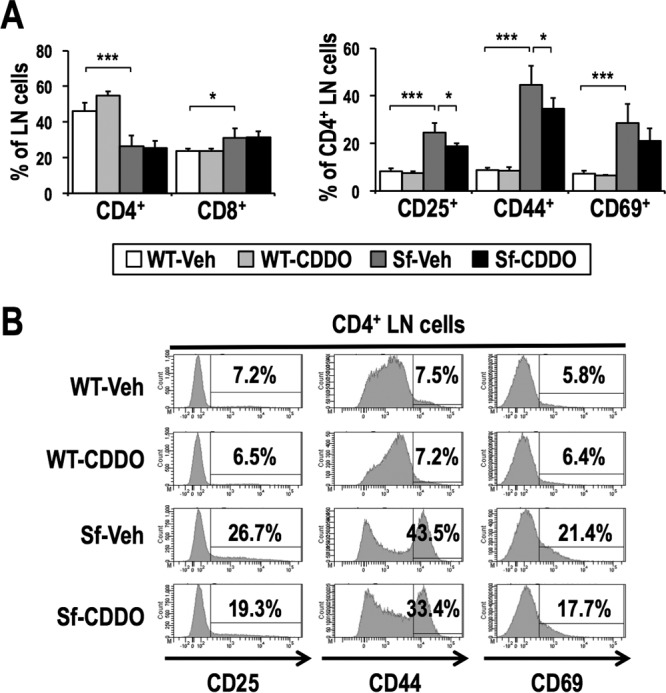
CDDO-Im treatment partially inhibits activation of T cells in Sf mice. (A) Frequencies of T cell populations in the LNs of vehicle-treated WT (n = 5), CDDO-Im-treated WT (n = 4), vehicle-treated Sf (n = 7), and CDDO-Im-treated Sf (n = 7) mice. Percentages of CD4 and CD8 single-positive cells in LN cells (left) and percentages of CD25+, CD44+, and CD69+ cells in CD4 single-positive cells (right) are shown. The values indicate the means and SD. (B) Representative histogram data for CD4 single-positive LN cells from WT and Sf mice treated with vehicle or CDDO-Im, along with percentages of CD25+, CD44+, and CD69+ cells. *, P < 0.05; ***, P < 0.0005.
FIG 13.

CDDO-Im treatment reduces IFN-γ production. (A) Frequencies of effector T cell subsets in the LNs of vehicle-treated WT (n = 5), CDDO-Im-treated WT (n = 4), vehicle-treated Sf (n = 7), and CDDO-Im-treated Sf (n = 7) mice. The percentages of the IFN-γ+, IL-4+, IL-17A+, and IL-10+ fractions of CD4 single-positive cells and of the IFN-γ+ fraction of CD8 single-positive cells are shown. The values represent the means and SD. (B) Representative contour plot data for the IFN-γ+, IL-4+, IL-17A+, and IL-10+ fractions of CD4 single-positive LN cells and the IFN-γ+ fraction of CD8 single-positive LN cells, with the percentages of cytokine-positive fractions shown. (C) Ifng gene expression in the spleens of vehicle-treated WT (n = 4), CDDO-Im-treated WT (n = 4), vehicle-treated Sf (n = 4), and CDDO-Im-treated Sf (n = 4) mice. The expression levels were normalized to Gapdh expression, and the expression level in vehicle-treated WT mice was set to 1. The values represent the means and SD. NS, not significant; *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
DISCUSSION
In this study, we found that systemic activation of NRF2 improves the inflammatory milieu in the context of Treg-deficient autoimmunity in Sf mice. Activation of effector T cells in Sf mice was suppressed in the Keap1 knockdown background, indicating that NRF2 mitigates the autoimmune response in a Treg-independent manner. Considering that the first step to correct autoimmunity by restoring self-tolerance has been proposed to be the suppression of the secretion of proinflammatory cytokines that often inhibit Treg activity (17–19), NRF2, which exhibits Treg-independent anti-inflammatory effects, may serve as a beneficial therapeutic target for initial intervention. Although we attempted to identify a cell lineage responsible for the NRF2-mediated suppression of the autoimmune response, none of the cell lineage-specific NRF2 activation results showed sufficient anti-inflammatory effects comparable to those achieved by the systemic activation of NRF2. Thus, we propose that NRF2-mediated anti-inflammatory effects in the context of autoimmunity, in which Treg activities are suppressed, require the integrity of NRF2-mediated functional modulations of multiple tissues and cell lineages.
