

ABSTRACT
Antigen presentation by dendritic cells (DCs) stimulates naive CD4+ T cells, triggering T cell activation and the adaptive arm of the immune response. Newly synthesized major histocompatibility complex class II (MHC-II) molecules accumulate at MHC-II-enriched endosomal compartments and are transported to the plasma membrane of DCs after binding to antigenic peptides to enable antigen presentation. In DCs, MHC-II molecules are included in tetraspanin-enriched microdomains (TEMs). However, the role of tetraspanin CD9 in these processes remains largely undefined. Here, we show that CD9 regulates the T cell-stimulatory capacity of granulocyte-macrophage colony-stimulating factor (GM-CSF)-dependent bone marrow-derived DCs (BMDCs), without affecting antigen presentation by fms-like tyrosine kinase 3 ligand (Flt3L)-dependent BMDCs. CD9 knockout (KO) GM-CSF-dependent BMDCs, which resemble monocyte-derived DCs (MoDCs), induce lower levels of T cell activation than wild-type DCs, and this effect is related to a reduction in MHC-II surface expression in CD9-deficient MoDCs. Importantly, MHC-II targeting to the plasma membrane is largely impaired in immature CD9 KO MoDCs, in which MHC-II remains arrested in acidic intracellular compartments enriched in LAMP-1 (lysosome-associated membrane protein 1), and MHC-II internalization is also blocked. Moreover, CD9 participates in MHC-II trafficking in mature MoDCs, regulating its endocytosis and recycling. Our results demonstrate that the tetraspanin CD9 specifically regulates antigenic presentation in MoDCs through the regulation of MHC-II intracellular trafficking.
KEYWORDS: CD9, MHC-II, monocyte-derived dendritic cells, antigen presentation, tetraspanin-enriched microdomain
INTRODUCTION
Dendritic cells (DCs) are the most efficient antigen (Ag)-presenting cells (APCs) to stimulate naive CD4+ T cells, leading to the initiation of the adaptive immune response. Ag presentation involves the internalization and processing of exogenous Ags to produce immunogenic peptides that bind to major histocompatibility complex class II (MHC-II) molecules. Newly synthesized MHC-II molecules are targeted to MHC-II-enriched endosomal compartments (MIICs), where they couple to antigenic peptides (1, 2). The accessory molecules class II-associated invariant chain (Ii) and DM (HLA-DM in humans [mouse homolog, H2-DM]) control the assembly of MHC-II molecules and MHC-II-restricted Ag processing (2, 3). DM interacts with MHC-II molecules at the MIIC (4), which displays acidic pH (5) and is enriched in conventional late endocytic markers, such as LAMP-1 (lysosome-associated membrane protein 1) (6). MIIC also includes resident proteases, such as cathepsins or GILT (gamma interferon [IFN-γ]-induced lysosomal thiol reductase) (7, 8), which are responsible for Ag proteolytic processing following Ag uptake by different routes depending on the APC (9).
After their final assembly at MIICs, stable peptide-bound MHC-II complexes egress to the DC surface. In maturing DCs, peptide–MHC-II complexes are transported to the plasma membrane by tubular extensions that emanate from MIICs (10–12). The formation of these MHC-II-enriched membrane tubules requires microtubules and microtubule-based motors such as dynein and kinesin (13–15), as well as cholesterol and lipid rafts (16–18). Once at the plasma membrane, peptide–MHC-II complexes are actively endocytosed and are recycled back to the cell surface. Endocytosis is partially due to the presence of a conserved dileucine motif in the cytoplasmic region of the MHC-II β chain (19), which could drive its internalization in a clathrin- and dynamin-independent manner (20). In immature DCs, MHC-II endocytosis and sorting at endosomes and multivesicular bodies (MVB) can be regulated through ubiquitination by the ubiquitin E3 ligase MARCH-I (21–25). Upon DC maturation, MARCH-I expression is downregulated, resulting in less-efficient MHC-II ubiquitination, and this would allow the steep increase in MHC-II presence at the cell surface (22, 25). However, other studies did not confirm the role of ubiquitination in peptide-bound MHC-II endocytosis (21, 23, 26). The precise mechanisms that regulate MHC-II internalization and recycling therefore remain undefined. Endocytosed molecules can be recycled from early endosomes (27, 28) or can be sorted into the luminal vesicles of MVB and then either be targeted for degradation by fusion with lysosomes (25) or sorted into exosomes (29). MHC-II molecules that have been internalized from the plasma membrane to early endosomes can bind to antigenic peptides generated in less-acidic and proteolytic compartments, through proteolysis by the proteasome, by autophagosomes, or by endosome resident proteases (30–33). Therefore, MHC-II surface expression levels result from the balance between postsynthesis plasma membrane targeting and endocytosis for recycling or degradation.
Tetraspanins are membrane proteins responsible for the organization of membrane macrocomplexes called tetraspanin-enriched microdomains (TEMs). Tetraspanins interact with other members of the superfamily, other transmembrane receptors, lipids, signaling molecules, and cytoskeletal components (34, 35). They modulate the function of their associated membrane partners, playing important roles in a wide variety of physiological and pathological processes (36–39). TEMs regulate Ag recognition and presentation and T cell activation and proliferation, as well as leukocyte extravasation (40). These microdomains contain several immune receptors, such as the T cell receptor (TCR) in T cells, the B cell receptor (BCR) in B cells, and pattern recognition receptors and MHC-II in APCs (38). Several tetraspanins associate with MHC-I and MHC-II at the surface of APCs (41–43). Tetraspanins CD63 and CD82 participate in Ag processing. They are both present in MIIC, interacting with MHC-II and other HLA molecules (41, 42, 44). Ag presentation can also be modulated by several tetraspanins. DCs from CD151−/−, CD37−/−, and Tssc6−/− single knockout and CD37−/− Tssc6−/− double knockout mice induce hyperstimulation of T cells (45, 46). CD81 is important for DC cross-presentation of bacterial Ag to cytotoxic T cells (47). CD63 downregulation in human B lymphocytes enhances CD4+ T lymphocyte activation (48). In DCs, whereas CD151 regulates T cell costimulation (46), CD37 interacts with MHC-II (49), limiting MHC-dependent Ag presentation (46, 50).
In DCs, according to one report, CD9 seems to mediate MHC-II clustering (51); however, other reports did not observe any effect on MHC-II multimerization (52). Therefore, the role of CD9 in the regulation of MHC-II expression and Ag presentation by DCs remains controversial. In this study, we demonstrated that CD9 is important for the T cell-stimulatory capacity of monocyte-derived DCs (MoDCs), through the regulation of MHC-II intracellular trafficking and expression at the cell surface.
RESULTS
CD9-deficient MoDCs display impaired T cell-stimulatory capacity.
To investigate the role of CD9 during Ag presentation, we compared the levels of T cell-stimulatory activity of wild-type (WT) and CD9 knockout (KO) DCs, using Ag-specific CD4+ T cells. First, we used granulocyte-macrophage colony-stimulating factor (GM-CSF)-dependent bone marrow-derived DCs (BMDCs), which were characterized as CD11c+ MHC-II+ Ly6G/Ly6C−. Lipopolysaccharide (LPS)-matured BMDCs were pulsed with MHC-II-restricted ovalbumin (OVA) peptide and cocultured with naive OVA-specific OT-II CD4+ T cells. CD9 KO DCs induced lower T cell activation than WT DCs, as measured by membrane expression levels of the T cell activation receptor CD25 (Fig. 1A). Accordingly, CD9 KO BMDCs induced less T cell proliferation than WT DCs (Fig. 1B). The percentage of OT-II T cells expressing IFN-γ was also significantly diminished after stimulation with CD9 KO DCs compared to T cells activated with WT DCs (Fig. 1C). The stimulatory potential of CD9 KO DCs was further investigated in vivo. OVA peptide-loaded WT or CD9 KO BMDCs were subcutaneously injected into WT mice, and OT-II CD4+ T cells labeled with cell violet tracer were intravenously transferred into the recipient mice 24 h later. After 2 and 4 days, respectively, activation and proliferation of adoptively transferred T cells were analyzed in draining lymph nodes by flow cytometry. Similarly to the in vitro results, CD9 KO BMDCs displayed a lower T cell-stimulatory potential than WT DCs, as revealed by the lower CD25 expression levels (Fig. 1D) and the reduced cell proliferation of transferred OT-II T cells (Fig. 1E). Since GM-CSF-dependent BMDCs were previously characterized as resembling inflammatory MoDCs (53, 54), we also investigated the role of CD9 for Ag presentation by fms-like tyrosine kinase 3 ligand (Flt3L)-dependent conventional DCs. BMDCs were generated in the presence of the Flt3 ligand, which is necessary for development of DCs (54), and WT or CD9 KO OVA peptide-loaded CD11c+ B220− Flt3L-dependent conventional DCs were subcutaneously injected into WT mice. OT-II CD4+ T cells labeled with cell violet tracer were intravenously transferred 24 h later into the recipient mice, and proliferation of adoptively transferred T cells was analyzed in draining lymph nodes by flow cytometry after 4 days. Unexpectedly, CD9 deficiency in Flt3L DCs did not affect T cell proliferation in vivo (Fig. 1F), suggesting that CD9 might play different roles in different DC subpopulations. Together, our results show that the absence of CD9 expression led to impaired Ag-specific CD4+ T cell responses specifically in GM-CSF-dependent BMDCs both in vitro and in vivo. Since these cells were previously described to resemble MoDCs (53, 54), we use that term here to refer to GM-CSF-dependent BMDCs.
FIG 1.

