

Abstract
Chronic rhinosinusitis (CRS) is a troublesome, chronic inflammatory disease that affects over 10% of the adult population, causing decreased quality of life, lost productivity, and lost time at work and leading to more than a million surgical interventions annually worldwide. The nose, paranasal sinuses, and associated lymphoid tissues play important roles in homeostasis and immunity, and CRS significantly impairs these normal functions. Pathogenic mechanisms of CRS have recently become the focus of intense investigations worldwide, and significant progress has been made. The two main forms of CRS that have been long recognized, with and without nasal polyps, are each now known to be heterogeneous, based on underlying mechanism, geographical location, and race. Loss of the immune barrier, including increased permeability of mucosal epithelium and reduced production of important antimicrobial substances and responses, is a common feature of many forms of CRS. One form of CRS with polyps found worldwide is driven by the cytokines IL-5 and IL-13 coming from Th2 cells, type 2 innate lymphoid cells, and probably mast cells. Type 2 cytokines activate inflammatory cells that are implicated in the pathogenic mechanism, including mast cells, basophils, and eosinophils. New classes of biological drugs that block the production or action of these cytokines are making important inroads toward new treatment paradigms in polypoid CRS.
Keywords: acantholysis, acanthosis, allergy, chronic rhinosinusitis, eosinophilic inflammation, epithelial barrier, epithelial-to-mesenchymal transition (EMT), glandular hyperplasia, nasal polyp, sinus disease
INTRODUCTION
Chronic rhinosinusitis (CRS) is a disease of inflammation of the nose and paranasal sinuses and upper airways characterized by 12 weeks of persistent symptoms including congestion, stuffiness, nasal discharge, pain or facial pressure, impairment or loss of the sense of smell (anosmia), cough, and fatigue. Although heterogeneous, investigators recognize two main forms, one with nasal polyps (CRSwNP) and one without (CRSsNP) (1, 2). Unremitting inflammation can last for years, and CRS affects approximately 12% of the population. Intranasal corticosteroids have limited effectiveness in CRS. It is accepted that the nasal microbiome is disturbed in CRS, and certain organisms, such as fungal species or Staphylococcus aureus, have been implicated in driving inflammation in some patients. Sinus disease (both acute and chronic) is the number one justification for the use of antibiotics, though these drugs have efficacy in only a subset of patients. After failure of medical management, patients often resort to surgical approaches to manage disease. Surgical removal of sinus tissues can aid drainage of sinuses filled with fluid (3). Tissues removed during surgery are of considerable value in laboratory-based investigations of pathologic mechanisms.
OVERVIEW OF FORMS OF CRS, EPIDEMIOLOGY, NATURAL HISTORY, AND COST
Heterogeneity in the pathology of CRS influences clinical phenotype, responses to treatment, and outcomes. CRSsNP comprises more than two-thirds of cases and is less likely to be managed by surgical intervention, whereas CRSwNP represents 20–25% of cases. Polyps are outgrowths of edematous inflammatory tissue that have grown into the middle meatus (see Figure 1). CRSwNP produces less pain and more facial pressure; it is more likely to cause anosmia, less likely to respond to antibiotics, and more likely to respond to corticosteroids (1, 2). A minor form of CRS is hyperplastic CRS, which has pathologic similarities to CRSwNP but without outgrowth of polyps. Another minor form of CRS is allergic fungal sinusitis (AFS), usually characterized by florid growth of a fungal organism, such as Aspergillus or Alternaria spp., resulting in local immune responses with eosinophilia and production of a characteristic thick mucin. Nasal polyp growth occurs in cystic fibrosis or as antrochoanal polyps, but these diseases are not referred to as CRS and are not considered in this review. A particularly severe form of CRS is known as aspirin-exacerbated respiratory disease (AERD), which is characterized by nasal polyps, asthma, and sensitivity to ingestion of aspirin or other COX1 inhibitors (4).
Figure 1.