It should be noted that CDDO-Im treatment of Sf mice only partially improved autoimmune inflammation compared to that with the genetically induced NRF2 activation in Sf::Keap1FA/− mice. This result may be due to the delayed activation of NRF2 by CDDO-Im treatment. NRF2 was already fully activated at the time of birth in Sf::Keap1FA/− mice, whereas CDDO-Im administration was initiated on day 7 after birth in this study due to technical difficulties in treating pups before 7 days of age. Thus, an earlier initiation of CDDO-Im treatment might have improved its anti-inflammatory effects.
Among the various parameters of the autoimmune response, decreased production of IFN-γ by T cells and its consequent decrease in the serum were the clearest effects of NRF2 activation in Sf mice. IFN-γ production by T cells was reduced by the systemic activation of NRF2 (both genetic and pharmacological) and by T cell-specific activation of NRF2, indicating that NRF2 inhibits IFN-γ production in T cells in a cell-autonomous manner. This result is consistent with a previous report revealing that NRF2 activation in T cells shifts the Th1/Th2 balance toward Th2-skewed immunity (41). Because IFN-γ has been reported to enhance the autoimmune response through increasing the numbers of activated effector T and B cells and the production of autoantibodies (42, 43), a reduction of IFN-γ contributes, at least partially, to the attenuation of the autoimmune response by NRF2. Consistent with this idea, a previous study showed that deletion of the Ifng gene prolongs the life span of Sf mice to 6 to 7 weeks and alleviates inflammation in the skin, lungs, and liver (36). Considering that T cell-specific and pharmacological NRF2 activation did not exert sufficient anti-inflammatory effects in Sf mice despite the reduced production of IFN-γ by T cells, we surmise that additional factors are required that indirectly lead to the decreased production of IL-4 and IL-17A in T cells.
Previous reports showed that NRF2 in macrophages and dendritic cells modulates their proinflammatory cytokine production and antigen-presenting activity (3, 5, 39, 40). However, in our experimental setting, NRF2 activation in macrophages or dendritic cells alone did not attenuate the inflammatory response. Because it was recently reported that depletion of B cells ameliorates multiorgan inflammation in Sf mice (44), it is worthwhile to study the significance of NRF2's contribution in B cells.
Another possible mechanism for the NRF2-mediated anti-inflammatory effects may be a decrease in the release of damage-associated molecular patterns (DAMPs) from tissue cells that are attacked by autoreactive immune cells. DAMPs recruit inflammatory cells and induce further inflammatory reactions (45). DAMPs are known to be involved in the pathogenesis of systemic autoinflammatory diseases, and the secretion of DAMPs is associated with oxidative stress (46, 47). Considering the enhancement of cell survival and cellular antioxidant capacity by NRF2, NRF2 is likely to inhibit the secretion of DAMPs by reducing cell damage from the autoimmune response and to contribute to the alleviation of systemic inflammation. Further investigation is required to elucidate the molecular mechanisms responsible for NRF2-dependent suppression of the autoimmune response.
Because NRF2 activation is effective for prevention and treatment of various pathological conditions caused by excessive oxidative and electrophilic stresses, clinical applications of NRF2 inducers to adjust the redox balance are in progress. Our current results showing the NRF2-mediated inhibition of effector T cell activities suggest that precautions are necessary in applying NRF2 inducers, especially for immunocompromised patients. We have to be aware of their possible immunosuppressive effects and to pay careful attention to infectious complications of treatment with NRF2 inducers.
Foxp3 mutations have been found in human populations and identified as major genetic alterations causing IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome, which is characterized by the development of multiple autoimmune disorders and consequent fatal inflammation (48, 49). Because Sf mice are regarded as a mouse model of IPEX syndrome, our results strongly suggest that NRF2 inducers are promising therapeutic agents for this intractable congenital disorder, in addition to other, more common autoimmune diseases. Thus, NRF2 inducers expand the therapeutic options for the control of inflammation caused by autoimmune diseases.
MATERIALS AND METHODS
Mice.