MoDC stimulatory capacity is dependent on CD9 expression. (A to C) LPS-matured WT or CD9 KO GM-CSF-dependent BMDCs were loaded with OVA peptide and cocultured with naive CD4+ OT-II T cells. After 72 h, T cell activation and proliferation were analyzed by flow cytometry. (A) CD25 membrane expression in CD4+ T cells. MFI, mean fluorescence intensity. (B) Percentages of proliferated T cells. (C) Percentages of IFN-γ+ CD4+ T cells. Data are means ± standard errors of the means (SEM) of results from three independent experiments analyzed by Student's t test (A and C) or two-way ANOVA with Bonferroni's post hoc multiple-comparison test (B). (D and E) LPS-matured OVA peptide-loaded WT or CD9 KO GM-CSF-dependent BMDCs were subcutaneously injected into WT mice, and 24 h later, OT-II CD4+ T cells labeled with cell violet tracer were intravenously transferred into the recipient mice. After a further 2 and 4 days, respectively, activation and proliferation of adoptively transferred CD4+ T cells were analyzed in the draining lymph nodes by flow cytometry. (D) CD25 membrane expression in CD4+ T cells. (E) Percentages of T cells. Data represent mean fold changes ± SEM from two independent experiments analyzed by Student's t test (D) and means ± SEM from one experiment representative of four with n = 5 mice per genotype analyzed by two-way ANOVA with Bonferroni's post hoc multiple-comparison test (E). (F) Experiments similar to those described for panel E were performed using LPS-matured OVA peptide-loaded WT or CD9 KO CD11c+ B220− Flt3L-dependent conventional BMDCs. Data represent means ± SEM of percentages of T cells from one experiment representative of three with n = 5 mice per genotype analyzed as described for panel E. (G) LPS-matured MoDCs were allowed to migrate toward CCL21 in a Transwell migration assay, and the percentage of cells at the lower chamber was measured by flow cytometry. Data are means ± SEM of results from four independent experiments. (H) In vivo migration of skin DCs to inguinal lymph nodes was measured by flow cytometry after FITC-painting sensitization of the abdominal skin of mice. Data are means ± SEM of percentages of FITC+ CD11c+ cells from two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To determine whether CD9 could be involved in the regulation of other MoDC functions, we assessed the effect of CD9 deficiency on DC migration. Migration of CD9 KO MoDCs was not affected during in vitro chemotaxis to CCL21 (Fig. 1G). CD9 KO DCs expressed levels of the chemokine receptor CCR7, which drives CCL21 migration, that were similar to those seen with WT DCs (see Fig. S1A in the supplemental material). In addition, no differences were found in terms of expression of the adhesion molecule ICAM-1 or of the integrins CD49d and CD11b in MoDCs and splenic DCs of the two genotypes (Fig. S1B to F). We next investigated whether the absence of CD9 could affect the presence of different DC subpopulations in lymphoid organs. We found that the absolute numbers and the proportions of plasmacytoid DCs (CD11c+ B220+ cells) and conventional DCs (CD11c+ B220− cells) in the spleen, lymph nodes, and thymus were similar in CD9 KO and WT mice (Fig. S1G and data not shown). Flt3L-dependent WT and CD9 KO BMDCs also displayed similar levels of in vitro differentiation (data not shown). Moreover, assessment of DC migration in vivo using the fluorescein isothiocyanate (FITC)-skin painting assay showed similar percentages of FITC+ CD11c+ cells in skin-draining lymph nodes from CD9 KO and WT mice (Fig. 1H). Hence, CD9 deficiency in DCs did not affect their in vitro or in vivo migratory responses.
CD9 deficiency in MoDCs does not affect IS formation.
During Ag presentation, T cells form stable cell-cell contacts with DCs that are known as immunological synapses (IS). The IS structure is highly organized, with specific redistribution of receptors, such as the TCR/CD3 at the T cell side and MHC-II and costimulatory molecules at the APC side, as well as accumulation of adhesion molecules and cytoskeleton components (55–57). At the T cell side, CD9 is important for integrin-mediated signaling at the IS, regulating T cell activation (58). We investigated whether CD9 could also be important for the IS organization at the APC side. Conjugation of naive OT-II CD4+ T cells with LPS-matured OVA-loaded WT or CD9 KO DCs was analyzed by time-lapse confocal microscopy. No differences were observed either in the total percentage of conjugates or in the duration of short-lived (<20 min), medium-lived (20 to 30 min), and long-lived (>30 min) T cell-APC contacts (Fig. 2A and B). DC adhesion to T cells appeared unaffected by CD9 deficiency during IS formation, since the results with respect to relocalization to the IS of the adhesion molecule ICAM-1 (Fig. 2C) or of F-actin (Fig. 2D) were similar in WT and CD9 KO DCs conjugated with OT-II CD4+ T cells.
FIG 2.
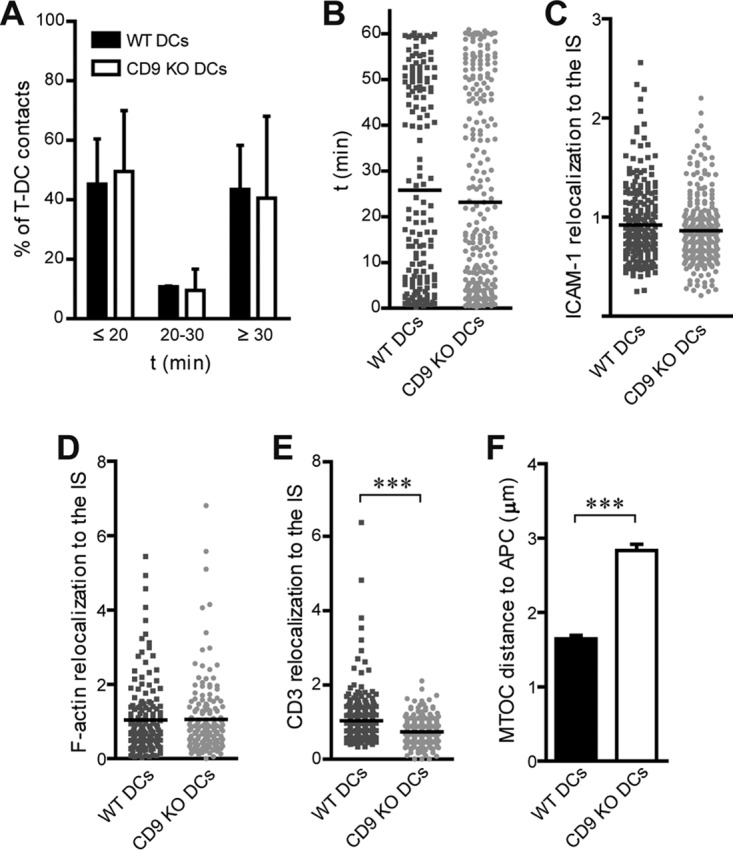
CD9 in MoDCs does not modulate IS formation. (A and B) CD4+ OT-II T cells and WT and CD9 KO LPS-matured OVA-loaded MoDCs were labeled with different cell dyes, allowed to form cell-cell conjugates, and then monitored by time-lapse confocal microscopy. (A) Percentages of cell-cell contacts. (B) Duration of cell-cell contacts. Data from two independent experiments are shown as means ± SEM (A) or with each symbol representing an individual cell-cell conjugate (n ≥ 150) (B). (C to F) LPS-matured OVA-loaded WT or CD9 KO MoDCs were cocultured with naive CD4+ OT-II T cells for 5 min, plated onto PLL-coated coverslips for 30 min, and then fixed and permeabilized. Cells were stained for antibodies against ICAM-1 (C), CD3 (E), or tubulin (F) or with phalloidin to detect F-actin (D) and were analyzed by confocal microscopy. Protein relocalization to T cell-DC contacts was determined by analyzing the fluorescence intensity signal at the cell-cell contact with respect to the fluorescence intensity signal at the rest of cell membranes, using the Synapsemeasures plug-in in ImageJ. Data were pooled from three independent experiments and analyzed by Student's t test. In panels C to E, each symbol represents an individual cell-cell conjugate and horizontal bars indicate the means (for panel C, n ≥ 250; for panel D, n ≥ 150; for panel E, n ≥ 300). In panel F, data are shown as means ± SEM (n ≥ 200). ***, P < 0.001.
In agreement with the reduced T cell-stimulatory capacity of CD9-deficient DCs, CD4+ T cells conjugated with CD9 KO DCs exhibited significantly lower CD3 redistribution to the IS than those conjugated with WT DCs (Fig. 2E). CD9-deficient DCs also induced an impaired translocation of the microtubule-organizing center (MTOC) in T cells, which displayed an increased MTOC distance from the APC surface (Fig. 2F). These results further indicate the importance of CD9 for Ag presentation by MoDCs but also indicate that this phenotype is not related to a regulation of IS formation on the APC side.
MHC-II surface expression is impaired in CD9 KO MoDCs.