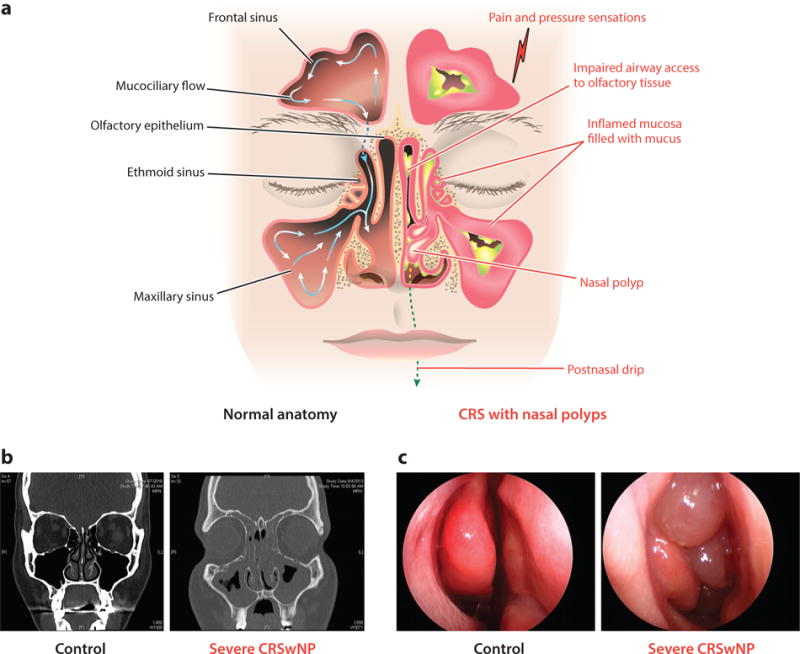
Panel a, overview of gross anatomical and inflammatory changes associated with chronic rhinosinusitis and illustration of the mucociliary flow pattern within the sinuses. Panel b, computed tomography images of a healthy person and one with severe chronic rhinosinusitis with nasal polyps (CRSwNP). Panel c, endoscopic images from a healthy person and from one with severe CRSwNP.
The annual cost of managing CRS in the United States alone is approximately $8 billion, and the aggregate number of surgeries for CRS in general is nearly 500,000 per year (5). Up to one-third of patients require multiple surgeries, sometimes as many as a dozen surgeries over an extended period of time as the inflammatory tissue regrows. Based on incidence and prevalence, it can be concluded that CRS is a relapsing, remitting disease (2, 6). More men suffer from CRSwNP than women, although more women suffer from AERD than men (7).
NORMAL FUNCTION OF THE NOSE AND SINUSES
The nose protects the lungs by filtering and humidifying air prior to entry to the lungs (8). Dense collections of vascular sinusoids and distensible blood vessels termed capacitance vessels vary flow from one nasal passage to the other in a nasal cycle. Lateral or anterior serous glands generate secretions. The nose senses airborne volatile compounds via pattern recognition and olfactory receptors. Olfaction can be essential in avoiding exposure to toxins and pathogens in the air or food. The filtering function of the nose initiates immune sensitization to potential pathogens. Waldeyer’s ring, a structural constellation that includes lymphoid tissue in the tonsils and the adenoid and submandibular lymph nodes, is an important sentinel portal for sensitization by respiratory pathogens (9). The pharyngeal tonsils (i.e., adenoids) are located in the roof of the nasopharynx, the tubal tonsils are at the outflow of the Eustachian tubes, and the palatine tonsils are located in the oropharynx. The mucosal epithelium of tonsils contains M cells that transmit antigens to the lymphoid structures. As seen in Figure 1, an orderly flow of mucosal surface fluid and mucus within the sinuses drains into the ostiomeatal complex; nasal and sinus fluids are then swallowed. Trapped antigens enter the lymphatic systems and reach lymph nodes in the occipital, cervical, parotid, submental, and submandibular regions where they prime and expand populations of B and T lymphocytes that can later populate the alimentary or respiratory tracts. Epithelial cells on the mucosae as well as glands create an antimicrobial mucous blanket containing immunoglobulins, enzymes, antimicrobial peptides, and other factors important in innate host defense (10).