Scurfy (Foxp3sf/X) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Two types of Keap1 conditional knockout mice were used in this study (50). Keap1FA is a knockdown allele in which Keap1 expression is decreased (27), whereas Keap1FB produces normal amounts of the Keap1 transcript (38). To establish Keap1 knockdown mice, Keap1FA was used in combination with the Keap1-null allele (Keap1−) (25). To conduct cell lineage-specific disruption of Keap1, Keap1FB was used. Male Keap1FA/FA mice (26) and Keap1FA/− mice were mated with female Foxp3sf/X mice to generate Foxp3sf/Y::Keap1FA/−(Sf::Keap1FA/−) mice. Keap1FB/FB mice (38) and Lck-Cre (51), CD11c-Cre (52), and LysM-Cre (53) mice were mated to obtain Keap1 conditional knockout mice with Keap1 deletion in T cells (Keap1-TKO), dendritic cells (Keap1-DKO), and myeloid cells (Keap1-MKO), respectively. Lck-Cre mice were kindly provided by Junji Takeda. CD11c-Cre mice and LysM-Cre mice were purchased from the Jackson Laboratory. All the mice used in this study were in the C57BL/6 background. The mice were used for analyses 15 to 19 days after birth, unless otherwise indicated. All genotypes were determined using tail genomic DNA. Peripheral blood was collected from the superficial temporal vein and analyzed by use of a hemocytometer (Nihon Kohden Co., Tokyo, Japan).
All animals were housed under specific-pathogen-free conditions according to the regulations of the standards for human care and use of laboratory animals of Tohoku University and the guidelines for proper conduct of animal experiments of the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Multiplex assays.
Collected blood was allowed to clot for 30 min at room temperature, and the serum was separated by centrifugation at 1,200 × g for 20 min and stored at −80°C until further use. For quantification of serum cytokines, a LEGENDplex kit (BioLegend, San Diego, CA) was used according to the manufacturer's instructions. The data were analyzed using LEGENDplex data analysis software (BioLegend). All samples were assayed in duplicate, and all presented values are the means of duplicate measurements.
Flow cytometry.
Bilateral axillary, brachial, and inguinal LNs were collected and homogenized in phosphate-buffered saline (PBS) supplemented with 3% heat-inactivated fetal bovine serum. The LN cells were stained with monoclonal antibodies against CD4 (GK1.5), CD8a (53-6.7), CD25 (PC61), CD44 (IM7), and CD69 (H1.2F3) (BioLegend or eBiosciences, San Diego, CA). The cells were then stained with propidium iodide for the exclusion of dead cells. For intracellular cytokine staining, LN cells (5 × 106 cells/ml) were stimulated in vitro with phorbol 12-myristate 13-acetate (50 ng/ml) and ionomycin (1 μg/ml) in the presence of monensin (3 μM) for 3 h in 24-well plates. The stimulated cells were stained with antibodies against surface antigens as described above, followed by fixation and permeabilization with fixation buffer (BioLegend) and intracellular staining permeabilization wash buffer (BioLegend), respectively. The fixed and permeabilized cells were further incubated with antibodies against IFN-γ (XMG1.2), IL-4 (11B11), IL-17A (TC11-18H10.1), and IL-10 (JES5-16E3) (BioLegend). The stained cells were analyzed with a FACSAria II or LSR Fortessa flow cytometer (BD Biosciences).
Quantitative real-time PCR analysis.
Tissues were homogenized in Isogen (Nippon Gene, Tokyo, Japan) to purify total RNAs, and cDNAs were synthesized with ReverTra Ace qPCR RT master mix with gDNA Remover (Toyobo, Osaka, Japan) according to the manufacturer's instructions. RNAs from the sorted cells were isolated using a ReliaPrep RNA cell miniprep system (Promega, Madison, WI), and cDNAs were synthesized with SuperScript III reverse transcriptase (Invitrogen, Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Quantitative PCR was performed on an Applied Biosystems 7300 sequence detector system, using Thunderbird SYBR qPCR mix (Toyobo) for the SYBR green system and Thunderbird probe qPCR mix (Toyobo) for the TaqMan probe system. The primer sequences are shown in Table S5 in the supplemental material.
Immunoblot analysis.
Tissues were lysed and sonicated in SDS sample buffer (25 mM Tris-HCl, pH 6.8, 5% glycerol, 1% SDS, 0.05% bromophenol blue, 20% 2-mercaptoethanol), and the lysates were heated at 95°C for 5 min. The samples were then subjected to immunoblot analysis using an anti-NRF2 antibody (clone 103; 1:500) (54), an anti-KEAP1 antibody (clone 111; 1:200) (55), and an anti-α-tubulin antibody (clone DM1A; 1:10,000) (Sigma-Aldrich).
Histological analysis.