Proper Ag presentation depends on the expression of MHC-II and costimulatory molecules by DCs (59). CD9 KO DCs exhibited normal expression levels of costimulatory molecules CD86, CD80, and CD40 both before and after LPS maturation (Fig. 3A; see also Fig. S1H to J). The proportions of CD11c+ DCs were also similar for WT and CD9 KO DCs, which displayed levels of 92.3% (immature WT), 93.4% (immature CD9 KO), 92.4% (mature WT), and 93.5% (mature CD9 KO) (Fig. 3A). Interestingly, immature CD9 KO MoDCs showed reduced MHC-II surface expression levels compared to WT cells (Fig. 3A). MHC-II levels increased upon maturation with LPS as expected, but the diminished surface expression remained in the absence of CD9 (Fig. 3A). In agreement with the reduction in surface MHC-II levels, mature CD9 KO MoDCs showed less binding of biotinylated OVA peptides than WT controls (Fig. 3B). However, the total amounts of MHC-II in the two genotypes were similar in either immature or LPS-matured cells, as detected by flow cytometry of permeabilized cells (Fig. 3C) and by immunoblotting of whole-cell lysates (Fig. 3D). Moreover, treatment of WT and CD9 KO DCs with cycloheximide (CHX) to inhibit protein synthesis showed that the levels of MHC-II turnover were similar in immature or mature cells (Fig. 3E). These results suggest that the reduced MHC-II surface expression observed in CD9 KO MoDCs was not related to impairment in protein synthesis or degradation.
FIG 3.

CD9 modulates MHC-II surface expression in MoDCs. (A) Immature and LPS-matured WT and CD9 KO MoDCs were stained for CD86, CD11c, and MHC-II antibodies and analyzed by flow cytometry. Charts present immature (left) or LPS-matured (center) WT (black) and CD9 KO (red) DCs, with CD86 and CD11c immunolabeling as dot plots (top) and histograms of MHC-II immunolabeling (bottom). Cells labeled with isotype control antibodies are in gray. The graph (right) shows the mean fold change ± SEM of results from four independent experiments analyzed by one-way ANOVA with Tukey's post hoc multiple-comparison test. (B) LPS-matured WT and CD9KO MoDCs were incubated with different concentrations of biotinylated OVA peptides (pOVA) for 1 h at 4°C, washed, stained for CD11c antibody and fluorescence-coupled streptavidin, and analyzed by flow cytometry. Data represent mean fold changes ± SEM of results from three independent experiments analyzed by one-way ANOVA with Tukey's post hoc multiple-comparison test. (C) Immature and LPS-matured WT and CD9 KO MoDCs stained for CD11c and MHC-II antibodies were fixed, permeabilized, and analyzed as described for panel A. (D) Whole-cell lysates from immature and LPS-matured WT and CD9 KO MoDCs were analyzed for MHC-II expression after immunoblotting. Tubulin was used as a loading control. Blots shown (top) are from a representative experiment. The graph (bottom) shows the mean fold changes ± SEM of the MHC-II/tubulin signal ratio from three independent experiments analyzed by one-way ANOVA with Tukey's post hoc multiple-comparison test. (E) Immature (top) and LPS-matured (bottom) WT and CD9 KO MoDCs were treated with 12.5 μg/ml of cycloheximide (CHX) for different times at 37°C and washed, and whole-cell lysates were analyzed for MHC-II expression after immunoblotting. ERM was used as a loading control. WT and CD9 KO blots belonged to the same membrane; the middle irrelevant lanes were removed. The blots shown are from one experiment representative of two. *, P < 0.05; ***, P < 0.001; ns, not significant.
CD9 regulates MHC-II egress from acidic compartments in immature MoDCs.
To study MHC-II intracellular localization in the absence of CD9, we incubated the DCs with biotinylated-MHC-II antibodies coupled to the pHrodo red avidin probe. This pH-sensitive dye allows visualization of the conjugated antibody in acidic compartments, since it is nonfluorescent in neutral pH and fluoresces as the pH decreases. Immature CD9 KO MoDCs showed an increased number of pHrodo+ vesicles, as well as an increase in pHrodo fluorescence intensity, in comparison with immature WT DCs (Fig. 4A), indicating that MHC-II accumulation in acidic compartments is enhanced in the absence of the tetraspanin. However, no such differences were observed in LPS-matured WT and CD9 KO MoDCs (Fig. 4B).
FIG 4.

MHC-II egress from acidic compartments in immature MoDCs depends on CD9 expression. (A and B) Immature and LPS-matured WT and CD9 KO MoDCs were incubated with anti-MHC-II antibodies coupled to pHrodo red avidin for 1 h at 4°C, washed, and then incubated for an additional hour at 37°C. Cells were fixed and analyzed by confocal microscopy. Graphs show the number of pHrodo+ spots/cell (top) and the pHrodo fluorescence intensity (bottom). Data are means ± SEM of results from three independent experiments analyzed by Student's t test. Representative images from immature WT (top) and CD9 KO (bottom) DCs are maximal projections of confocal stacks and show the pHrodo signal (red) either alone or merged with bright-field and nuclei (blue) channels. Bars, 10 μm. (C) Immature WT (top) and CD9 KO (bottom) MoDCs were plated onto PLL-coated coverslips, fixed, permeabilized, stained with antibodies against MHC-II (green) and LAMP-1 (red), and analyzed by confocal microscopy. Nuclei are in blue; one single confocal stack is shown. Bar, 10 μm. Graphs show the quantification of LAMP-1–MHC-II colocalization in three-dimensional (3D) stack confocal microscopy images by the use of Pearson's (left graph) and Mander's (right graph) coefficients. Data are means ± SEM of results from three independent experiments (n ≥ 130 cells) analyzed by Student's t test. (D) LPS-matured WT (top) and CD9 KO (bottom) MoDCs were analyzed as described for panel C. Data are means ± SEM of results from three independent experiments (n ≥ 40 cells). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further investigate the subcellular localization of MHC-II in CD9-deficient immature DCs, cells were plated onto poly-l-lysine (PLL)-coated coverslips for 30 min, fixed, permeabilized, stained with antibodies against MHC-II and markers of different intracellular vesicles, and analyzed by confocal microscopy. We quantified MHC-II colocalization with EEA-1 (early endosomal antigen 1), which is a marker of early endosomes, HGS/HRS (hepatocyte growth factor-regulated tyrosine kinase substrate) and CD63, markers of MVB, and LAMP-1, a marker of late endocytic vesicles such as the MIIC. Quantification was performed by measuring the amount of MHC-II fluorescent signal that coincided with the fluorescent signal of each of the different intracellular markers used (Mander's coefficient) and the correlative variation between the two signals (Pearson's coefficient). No differences were observed between WT and CD9 KO immature DCs regarding the partial colocalization of MHC-II with EEA1 (Fig. S2A), HGS/HRS (Fig. S2B), or CD63 (Fig. S2C). Moreover, the total amounts of EEA1, HGS/HRS, or CD63 were similar between WT and CD9 KO immature DCs (Fig. S2D to F). Interestingly, in agreement with the enhanced accumulation in acidic compartments, CD9 deletion in immature MoDCs induced an increase in MHC-II colocalization with LAMP-1 (Fig. 4C), suggesting that CD9 specifically regulates MHC-II localization at the MIIC. However, CD9 did not affect the total amount of LAMP-1 or the lysosomal content measured with Lysotracker Red, a freely permeable cellular dye that selectively accumulates at cellular compartments with low internal pH (Fig. S2G and H), indicating that the MIIC composition was not affected by CD9 deficiency.
MIIC displays acidic pH (5), and both MHC-II peptide loading and MHC-II degradation induced by the fusion of MIIC with lysosomes are pH dependent. These processes can be inhibited by treatment with ammonium chloride (NH4Cl) (60), which dissipates the pH gradient across acidic vesicles. Treatment of WT and CD9 KO DCs with NH4Cl slightly enhanced surface and total MHC-II levels in immature cells from both genotypes (Fig. S3A and B). However, the reduction in MHC-II surface levels with respect to WT cells was still observed in the absence of CD9, with no differences in the total levels of the protein (Fig. S3A and B), indicating that MHC-II surface regulation by CD9 is independent of MIIC acidification. To analyze whether CD9 could regulate antigen proteolytic processing, immature WT and CD9 KO DCs were incubated for different times with DQ-OVA, a self-quenched full-length OVA conjugated with a pH-insensitive fluorescent dye that exhibits fluorescence only upon degradation. Analysis of the fluorescence derived from DQ-OVA intracellular processing showed no differences in the percentages of fluorescent WT and CD9 KO immature cells (Fig. S3C), indicating that Ag processing was unaffected in the CD9 KO DCs.
In agreement with the normal accumulation of MHC-II at the MIIC in CD9-deleted LPS-matured MoDCs (Fig. 4B), MHC-II colocalization with LAMP-1 was similar to that seen in WT cells (Fig. 4D). We also studied MHC-II colocalization with EEA1, HGS/HRS, and CD63, but no significant differences were observed between CD9 KO and WT mature DCs (Fig. S2A to C). The total amount of these proteins or of the lysosomal content in CD9 KO mature DCs was also comparable to that seen with WT cells (Fig. S2D to H). Treatment with NH4Cl did not affect the CD9 KO phenotype in mature DCs, which still displayed reduced surface MHC-II expression and similar levels of total MHC-II (Fig. S3D and E). Therefore, CD9 regulation of MHC-II surface expression in mature MoDCs seems to depend on a different mechanism.
CD9 regulates MHC-II internalization and recycling in mature MoDCs.
Once at the plasma membrane, MHC-II molecules are actively endocytosed (9). Therefore, we have investigated the role of CD9 in MHC-II internalization. After binding of biotinylated-MHC-II antibodies at 4°C, MoDCs were incubated for different times at 37°C to allow antibody internalization, and then the surface levels of MHC-II were measured by flow cytometry. Interestingly, deficiency in CD9 abolished MHC-II endocytosis in both immature MoDCs (Fig. 5A) and LPS-matured MoDCs (Fig. 5B), indicating that CD9 is essential for this process. In addition, to directly follow the internalization of MHC-II molecules, we covalently tagged biotin to the plasma membrane of mature DCs and then followed the transport of MHC-II molecules by biochemical approaches (protocol adapted from reference 21). After DC biotinylation at 4°C, cells were incubated for different times at 37°C to allow protein internalization. The remaining surface biotin was removed by washing the cells with MESNA (sodium 2-mercaptoethane sulfonic acid); the cells were then lysed, and MHC-II molecules were immunoprecipitated. Internalized biotinylated MHC-II proteins were visualized by immunoblotting after membrane incubation with streptavidin-horseradish peroxidase (HRP). Figure 5C shows that acidic washing with MESNA efficiently removed noninternalized biotin in cells kept at 4°C (Fig. 5C; compare 0 with 0 +MESNA). Incubation of LPS-matured WT DCs at 37°C allowed MHC-II internalization, with increasing amounts of intracellular biotinylated MHC-II proteins (Fig. 5C, WT +LPS +MESNA). In contrast, MHC-II internalization was greatly impaired in the absence of CD9 (Fig. 5C, CD9 KO +LPS +MESNA), confirming that CD9 is important for MHC-II internalization in mature MoDCs.
FIG 5.

CD9 regulates MHC-II internalization and recycling in mature MoDCs. (A and B) Immature (A) and LPS-matured (B) WT and CD9 KO MoDCs were incubated with biotinylated MHC-II antibodies for 1 h at 4°C, washed, and incubated for different times at 37°C, and then MHC-II surface expression was detected by flow cytometry after streptavidin labeling in CD11c+ cells. Data represent mean fold changes ± SEM of results from three independent experiments performed in triplicate and were analyzed by two-way ANOVA with Bonferroni's post hoc multiple-comparison test. (C and D) Measurement of MHC-II internalization (C) and recycling (D) in mature WT and CD9 KO MoDCs by cell biotinylation assay. After incubation with biotin (blue in the schemes) at 4°C, cells were kept at 37°C (internalization), and the remaining surface biotins were removed by MESNA washing. (D) For recycling experiments, cells were further incubated for different times at 37°C (recycling), and surface biotins removed by MESNA washing. Cell lysates were immunoprecipitated with an anti-MHC-II antibody (M5/114), and biotinylated proteins were detected after membrane incubation with streptavidin-HRP (StrepHRP). Membranes were reprobed with MHC-II antibody for loading measurement. The blots shown are from representative experiments. (C) Only a fractional amount from cells incubated at 4°C in the absence of MESNA (0 min) was loaded. The graph shows the biotinylated MHC-II/total MHC-II signal ratio. The graph shows data determined as follows: [1 − (biotinylated MHC-II/total MHC-II signal ratio)]. Data represent mean fold changes ± SEM of results from four (C) and three (D) independent experiments analyzed by two-way ANOVA with Bonferroni's post hoc multiple-comparison test. **, P < 0.01; ***, P < 0.001.
To investigate the mechanism by which CD9 regulates MHC-II endocytosis, we analyzed MHC-II ubiquitination, which has been suggested to drive MHC-II internalization (22, 25) and sorting to MVB (22, 26). WT and CD9 KO immature or LPS-matured MoDCs were first treated with MG132, which mainly blocks ubiquitin-dependent sorting of membrane proteins at MVB (61), although it can also inhibit lysosomal proteases (62). After MG132 treatment, CD9 KO immature or mature DCs still exhibited reduced MHC-II surface expression in comparison with WT cells, with no differences in the total levels of the protein (Fig. S4A and B). Next, we directly assessed MHC-II ubiquitination by immunoprecipitating MHC-II and probing the membrane with anti-ubiquitin antibody. The amounts of ubiquitinated MHC-II molecules in CD9 KO and WT DCs were similar (Fig. S4C). Moreover, no differences were observed in the mRNA levels of the ubiquitin ligase MARCH-I in immature WT and CD9 KO BMDCs (Fig. S4D). These results indicate that the role of CD9 in the regulation of MHCII endocytosis is independent of MHC-II ubiquitination.
Most of the MHC-II molecules in immature DCs are targeted for degradation, whereas many proteins in mature DCs efficiently recycle back to the cell surface (2, 9, 21). We therefore investigated whether CD9 could regulate MHC-II recycling in mature MoDCs, using a biotinylation assay. After biotinylation at 4°C, incubation at 37°C, and removal of surface biotin by MESNA washing, the cells were further incubated at 37°C for different times to allow protein recycling to the plasma membrane. Then, surface biotins were removed from the cell surface by MESNA washing, cells were lysed, MHC-II molecules were immunoprecipitated, and biotinylated MHC-II proteins were visualized with streptavidin-HRP blotting. In LPS-matured WT DCs, the amount of intracellular biotinylated MHC-II proteins decreased with the duration of incubation, showing that MHC-II molecules had been recycled back to the plasma membrane and that the biotins were washed away by the acidic treatment (Fig. 5D, WT + LPS + MESNA). Data were quantified by representing the amount of internalized MHC-II reappearing at the cell surface (recycled MHC-II). In CD9 KO cells kept at 4°C (0 min; no recycling allowed), smaller amounts of biotinylated MHC-II were detected (Fig. 5D; compare WT and CD9 KO at 0 min). This is in agreement with the reduction in protein internalization observed in the absence of CD9. Strikingly, CD9 deficiency prevented MHC-II recycling (Fig. 5D, CD9 KO +LPS +MESNA). Together, our results show that CD9 is important for both MHC-II internalization and recycling.
DISCUSSION
In this study, we showed that deficiency in tetraspanin CD9 impairs the T cell-stimulatory capacity of GM-CSF-dependent BMDCs both in vitro and in vivo without affecting the DC migratory response. Unexpectedly, we showed that CD9 deficiency did not affect Ag presentation by Flt3L-dependent conventional BMDCs. Recently, it has been suggested that BMDC cultures generated with GM-CSF are heterogeneous, containing CD11c+ MHC-II+ conventional DCs and monocyte-derived macrophages (63). GM-CSF-dependent BMDCs resemble monocyte-derived DCs (MoDCs), which constitute specialized inflammatory DCs, although monocytes can also be precursors of different DC subsets under steady-state conditions (53, 54). The role of CD9 in MHC-II expression and Ag presentation has been a matter of debate. Upon DC maturation, recruitment of the peptide–MHC-II complex to membrane microdomains, such as lipid rafts (64) and TEMs (65), was suggested to be important for Ag presentation and subsequent T cell activation (66). TEMs include Ii-associated MHC-II molecules, HLA-DR, HLA-DM, and MHC-II loaded with restricted antigenic peptide repertoires (65, 67). Previous work also suggested that CD9 regulates MHC-II clustering at the DC surface, enhancing the T cell-stimulatory capacity of APCs (51); however, others could not detect differences in the formation of I-A/I-E multimers in CD9 KO BMDCs (52). Importantly, our report provides new clues that help to clarify this controversy; CD9 regulates Ag presentation by specific DC subsets.
The mechanism by which CD9 specifically modulates the function of MoDCs (GM-CSF-dependent BMDCs) involves the control of MHC-II surface expression, with no changes in the expression of costimulatory molecules. Interestingly, plasmacytoid DCs, which have limited T cell-stimulatory potential and reduced MHC-II surface expression in comparison to conventional DCs (68), display no CD9 surface expression (69). Our results clearly indicate that, in both immature and mature MoDCs, CD9 regulates MHC-II intracellular trafficking. The absence of the tetraspanin limits MHC-II surface expression without affecting the formation of the IS between DC and Ag-specific T cells. IS formation depends on specific Ag recognition by T cells and on cell-cell adhesion driven by the interaction between adhesion receptors and integrins (70–72). Although the absence of CD9 in MoDCs did not alter the expression of adhesion molecules or the IS formation, it significantly impaired CD4+ T cell activation, suggesting that CD9 regulates Ag presentation. This effect does not seem to be related to an impairment of MHC-II coupling to antigenic peptides, since it was previously shown that CD9 knockdown in DC does not affect MHC-II peptide loading (44). In this regard, we did not observe differences in Ag proteolytic processing in CD9 KO MoDCs. The decrease in MHC-II surface expression observed in CD9-deficient DCs could also be related to an increase in MHC-II sorting into exosomes. Indeed, MHC-II colocalizes with CD9 in MVB of mature DCs, and MHC-II sorting into exosomes involves detergent-resistant membranes (73). However, we did not observe significant differences in the total expression levels of MHC-II or in the turnover of the protein, suggesting that CD9 regulation of MHC-II surface expression is related neither to an increase in MHC-II secretion through sorting into exosomes nor to an increase in protein degradation.
Importantly, our data show that CD9 deletion in immature MoDCs triggers MHC-II accumulation in acidic intracellular compartments. The MIIC, which displays acidic pH and is enriched in LAMP-1 (5, 6), contains luminal vesicles that dynamically form and disappear as a result of retrofusion with the limiting outer membrane (8). MHC-II and HLA-DM can be detected on both membranes (8). These molecules associate with tetraspanins CD63 and CD82 on both MIIC subdomains (41, 42, 44), with CD63 stably interacting with MHC-II and CD82 with HLA-DM (44). CD63 regulates peptide-loaded MHC-II surface expression and could act as a cochaperone (44). We have investigated the colocalization of MHC-II with different markers of intracellular vesicles. Although no differences of colocalization with CD63 were observed in the absence of CD9, MHC-II colocalization with LAMP-1, which is enriched at the MIIC limiting membrane (8), was significantly increased. Egress of peptide–MHC-II complexes from MIIC to the cell surface involves tubular membrane extensions in maturing DCs (10–12). In immature DCs, this process remains largely undefined. Our results indicate that CD9 is an important regulator of MHC-II transport from MIIC to the surface of immature MoDCs. In DCs, surface exposure of peptide–MHC-II molecules is transient, due to rapid internalization and sorting to intracellular compartments (2). Importantly, CD9 deficiency abolishes MHC-II internalization in immature MoDCs. Together, our results suggest that CD9 is important for MHC-II trafficking; in its absence, MHC-II molecules are not transported properly to the surface of immature MoDCs, remaining arrested at the MIIC, and endocytosis of the MHC-II molecules that can reach the membrane in a CD9-independent process is prevented.