The skin, liver, and lungs were fixed with Mildform 10 N (Wako) and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin (HE).
CDDO-Im treatment of Sf mice.
WT and Sf mice were injected intraperitoneally with 5 μmol/kg CDDO-Im (the total volume was set to 20 μl/g [for body weights of <12.5 g] or 10 μl/g [for body weights of >12.5 g]) or the same volume of vehicle (PBS with 10% dimethyl sulfoxide and 10% Cremophor-EL) every other day starting 7 or 8 days after birth. Mice were analyzed one day after the fourth administration. For survival analysis, treatment was continued throughout the observation period.
Statistical analysis.
Quantitative data are presented as means ± standard deviations (SD) and were analyzed using Student's t tests. P values of <0.05 were considered statistically significant. Survival rates were analyzed using the log rank test with the Bonferroni correction for multiple comparisons. Bonferroni-adopted P values of <0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nao Ohta, Eriko Naganuma, and the Biomedical Research Core of the Tohoku University Graduate School of Medicine for their technical support.
This work was supported by JSPS KAKENHI grants 15H04692 (H.M.), 16K15228 (H.M.), and 15K18999 (S.M.), the Uehara Memorial Foundation (H.M.), the Mitsubishi Foundation (H.M.), the Naito Foundation (H.M.), a research grant from the Princess Takamatsu Cancer Research Fund (grant 15-24728 to H.M.), a GSK Japan Research Grant (S.M.), and a Core Research for Evolutional Science and Technology grant from the AMED (H.M. and M.Y.).
T.S., S.M., and H.M. designed the research study; S.S.B. and S.S. provided key experimental materials; T.S. and S.M. conducted the experiments; T.S., S.M., S.S.B., S.S., H.H., M.Y., and H.M. analyzed the data; and T.S., S.M., and H.M. wrote the early draft of the paper.
We declare that we have no conflicts of interest.
Funding Statement
The funding sources had no role in the design of the study, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00063-17.
REFERENCES
- 1.Motohashi H, Yamamoto M. 2004. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Taguchi K, Motohashi H, Yamamoto M. 2011. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, Yamamoto M. 2016. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 7:11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishii Y, Itoh K, Morishima Y, Kimura T, Kiwamoto T, Iizuka T, Hegab AE, Hosoya T, Nomura A, Sakamoto T, Yamamoto M, Sekizawa K. 2005. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J Immunol 175:6968–6975. doi: 10.4049/jimmunol.175.10.6968. [DOI] [PubMed] [Google Scholar]
- 5.Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, Biswal S. 2011. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med 184:928–938. doi: 10.1164/rccm.201102-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. 2006. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res 66:11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 7.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. 2005. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun CC, Li SJ, Yang CL, Xue RL, Xi YY, Wang L, Zhao QL, Li DJ. 2015. Sulforaphane attenuates muscle inflammation in dystrophin-deficient mdx mice via NF-E2-related factor 2 (Nrf2)-mediated inhibition of NF-κB signaling pathway. J Biol Chem 290:17784–17795. doi: 10.1074/jbc.M115.655019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Wruck CJ, Fragoulis A, Gurzynski A, Brandenburg LO, Kan YW, Chan K, Hassenpflug J, Freitag-Wolf S, Varoga D, Lippross S, Pufe T. 2011. Role of oxidative stress in rheumatoid arthritis: insights from the Nrf2-knockout mice. Ann Rheum Dis 70:844–850. doi: 10.1136/ard.2010.132720. [DOI] [PubMed] [Google Scholar]
- 10.Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S. 2001. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int 60:1343–1353. doi: 10.1046/j.1523-1755.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- 11.Jiang T, Tian F, Zheng H, Whitman SA, Lin Y, Zhang Z, Zhang N, Zhang DD. 2014. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int 85:333–343. doi: 10.1038/ki.2013.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164. [PubMed] [Google Scholar]