Upon maturation, MHC-II surface expression is greatly increased (59, 74). Our results show that both genotypes of LPS-matured MoDCs upregulated MHC-II, but a reduced level of MHC-II surface expression was still observed in the absence of CD9. Unlike immature cells, mature CD9 KO MoDCs did not show any significant difference in MHC-II accumulation at LAMP-1+ acidic compartments. During DC maturation, MHC-II trafficking is tightly regulated, and DCs mostly use newly synthesized molecules to present antigens (75). MHC-II molecules synthesized before activation, when they are mainly bound to self-peptides, are mostly targeted for degradation to prevent autoimmunity (2). Our results therefore indicate that, in mature MoDCs, CD9 does not regulate egress of MHC-II molecules from the MIIC to the plasma membrane, which would mainly occur through a CD9-independent mechanism. In mature DCs, while endocytosis of peptide–MHC-II complexes is similar to that seen in immature cells, recycling is highly stimulated by activation, contributing to increases in the levels of surface MHC-II (21). We have measured MHC-II internalization in mature MoDCs, and, similarly to what we had observed in immature MoDCs, CD9 KO blocks MHC-II endocytosis. Interestingly, CD9 deficiency greatly impairs MHC-II recycling in mature MoDCs. Taken together, our results suggest that in mature MoDCs, CD9 is essential for MHC-II trafficking from the surface to endosomes and back to the surface. The amount of surface MHC-II in mature CD9 KO MoDCs might be the consequence of egress from MIIC to the plasma membrane driven by a CD9-independent mechanism, which would be increased upon maturation of DCs.
Several tetraspanins regulate the intracellular trafficking of their molecular partners, controlling expression at the plasma membrane or sorting into intracellular vesicles (76, 77). Our results show that CD9 controls different steps of MHC-II intracellular trafficking in MoDCs, regulating egress from MIIC to the surface in immature cells, internalization in immature and mature MoDCs, and recycling in mature cells. This work provides new insights into the regulation of the presence of peptide–MHC-II complexes at the MoDC surface which could be applied to boost the efficiency of antigen presentation or to impair the presentation of self-antigens involved in autoimmunity.
MATERIALS AND METHODS
Mice.
CD9−/− mice were described previously (78). Experiments were performed using CD9−/− mice in a C57BL/6 genetic background, and wild-type (WT) mice were littermates that were sex and age matched. OT-II transgenic mice express a TCR specific for OVA peptide 323–339. Mice were housed under specific-pathogen-free conditions at the Centro Nacional de Investigaciones Cardiovasculares, and the experiments were approved by the Centro Nacional de Investigaciones Cardiovasculares Ethical Committee for Animal Welfare and by the Spanish Ministry of Agriculture, Food and the Environment. Animal procedures conformed to European Union directive 2010/63EU and recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes, enforced in Spanish law under Real Decreto 1201/2005.
Generation of BMDCs.
Bone marrow-derived DCs (BMDCs) were obtained from bone marrow cell suspensions after culture on nontreated cell culture dishes in complete RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 mg/ml penicillin, 100 mg/ml streptomycin, 50 mM 2-mercaptoethanol (2-ME), and either 20 ng/ml GM-CSF or 100 ng/ml Flt3L (PeproTech, London, United Kingdom). Cells were collected at days 8 and 9, and BMDCs were characterized using antibodies against CD11c, MHC-II, CD11b, B220, CD80, CD86, and Ly6G/Ly6C. Maturation was induced after overnight incubation with LPS from Escherichia coli O111:B4 (Sigma-Aldrich) (1 μg/ml).
T cell stimulation assays.
Naive CD4+ OT-II T cells were isolated by negative selection in an autoMACS Pro separator (Miltenyi Biotec) according to the manufacturer's instructions. Selected cells were >95% CD4+ Vα2+. For in vitro T cell stimulation, naive OT-II cells were labeled with CellTrace carboxyfluorescein succinimidyl ester (CFSE) cell proliferation probe (Molecular Probes) (0.5 μM) and cocultured for 72 h with LPS-matured BMDCs loaded with the OVA peptide (Genscript) (10 μg/ml) in a 10:1 T cell/DC ratio. T cells were analyzed for cell activation by measurement of CD25 surface expression and for proliferation by CFSE dilution assay. Additionally, IFN-γ expression was analyzed after restimulation with phorbol myristate acetate (PMA) and ionomycin (Sigma) during 6 h in the presence of BD GolgiPlug (BD Biosciences) to block cytokine secretion. Cells were stained for detection of IFN-γ expression after fixation and permeabilization using a BD Cytofix/Cytoperm kit (BD Biosciences).
For in vivo T cell stimulation assays, LPS-matured OVA peptide-loaded WT or CD9 KO BMDCs were subcutaneously injected in the footpads of recipient C57BL/6 mice. CellTrace violet (Thermofisher)-labeled OT-II T cells were intravenously transferred 24 h later into the recipient mice. After a further 2 and 4 days, respectively, activation and proliferation of adoptively transferred T cells were analyzed in popliteal draining lymph nodes.
Cell migration assays.
In vitro Transwell migration assays were performed in Transwell plates (CoStar; Corning), in which DCs were seeded into the upper chamber with different concentrations of CCL21 (R&D Systems) at the lower chamber. After 2 h, the presence of DCs in the lower chamber was analyzed by flow cytometry to determine the percentage of migrated cells.
In vivo cell migration was assessed by FITC-skin sensitization migration assay. Abdominal skin of mice was painted with 1% FITC (Sigma-Aldrich) diluted in acetone and dibutyl phtalate. After 24 h, inguinal lymph nodes were collected and analyzed by flow cytometry to detect the presence of FITC+ CD11c+ cells.
Flow cytometry.
Cells were stained with appropriate primary antibodies followed by species-matching secondary antibodies. For detection of intracellular proteins, cells were fixed and permeabilized using a BD Cytofix/Cytoperm kit. Data were acquired with a FACSCanto II flow cytometer (BD) and were then analyzed with BD FACSDiva (BD) or FlowJo software (FlowJo LLC). The antibodies used were as follows: anti-MHC-II (I-A/I-E, clone M5/114) and CD16/CD32 (2.4G2) from Tonbo; allophycocyanin-conjugated MHC-II and biotin-conjugated CCR7 antibodies from eBiosciences; and biotin-conjugated anti-CD9, ICAM-1, Mac-1, MHC-II, VLA-4, CD80, CD86, and CD40, phycoerythrin (PE)-conjugated anti-CD11c and CD4, FITC-conjugated anti-MHC-II, Vα2, and B220, and allophycocyanin-conjugated anti-Gr1, CD25, IFN-γ, and streptavidin-peridinin chlorophyll protein (PerCP) antibodies from BD Biosciences.
For quantification of binding to MHC-II, biotin-labeled OVA peptides (323–339; AnaSpec) were incubated for 1 h at 4°C. For quantification of antigen processing, DCs were incubated with 45 μg of DQ-OVA (Invitrogen) for different times, washed, and analyzed by flow cytometry. Lysotracker Red (Invitrogen) was added to cells according to the manufacturer's protocol. Cells were incubated with different concentrations of ammonium chloride (NH4Cl; Sigma) for 1 h at 37°C or MG132 (Sigma) for 4 h at 37°C. For the fluorescence-activated cell sorter (FACS)-based internalization assay, cells were incubated for 30 min at 37°C in incomplete medium and then incubated with biotinylated MHC-II antibodies (BD Biosciences) (10 μg/ml) for 1 h at 4°C, washed, and incubated for different times at 37°C. After washing, cells were labeled with LIVE/DEAD fixable stain (Invitrogen), incubated with Fc block (Tonbo) for 15 min, and incubated with antibodies against CD11c and MHC-II. To visualize intracellular proteins, cells were fixed and permeabilized using a BD Cytofix/Cytoperm kit before immune staining was performed with EEA1, HGS/HRS, CD63, or LAMP-1 antibodies.
Fluorescence confocal microscopy.
For analysis of T cell-APC cognate interactions, DCs were mixed with OT-II CD4+ T cells (1:1) previously labeled with the cell tracer CMAC (7-amino-4-chloromethylcoumarin). For measurement of live T cell-APC interactions, DCs labeled with DiO or DiD lipophilic tracers (Invitrogen) were resuspended in Hanks balanced salt solution (HBSS) (Lonza)–2% FBS–20 mM HEPES (Lonza) and were plated onto poly-l-lysine (PLL; Sigma)-coated 35-mm-diameter culture dishes (MatTek) for at least 15 min at 37°C. OT-II T cells labeled with CMAC and the Dil lipophilic tracer (Invitrogen) were resuspended in HBSS–2% FBS–20 mM HEPES and were added to DCs at the microscope, which was fully covered by an acrylic box to allow time-lapse confocal microscopy acquisition at 37°C and 5% CO2.
For analysis of MHC-II accumulation in acidic compartments, pHrodo red avidin (Invitrogen) was coupled to biotin-conjugated MHC-II (I-A/I-E) antibodies for 1 h at 4°C in live imaging solution (Invitrogen) with 1% bovine serum albumin (BSA). DCs plated onto coverslips were incubated with the antibody-pHrodo conjugate solution for 1 h at 37°C, washed, and fixed.
For immunostaining, cells were plated onto PLL-coated coverslips (20 μg/ml), incubated for 30 min at 37°C, fixed in 4% paraformaldehyde (PFA), permeabilized in phosphate-buffered saline (PBS)–0.1% Triton X-100 for 5 min when appropriate, stained with appropriate primary antibodies followed by species-matching secondary antibodies, incubated with DAPI (4′,6-diamidino-2-phenylindole), and mounted in Prolong antifade medium (Invitrogen). The antibodies used were as follows: CD63 and EEA1 (Santa Cruz); Alexa Fluor 488-conjugated α-tubulin (Sigma); HGS/HRS (Cell Signaling); and biotin-conjugated ICAM-1, FITC-conjugated or biotin-conjugated MHC-II, and Alexa Fluor 647-conjugated LAMP-1 and CD3 (BD Pharmingen). Phalloidins (Alexa Fluor 488-phalloidin and Alexa Fluor 647-phalloidin) were from Invitrogen.
Confocal images were obtained with a Leica TCS-SP5 confocal scanning laser unit attached to an inverted epifluorescence DMI6000B microscope fitted with an HCX PL APO lambda blue 63×/1.4-numerical-aperture (NA) oil immersion objective, using Las-AF acquisition software (Leica Microsystems), or, alternatively, with a Zeiss LSM700 confocal scanning laser unit attached to an inverted epifluorescence microscope (Observer.Z1) fitted with a Pan APO Chromat 63×/1.4-NA oil immersion objective, using ZEN 2009 acquisition software (Carl Zeiss Microscopy GmbH). Images were analyzed with Leica LAS-AF, Metamorph (Molecular Devices), or ImageJ (NIH).
Quantification of protein relocalization to the IS was performed with the Synapsemeasure plug-in in Image J as described previously (79). For the quantification, briefly, background signal was subtracted from all other measurements, and the fluorescence intensity signals were measured in selected regions with similar areas. For the analysis of protein accumulation at the T cell side of the IS, the level of signal accumulation at the T cell-APC contact zone (IS) was determined relative to accumulation at the rest of the T cell surface, while in the analysis of protein accumulation at the APC side of the IS, signal accumulation at the IS was related to the accumulation seen on the rest of the APC surface. In the charts, each dot corresponds to a T cell-APC conjugate.
Measurements of protein colocalization (Pearson's and Mander's coefficients) were performed using Imaris (Bitplane), analyzing three-dimensional stack confocal microscopy images. The signals inside the nucleus were excluded from the analyses.
Cell biotinylation, immunoprecipitation, and immunoblotting.
The cell biotinylation assay was adapted from the method described by Cho and collaborators (21). For internalization analysis, cells were washed in ice-cold HBSS, biotinylated with EZ-Link sulfo-NHS-SS-biotin {(sulfosuccinimidyl-2-[biotinamido]ethyl-1,3-dithiopropionate)} (Invitrogen) (1 mg/ml) for 30 min at 4°C, washed in HBSS, and incubated in growth medium for different times at 37°C to allow protein internalization. Cells were then washed in HBSS and incubated for 15 min at 4°C with 100 mM sodium 2-mercaptoethane sulfonic acid (MESNA; Sigma) mixed with 50 mM Tris-HCl (pH 8.6), 100 mM NaCl, 1 mM EDTA, and 0.2% BSA (to remove noninternalized biotin). Cells were washed again in HBSS, incubated with 120 mM iodoacetamide (Sigma), washed, and lysed with a mixture of 1% Triton X, 1 mM CaCl2, 1 mM MgCl2, and PBS containing protease inhibitors. For measurement of MHC-II recycling, after cell biotinylation, cells were incubated 30 min at 37°C to allow protein internalization. Cells were then incubated with MESNA to remove noninternalized biotin, washed, and incubated in growth medium for different times to allow protein recycling. Cells were then treated as described for the internalization protocol.
For the analysis of MHC-II ubiquitination, cells were lysed with a mixture of 1% Triton X, 25 mM N-ethylmaleimide, 10 mM iodoacetamide, and PBS containing protease inhibitors. Cell lysates were immunoprecipitated with antibodies against MHC-II (I-A/I-E, M5/114 clone) coupled to Dynabeads–protein G (Life Technologies) for 18 h at 4°C. After washing, beads were resuspended in Laemmli buffer and separated by SDS-PAGE. For internalization and recycling assays, biotinylated proteins were revealed using a Fujifilm LAS-4000 system after membrane incubation with streptavidin-HRP (Invitrogen). For ubiquitination assay, membranes were probed with antiubiquitin antibody (P4D1; Enzo Lifescience). Membranes were reprobed with anti-MHC-II (I-A/I-E, M5/114 clone) and peroxidase-conjugated secondary antibody. Band intensities were quantified using ImageGauge (Fujifilm), and results were normalized to the total protein expression.
For immunoblotting of whole-cell lysates, cells were lysed with a mixture of 1% Triton X-100, 1 mM CaCl2, 1 mM MgCl2, and PBS containing protease inhibitors. Cells were treated with 12.5 μg/ml of cycloheximide (CHX) for different times at 37°C, washed, and then lysed. Membranes were revealed after incubation with MHC-II (I-A/I-E, clone M5/114), α-tubulin (Sigma), or ezrin, radixin, and moesin (ERM) (kindly provided by H. Furthmayr, Stanford University, USA) (90:3) and peroxidase-conjugated secondary antibodies. Band intensities were quantified, and results were normalized with respect to the tubulin or ERM intensity signals.
RNA isolation, mRNA reverse transcription, and quantitative real-time PCR.
Total RNA was extracted with an RNeasy minikit (Qiagen). Purity and concentration were measured in a NanoDrop 1000 spectrophotometer (Thermo Scientific). cDNA was synthesized using a High Capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative PCR was performed with SYBR green PCR master mix (Applied Biosystems) and the following corresponding primers: for MARCH-I, 5′-AAGAGAGCCCACTCATCACACC-3′ (forward) and 5′-ATCTGGAGCTTTTCCCACTTCC-3′ (reverse); for β-actin, 5′-CAGAAGGAGATTACTGCTCTGGCT-3′ (forward) and 5′-TACTCCTGCTTGCTGATCCACATC-3′ (reverse); and for Ywhaz (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta), 5′-CGTTGTAGGAGCCCGTAGGTCAT-3′ (forward) and 5′-CGTTGTAGGAGCCCGTAGGTCAT-3′ (reverse). Data were acquired and analyzed using an ABI Prism 7900HT sequence detection system (Applied Biosystems) and Biogazelle QBasePlus software (Biogazelle). β-Actin and Ywhaz genes were used as endogenous controls for presentation of relative mRNA levels.
Statistical analysis.
All statistical analyses were performed with GraphPad Prism (GraphPad Software Inc.). P values were calculated using the two-tailed Student's t test or one-way or two-way analysis of variance (ANOVA) with Bonferroni's or Tukey's post hoc multiple-comparison test, as appropriate.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Yáñez-Mó (UAM, Spain), M. Vicente-Manzanares (UAM, Spain), and S. Iborra (CNIC, Spain) for critical reading of the manuscript. Microscopy was performed at CNIC Microscopy & Dynamic Imaging Unit.
This work was supported by grants to F.S.-M. (SAF2014-55579-R; INDISNET-S2011/BMD-2332; ERC-2011-AdG 294340-GENTRIS; PIE13/00041), to G.M.D.H. (PI11/00939), and to J.M.G.-G. (PI14/00526; CP11/00145; CPII16/00022) and was cofunded by Fondo Europeo de Desarrollo Regional (FEDER). J.M.G.-G. is supported by Miguel Servet Program (ISCIII) and Fundación Ramón Areces. The CNIC is supported by the Spanish Ministry of Economy, Industry and Competitiveness (MINECO) and by the Pro CNIC Foundation.
We declare that we have no competing financial interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00202-17.