- 13.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. 1995. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 14.Kriegel MA, Lohmann T, Gabler C, Blank N, Kalden JR, Lorenz HM. 2004. Defective suppressor function of human CD4+ CD25+ regulatory T cells in autoimmune polyglandular syndrome type II. J Exp Med 199:1285–1291. doi: 10.1084/jem.20032158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kekäläinen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pöntynen N, Talvensaari K, Perheentupa J, Miettinen A, Arstila TP. 2007. A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Immunol 178:1208–1215. doi: 10.4049/jimmunol.178.2.1208. [DOI] [PubMed] [Google Scholar]
- 16.Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, Amoura Z. 2011. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev 10:744–755. doi: 10.1016/j.autrev.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, Liu X, Xiao L, Chen X, Wan B, Chin YE, Zhang JZ. 2013. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat Med 19:322–328. doi: 10.1038/nm.3085. [DOI] [PubMed] [Google Scholar]
- 18.Carbone F, De Rosa V, Carrieri PB, Montella S, Bruzzese D, Porcellini A, Procaccini C, La Cava A, Matarese G. 2014. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat Med 20:69–74. doi: 10.1038/nm.3411. [DOI] [PubMed] [Google Scholar]
- 19.Miyara M, Ito Y, Sakaguchi S. 2014. TREG-cell therapies for autoimmune rheumatic diseases. Nat Rev Rheumatol 10:543–551. doi: 10.1038/nrrheum.2014.105. [DOI] [PubMed] [Google Scholar]
- 20.Noel S, Martina MN, Bandapalle S, Racusen LC, Potteti HR, Hamad AR, Reddy SP, Rabb H. 2015. T lymphocyte-specific activation of Nrf2 protects from AKI. J Am Soc Nephrol 26:2989–3000. doi: 10.1681/ASN.2014100978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. 2001. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27:68–73. [DOI] [PubMed] [Google Scholar]
- 22.Fontenot JD, Gavin MA, Rudensky AY. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 23.Kim JM, Rasmussen JP, Rudensky AY. 2007. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 24.Ju ST, Sharma R, Gaskin F, Kung JT, Fu SM. 2012. The biology of autoimmune response in the Scurfy mice that lack the CD4+Foxp3+ regulatory T-cells. Biology 1:18–42. doi: 10.3390/biology1010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M. 2003. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 26.Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler TW, Yamamoto M. 2006. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun 339:79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- 27.Taguchi K, Maher JM, Suzuki T, Kawatani Y, Motohashi H, Yamamoto M. 2010. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol Cell Biol 30:3016–3026. doi: 10.1128/MCB.01591-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki T, Seki S, Hiramoto K, Naganuma E, Kobayashi EH, Yamaoka A, Baird L, Takahashi N, Sato H, Yamamoto M. 2017. Hyperactivation of Nrf2 in early tubular development induces nephrogenic diabetes insipidus. Nat Commun 8:14577. doi: 10.1038/ncomms14577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blair PJ, Bultman SJ, Haas JC, Rouse BT, Wilkinson JE, Godfrey VL. 1994. CD4+CD8− T cells are the effector cells in disease pathogenesis in the scurfy (sf) mouse. J Immunol 153:3764–3774. [PubMed] [Google Scholar]
- 30.Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramsdell F. 1999. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol 162:2546–2554. [PubMed] [Google Scholar]
- 31.Sharma R, Sharma PR, Kim YC, Leitinger N, Lee JK, Fu SM, Ju ST. 2011. IL-2-controlled expression of multiple T cell trafficking genes and Th2 cytokines in the regulatory T cell-deficient scurfy mice: implication to multiorgan inflammation and control of skin and lung inflammation. J Immunol 186:1268–1278. doi: 10.4049/jimmunol.1002677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bacchetta R, Sartirana C, Levings MK, Bordignon C, Narula S, Roncarolo MG. 2002. Growth and expansion of human T regulatory type 1 cells are independent from TCR activation but require exogenous cytokines. Eur J Immunol 32:2237–2245. doi:. [DOI] [PubMed] [Google Scholar]
- 33.Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 34.Vieira PL, Christensen JR, Minaee S, O'Neill EJ, Barrat FJ, Boonstra A, Barthlott T, Stockinger B, Wraith DC, O'Garra A. 2004. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol 172:5986–5993. doi: 10.4049/jimmunol.172.10.5986. [DOI] [PubMed] [Google Scholar]