REFERENCES
- 1.Rocha N, Neefjes J. 2008. MHC class II molecules on the move for successful antigen presentation. EMBO J 27:1–5. doi: 10.1038/sj.emboj.7601945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.ten Broeke T, Wubbolts R, Stoorvogel W. 2013. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb Perspect Biol 5:a016873. doi: 10.1101/cshperspect.a016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Kaer L. 2001. Accessory proteins that control the assembly of MHC molecules with peptides. Immunol Res 23:205–214. doi: 10.1385/IR:23:2-3:205. [DOI] [PubMed] [Google Scholar]
- 4.Zwart W, Griekspoor A, Kuijl C, Marsman M, van Rheenen J, Janssen H, Calafat J, van Ham M, Janssen L, van Lith M, Jalink K, Neefjes J. 2005. Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity 22:221–233. doi: 10.1016/j.immuni.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Ziegler HK, Unanue ER. 1982. Decrease in macrophage antigen catabolism caused by ammonia and chloroquine is associated with inhibition of antigen presentation to T cells. Proc Natl Acad Sci U S A 79:175–178. doi: 10.1073/pnas.79.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calafat J, Nijenhuis M, Janssen H, Tulp A, Dusseljee S, Wubbolts R, Neefjes J. 1994. Major histocompatibility complex class II molecules induce the formation of endocytic MIIC-like structures. J Cell Biol 126:967–977. doi: 10.1083/jcb.126.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Honey K, Rudensky AY. 2003. Lysosomal cysteine proteases regulate antigen presentation. Nat Rev Immunol 3:472–482. doi: 10.1038/nri1110. [DOI] [PubMed] [Google Scholar]
- 8.van den Hoorn T, Paul P, Jongsma ML, Neefjes J. 2011. Routes to manipulate MHC class II antigen presentation. Curr Opin Immunol 23:88–95. doi: 10.1016/j.coi.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Roche PA, Furuta K. 2015. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 15:203–216. doi: 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barois N, de Saint-Vis B, Lebecque S, Geuze HJ, Kleijmeer MJ. 2002. MHC class II compartments in human dendritic cells undergo profound structural changes upon activation. Traffic 3:894–905. doi: 10.1034/j.1600-0854.2002.31205.x. [DOI] [PubMed] [Google Scholar]
- 11.Chow A, Toomre D, Garrett W, Mellman I. 2002. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature 418:988–994. doi: 10.1038/nature01006. [DOI] [PubMed] [Google Scholar]
- 12.Kleijmeer M, Ramm G, Schuurhuis D, Griffith J, Rescigno M, Ricciardi-Castagnoli P, Rudensky AY, Ossendorp F, Melief CJ, Stoorvogel W, Geuze HJ. 2001. Reorganization of multivesicular bodies regulates MHC class II antigen presentation by dendritic cells. J Cell Biol 155:53–63. doi: 10.1083/jcb.200103071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vyas JM, Kim YM, Artavanis-Tsakonas K, Love JC, Van der Veen AG, Ploegh HL. 2007. Tubulation of class II MHC compartments is microtubule dependent and involves multiple endolysosomal membrane proteins in primary dendritic cells. J Immunol 178:7199–7210. doi: 10.4049/jimmunol.178.11.7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Nispen tot Pannerden HE, Geerts WJ, Kleijmeer MJ, Heijnen HF. 2010. Spatial organization of the transforming MHC class II compartment. Biol Cell 102:581–591. doi: 10.1042/BC20100046. [DOI] [PubMed] [Google Scholar]
- 15.Wubbolts R, Fernandez-Borja M, Jordens I, Reits E, Dusseljee S, Echeverri C, Vallee RB, Neefjes J. 1999. Opposing motor activities of dynein and kinesin determine retention and transport of MHC class II-containing compartments. J Cell Sci 112(Pt 6):785–795. [DOI] [PubMed] [Google Scholar]
- 16.Bosch B, Heipertz EL, Drake JR, Roche PA. 2013. Major histocompatibility complex (MHC) class II-peptide complexes arrive at the plasma membrane in cholesterol-rich microclusters. J Biol Chem 288:13236–13242. doi: 10.1074/jbc.M112.442640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuipers HF, Biesta PJ, Groothuis TA, Neefjes JJ, Mommaas AM, van den Elsen PJ. 2005. Statins affect cell-surface expression of major histocompatibility complex class II molecules by disrupting cholesterol-containing microdomains. Hum Immunol 66:653–665. doi: 10.1016/j.humimm.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 18.Poloso NJ, Muntasell A, Roche PA. 2004. MHC class II molecules traffic into lipid rafts during intracellular transport. J Immunol 173:4539–4546. doi: 10.4049/jimmunol.173.7.4539. [DOI] [PubMed] [Google Scholar]
- 19.Zhong G, Romagnoli P, Germain RN. 1997. Related leucine-based cytoplasmic targeting signals in invariant chain and major histocompatibility complex class II molecules control endocytic presentation of distinct determinants in a single protein. J Exp Med 185:429–438. doi: 10.1084/jem.185.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walseng E, Bakke O, Roche PA. 2008. Major histocompatibility complex class II-peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J Biol Chem 283:14717–14727. doi: 10.1074/jbc.M801070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho KJ, Walseng E, Ishido S, Roche PA. 2015. Ubiquitination by March-I prevents MHC class II recycling and promotes MHC class II turnover in antigen-presenting cells. Proc Natl Acad Sci U S A 112:10449–10454. doi: 10.1073/pnas.1507981112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, Gatti E. 2008. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc Natl Acad Sci U S A 105:3491–3496. doi: 10.1073/pnas.0708874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, Uematsu M, Hasegawa T, Koseki H, Ohara O, Nakayama M, Toyooka K, Matsuoka K, Hotta H, Yamamoto A, Ishido S. 2007. Novel regulation of MHC class II function in B cells. EMBO J 26:846–854. doi: 10.1038/sj.emboj.7601556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin JS, Ebersold M, Pypaert M, Delamarre L, Hartley A, Mellman I. 2006. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature 444:115–118. doi: 10.1038/nature05261. [DOI] [PubMed] [Google Scholar]
- 25.van Niel G, Wubbolts R, Ten Broeke T, Buschow SI, Ossendorp FA, Melief CJ, Raposo G, van Balkom BW, Stoorvogel W. 2006. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity 25:885–894. doi: 10.1016/j.immuni.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Walseng E, Furuta K, Bosch B, Weih KA, Matsuki Y, Bakke O, Ishido S, Roche PA. 2010. Ubiquitination regulates MHC class II-peptide complex retention and degradation in dendritic cells. Proc Natl Acad Sci U S A 107:20465–20470. doi: 10.1073/pnas.1010990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pathak SS, Lich JD, Blum JS. 2001. Cutting edge: editing of recycling class II:peptide complexes by HLA-DM. J Immunol 167:632–635. doi: 10.4049/jimmunol.167.2.632. [DOI] [PubMed] [Google Scholar]
- 28.Sinnathamby G, Eisenlohr LC. 2003. Presentation by recycling MHC class II molecules of an influenza hemagglutinin-derived epitope that is revealed in the early endosome by acidification. J Immunol 170:3504–3513. doi: 10.4049/jimmunol.170.7.3504. [DOI] [PubMed] [Google Scholar]
- 29.Raposo G, Stoorvogel W. 2013. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffin JP, Chu R, Harding CV. 1997. Early endosomes and a late endocytic compartment generate different peptide-class II MHC complexes via distinct processing mechanisms. J Immunol 158:1523–1532. [PubMed] [Google Scholar]
- 31.Lich JD, Elliott JF, Blum JS. 2000. Cytoplasmic processing is a prerequisite for presentation of an endogenous antigen by major histocompatibility complex class II proteins. J Exp Med 191:1513–1524. doi: 10.1084/jem.191.9.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukherjee P, Dani A, Bhatia S, Singh N, Rudensky AY, George A, Bal V, Mayor S, Rath S. 2001. Efficient presentation of both cytosolic and endogenous transmembrane protein antigens on MHC class II is dependent on cytoplasmic proteolysis. J Immunol 167:2632–2641. doi: 10.4049/jimmunol.167.5.2632. [DOI] [PubMed] [Google Scholar]
- 33.Schmid D, Pypaert M, Munz C. 2007. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charrin S, le Naour F, Silvie O, Milhiet PE, Boucheix C, Rubinstein E. 2009. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem J 420:133–154. doi: 10.1042/BJ20082422. [DOI] [PubMed] [Google Scholar]
- 35.Yáñez-Mó M, Barreiro O, Gordon-Alonso M, Sala-Valdés M, Sánchez-Madrid F. 2009. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol 19:434–446. doi: 10.1016/j.tcb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Hemler ME. 2008. Targeting of tetraspanin proteins–potential benefits and strategies. Nat Rev Drug Discov 7:747–758. doi: 10.1038/nrd2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levy S, Shoham T. 2005. The tetraspanin web modulates immune-signalling complexes. Nat Rev Immunol 5:136–148. doi: 10.1038/nri1548. [DOI] [PubMed] [Google Scholar]
- 38.Rocha-Perugini V, Sanchez-Madrid F, Martinez Del Hoyo G. 2015. Function and dynamics of tetraspanins during antigen recognition and immunological synapse formation. Front Immunol 6:653. doi: 10.3389/fimmu.2015.00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Spriel AB, Figdor CG. 2010. The role of tetraspanins in the pathogenesis of infectious diseases. Microbes Infect 12:106–112. doi: 10.1016/j.micinf.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Tarrant JM, Robb L, van Spriel AB, Wright MD. 2003. Tetraspanins: molecular organisers of the leukocyte surface. Trends Immunol 24:610–617. doi: 10.1016/j.it.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 41.Engering A, Pieters J. 2001. Association of distinct tetraspanins with MHC class II molecules at different subcellular locations in human immature dendritic cells. Int Immunol 13:127–134. doi: 10.1093/intimm/13.2.127. [DOI] [PubMed] [Google Scholar]
- 42.Hammond C, Denzin LK, Pan M, Griffith JM, Geuze HJ, Cresswell P. 1998. The tetraspan protein CD82 is a resident of MHC class II compartments where it associates with HLA-DR, -DM, and -DO molecules. J Immunol 161:3282–3291. [PubMed] [Google Scholar]
- 43.Szöllósi J, Horejsí V, Bene L, Angelisová P, Damjanovich S. 1996. Supramolecular complexes of MHC class I, MHC class II, CD20, and tetraspan molecules (CD53, CD81, and CD82) at the surface of a B cell line JY. J Immunol 157:2939–2946. [PubMed] [Google Scholar]
- 44.Hoorn T, Paul P, Janssen L, Janssen H, Neefjes J. 2012. Dynamics within tetraspanin pairs affect MHC class II expression. J Cell Sci 125:328–339. doi: 10.1242/jcs.088047. [DOI] [PubMed] [Google Scholar]
- 45.Gartlan KH, Belz GT, Tarrant JM, Minigo G, Katsara M, Sheng KC, Sofi M, van Spriel AB, Apostolopoulos V, Plebanski M, Robb L, Wright MD. 2010. A complementary role for the tetraspanins CD37 and Tssc6 in cellular immunity. J Immunol 185:3158–3166. doi: 10.4049/jimmunol.0902867. [DOI] [PubMed] [Google Scholar]
- 46.Sheng KC, van Spriel AB, Gartlan KH, Sofi M, Apostolopoulos V, Ashman L, Wright MD. 2009. Tetraspanins CD37 and CD151 differentially regulate Ag presentation and T-cell co-stimulation by DC. Eur J Immunol 39:50–55. doi: 10.1002/eji.200838798. [DOI] [PubMed] [Google Scholar]
- 47.Martínez del Hoyo G, Ramírez-Huesca M, Levy S, Boucheix C, Rubinstein E, Minguito de la Escalera M, González-Cintado L, Ardavín C, Veiga E, Yáñez-Mó M, Sánchez-Madrid F. 2015. CD81 controls immunity to Listeria infection through Rac-dependent inhibition of proinflammatory mediator release and activation of cytotoxic T cells. J Immunol 194:6090–6101. doi: 10.4049/jimmunol.1402957. [DOI] [PubMed] [Google Scholar]
- 48.Petersen SH, Odintsova E, Haigh TA, Rickinson AB, Taylor GS, Berditchevski F. 2011. The role of tetraspanin CD63 in antigen presentation via MHC class II. Eur J Immunol 41:2556–2561. doi: 10.1002/eji.201141438. [DOI] [PubMed] [Google Scholar]
- 49.Angelisová P, Hilgert I, Horejsí V. 1994. Association of four antigens of the tetraspans family (CD37, CD53, TAPA-1, and R2/C33) with MHC class II glycoproteins. Immunogenetics 39:249–256. doi: 10.1007/BF00188787. [DOI] [PubMed] [Google Scholar]
- 50.Jones EL, Wee JL, Demaria MC, Blakeley J, Ho PK, Vega-Ramos J, Villadangos JA, van Spriel AB, Hickey MJ, Hammerling GJ, Wright MD. 2016. Dendritic cell migration and antigen presentation are coordinated by the opposing functions of the tetraspanins CD82 and CD37. J Immunol 196:978–987. doi: 10.4049/jimmunol.1500357. [DOI] [PubMed] [Google Scholar]
- 51.Unternaehrer JJ, Chow A, Pypaert M, Inaba K, Mellman I. 2007. The tetraspanin CD9 mediates lateral association of MHC class II molecules on the dendritic cell surface. Proc Natl Acad Sci U S A 104:234–239. doi: 10.1073/pnas.0609665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khandelwal S, Roche PA. 2010. Distinct MHC class II molecules are associated on the dendritic cell surface in cholesterol-dependent membrane microdomains. J Biol Chem 285:35303–35310. doi: 10.1074/jbc.M110.147793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Domínguez PM, Ardavín C. 2010. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev 234:90–104. doi: 10.1111/j.0105-2896.2009.00876.x. [DOI] [PubMed] [Google Scholar]
- 54.Merad M, Sathe P, Helft J, Miller J, Mortha A. 2013. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benvenuti F. 2016. The dendritic cell synapse: a life dedicated to T cell activation. Front Immunol 7:70. doi: 10.3389/fimmu.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de la Fuente H, Mittelbrunn M, Sanchez-Martin L, Vicente-Manzanares M, Lamana A, Pardi R, Cabanas C, Sanchez-Madrid F. 2005. Synaptic clusters of MHC class II molecules induced on DCs by adhesion molecule-mediated initial T-cell scanning. Mol Biol Cell 16:3314–3322. doi: 10.1091/mbc.E05-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hashimoto-Tane A, Saito T. 2016. Dynamic regulation of TCR-microclusters and the microsynapse for T cell activation. Front Immunol 7:255. doi: 10.3389/fimmu.2016.00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rocha-Perugini V, Gonzalez-Granado JM, Tejera E, Lopez-Martin S, Yanez-Mo M, Sanchez-Madrid F. 2014. Tetraspanins CD9 and CD151 at the immune synapse support T-cell integrin signaling. Eur J Immunol 44:1967–1975. doi: 10.1002/eji.201344235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villadangos JA, Schnorrer P, Wilson NS. 2005. Control of MHC class II antigen presentation in dendritic cells: a balance between creative and destructive forces. Immunol Rev 207:191–205. doi: 10.1111/j.0105-2896.2005.00317.x. [DOI] [PubMed] [Google Scholar]
- 60.Svensson M, Stockinger B, Wick MJ. 1997. Bone marrow-derived dendritic cells can process bacteria for MHC-I and MHC-II presentation to T cells. J Immunol 158:4229–4236. [PubMed] [Google Scholar]
- 61.Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH. 2002. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol 156:843–854. doi: 10.1083/jcb.200106056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fuertes G, Martin De Llano JJ, Villarroya A, Rivett AJ, Knecht E. 2003. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J 375:75–86. doi: 10.1042/bj20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, Reis e Sousa C. 2015. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity 42:1197–1211. doi: 10.1016/j.immuni.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 64.Meyer zum Bueschenfelde CO, Unternaehrer J, Mellman I, Bottomly K. 2004. Regulated recruitment of MHC class II and costimulatory molecules to lipid rafts in dendritic cells. J Immunol 173:6119–6124. doi: 10.4049/jimmunol.173.10.6119. [DOI] [PubMed] [Google Scholar]
- 65.Kropshofer H, Spindeldreher S, Rohn TA, Platania N, Grygar C, Daniel N, Wolpl A, Langen H, Horejsi V, Vogt AB. 2002. Tetraspan microdomains distinct from lipid rafts enrich select peptide-MHC class II complexes. Nat Immunol 3:61–68. doi: 10.1038/ni750. [DOI] [PubMed] [Google Scholar]
- 66.Anderson HA, Hiltbold EM, Roche PA. 2000. Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nat Immunol 1:156–162. doi: 10.1038/77842. [DOI] [PubMed] [Google Scholar]
- 67.Poloso NJ, Denzin LK, Roche PA. 2006. CDw78 defines MHC class II-peptide complexes that require Ii chain-dependent lysosomal trafficking, not localization to a specific tetraspanin membrane microdomain. J Immunol 177:5451–5458. doi: 10.4049/jimmunol.177.8.5451. [DOI] [PubMed] [Google Scholar]
- 68.Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O'Garra A, Biron C, Briere F, Trinchieri G. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 69.Mittelbrunn M, Martinez del Hoyo G, Lopez-Bravo M, Martin-Cofreces NB, Scholer A, Hugues S, Fetler L, Amigorena S, Ardavin C, Sanchez-Madrid F. 2009. Imaging of plasmacytoid dendritic cell interactions with T cells. Blood 113:75–84. doi: 10.1182/blood-2008-02-139865. [DOI] [PubMed] [Google Scholar]
- 70.Barreiro O, de la Fuente H, Mittelbrunn M, Sanchez-Madrid F. 2007. Functional insights on the polarized redistribution of leukocyte integrins and their ligands during leukocyte migration and immune interactions. Immunol Rev 218:147–164. doi: 10.1111/j.1600-065X.2007.00529.x. [DOI] [PubMed] [Google Scholar]
- 71.Springer TA, Dustin ML. 2012. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol 24:107–115. doi: 10.1016/j.ceb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yokosuka T, Saito T. 2010. The immunological synapse, TCR microclusters, and T cell activation. Curr Top Microbiol Immunol 340:81–107. [DOI] [PubMed] [Google Scholar]
- 73.Buschow SI, Nolte-'t Hoen EN, van Niel G, Pols MS, ten Broeke T, Lauwen M, Ossendorp F, Melief CJ, Raposo G, Wubbolts R, Wauben MH, Stoorvogel W. 2009. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 10:1528–1542. doi: 10.1111/j.1600-0854.2009.00963.x. [DOI] [PubMed] [Google Scholar]
- 74.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, Hornell TM, Mellins ED. 2005. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev 207:242–260. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 75.ten Broeke T, van Niel G, Wauben MH, Wubbolts R, Stoorvogel W. 2011. Endosomally stored MHC class II does not contribute to antigen presentation by dendritic cells at inflammatory conditions. Traffic 12:1025–1036. doi: 10.1111/j.1600-0854.2011.01212.x. [DOI] [PubMed] [Google Scholar]
- 76.Berditchevski F, Odintsova E. 2007. Tetraspanins as regulators of protein trafficking. Traffic 8:89–96. doi: 10.1111/j.1600-0854.2006.00515.x. [DOI] [PubMed] [Google Scholar]
- 77.Matthews AL, Noy PJ, Reyat JS, Tomlinson MG. 2016. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: the emerging role of tetraspanins and rhomboids. Platelets 2016:1–9. doi: 10.1080/09537104.2016.1184751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Le Naour F, Rubinstein E, Jasmin C, Prenant M, Boucheix C. 2000. Severely reduced female fertility in CD9-deficient mice. Science 287:319–321. doi: 10.1126/science.287.5451.319. [DOI] [PubMed] [Google Scholar]
- 79.Rocha-Perugini V, Zamai M, Gonzalez-Granado JM, Barreiro O, Tejera E, Yanez-Mo M, Caiolfa VR, Sanchez-Madrid F. 2013. CD81 controls sustained T cell activation signaling and defines the maturation stages of cognate immunological synapses. Mol Cell Biol 33:3644–3658. doi: 10.1128/MCB.00302-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.