- 35.Passerini L, Di Nunzio S, Gregori S, Gambineri E, Cecconi M, Seidel MG, Cazzola G, Perroni L, Tommasini A, Vignola S, Guidi L, Roncarolo MG, Bacchetta R. 2011. Functional type 1 regulatory T cells develop regardless of FOXP3 mutations in patients with IPEX syndrome. Eur J Immunol 41:1120–1131. doi: 10.1002/eji.201040909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma R, Sung SS, Gaskin F, Fu SM, Ju ST. 2012. A novel function of IL-2: chemokine/chemoattractant/retention receptor genes induction in Th subsets for skin and lung inflammation. J Autoimmun 38:322–331. doi: 10.1016/j.jaut.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Billiau A, Matthys P. 2009. Interferon-γ: a historical perspective. Cytokine Growth Factor Rev 20:97–113. doi: 10.1016/j.cytogfr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Blake DJ, Singh A, Kombairaju P, Malhotra D, Mariani TJ, Tuder RM, Gabrielson E, Biswal S. 2010. Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am J Respir Cell Mol Biol 42:524–536. doi: 10.1165/rcmb.2009-0054OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macoch M, Morzadec C, Génard R, Pallardy M, Kerdine-Römer S, Fardel O, Vernhet L. 2015. Nrf2-dependent repression of interleukin-12 expression in human dendritic cells exposed to inorganic arsenic. Free Radic Biol Med 88:381–390. doi: 10.1016/j.freeradbiomed.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Aw Yeang HX, Hamdam JM, Al-Huseini LM, Sethu S, Djouhri L, Walsh J, Kitteringham N, Park BK, Goldring CE, Sathish JG. 2012. Loss of transcription factor nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) leads to dysregulation of immune functions, redox homeostasis, and intracellular signaling in dendritic cells. J Biol Chem 287:10556–10564. doi: 10.1074/jbc.M111.322420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rockwell CE, Zhang M, Fields PE, Klaassen CD. 2012. Th2 skewing by activation of Nrf2 in CD4+ cells. J Immunol 188:1630–1637. doi: 10.4049/jimmunol.1101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farrar MA, Schreiber RD. 1993. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol 11:571–611. [DOI] [PubMed] [Google Scholar]
- 43.Baudino L, Azeredo da Silveria S, Nakata M, Izui S. 2006. Molecular and cellular basis for pathogenicity of autoantibodies: lessons from murine monoclonal autoantibodies. Springer Semin Immunopathol 28:175–184. doi: 10.1007/s00281-006-0037-0. [DOI] [PubMed] [Google Scholar]
- 44.Aschermann S, Lehmann CH, Mihai S, Schett G, Dudziak D, Nimmerjahn F. 2013. B cells are critical for autoimmune pathology in Scurfy mice. Proc Natl Acad Sci U S A 110:19042–19047. doi: 10.1073/pnas.1313547110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubartelli A, Lotze MT. 2007. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol 28:429–436. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Varga G, Gattorno M, Foell D, Rubartelli A. 2015. Redox distress and genetic defects conspire in systemic autoinflammatory diseases. Nat Rev Rheumatol 11:670–680. doi: 10.1038/nrrheum.2015.105. [DOI] [PubMed] [Google Scholar]
- 47.Yu Y, Tang D, Kang R. 2015. Oxidative stress-mediated HMGB1 biology. Front Physiol 6:93. doi: 10.3389/fphys.2015.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wildin RS, Smyk-Pearson S, Filipovich AH. 2002. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet 39:537–545. doi: 10.1136/jmg.39.8.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. 2001. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 50.Uruno A, Yagishita Y, Katsuoka F, Kitajima Y, Nunomiya A, Nagatomi R, Pi J, Biswal SS, Yamamoto M. 2016. Nrf2-mediated regulation of skeletal muscle glycogen metabolism. Mol Cell Biol 36:1655–1672. doi: 10.1128/MCB.01095-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahama Y, Ohishi K, Tokoro Y, Sugawara T, Yoshimura Y, Okabe M, Kinoshita T, Takeda J. 1998. Functional competence of T cells in the absence of glycosylphosphatidylinositol-anchored proteins caused by T cell-specific disruption of the Pig-a gene. Eur J Immunol 28:2159–2166. doi:. [DOI] [PubMed] [Google Scholar]
- 52.Caton ML, Smith-Raska MR, Reizis B. 2007. Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J Exp Med 204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8:265–277. doi: 10.1023/A:1008942828960. [DOI] [PubMed] [Google Scholar]
- 54.Maruyama A, Tsukamoto S, Nishikawa K, Yoshida A, Harada N, Motojima K, Ishii T, Nakane A, Yamamoto M, Itoh K. 2008. Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch Biochem Biophys 477:139–145. doi: 10.1016/j.abb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Watai Y, Kobayashi A, Nagase H, Mizukami M, McEvoy J, Singer JD, Itoh K, Yamamoto M. 2007. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 12:1163–1178. doi: 10.1111/j.1365-2443.2007.01118.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.