STRUCTURAL CHANGES IN THE NOSE AND SINUSES IN CRS WITH FOCUS ON POLYPS
The nose and sinuses undergo significant structural metamorphosis in CRS, as illustrated in Figure 1. The sinuses fill with inflammatory mucosal tissue as well as secretions. The olfactory zone is shown at the top of the superior meatus. Both obstruction and inflammatory events can lead to anosmia. Figure 1b shows opacification in a computed tomography (CT) scan of the sinuses and nasal vestibule of a CRS patient compared with a healthy subject; changes are quantified using an objective Lund-Mackay score (11). Endoscopic views in Figure 1c of a healthy control patient and a patient with CRSwNP show nasal polyps emerging from the ethmoid sinus through the ostium. In CRSsNP, sinus tissues and the nasal vestibule are swollen and copious secretions are observed, often with pus. Significant heterogeneity is observed macroscopically and microscopically, and underlying histopathologic mechanisms vary. Table 1 represents an attempt to summarize findings about the general histologic features and cytokine expression in CRSsNP and CRSwNP, the latter divided based on presence of eosinophils. Notably, studies of gene expression, histopathology, or physical structure generally utilize control tissues from healthy patients undergoing surgery for noninflammatory indications. Control tissues used are variable, including inferior turbinate, uncinate, or ethmoid tissues, making comparisons among studies challenging (see Figure 1). Distribution of the forms of disease described in Table 1 and the underlying pathologic manifestations vary by continent and race (see below).
Table 1.
Overview of chronic rhinosinusitis (CRS) heterogeneity and consensus of the literature regarding features, cells, and mediators present in tissues from patients with CRS without polyps (CRSsNP), eosinophilic CRS with polyps (E-CRS), and noneosinophilic CRS with polyps (NE-CRS). Note that the prevalence of the forms, features, cells, and mediators vary by geography and race
| CRSsNP | E-CRS | NE-CRS | |
|---|---|---|---|
| Feature | |||
| Fibrosis | Pronounced | Minimal | Minimal |
| Collagen deposition | Abundant | Minimal | Minimal |
| Edema | Minimal | Abundant | Significant |
| Glandular hyperplasia | Abundant | Outside of polyp | Outside of polyp |
| Pseudocyst formation | Not present | Present | Present |
| Cells | |||
| Eosinophils | In some patients | Abundant | Present |
| Neutrophils | Abundant | Some present | Many present |
| Mast cells | Slightly increased | Increased | Somewhat increased |
| B cells/plasma cells | Few present | Abundant | Abundant |
| Basophils | Not present | Abundant | In some patients |
| Mediators | |||
| IL-4, IL-5, IL-13 | In some patients | Significant expression | Minimal expression |
| IFN-γ | In some patients | Minimal expression | Minimal expression |
| IL-17 | In some patients | Expression in Asia | Expression |
| TGF-β | Increased expression | Minimal expression | Some expression |
| MMPs | MMP1, 2, 8, and 9 | MMP1, 2, 7, 8, and 9 | MMP1 |
Abbreviations: IFN-γ, interferon-γ; IL, interleukin; MMPs, matrix metalloproteinases; TGF-β, transforming growth factor β.
TISSUE REMODELING AND POLYP FORMATION IN CRS
Early theories proposed that polyps are adenomas, fibromas, mucosal exudates; cystic dilations of excretory ducts; or swelling due to blockage of glands or lymphatics (12). Alternative theories suggested lymphangitis from recurrent infection or glandular hyperplasia as core causes. Tos and colleagues (12) described a model based on epithelial rupture and necrosis leading to protrusions from the lamina propria and epithelial repair. Many of these early theories identified mechanistic elements still held as important. Recent molecular studies identify emerging endotypes based on cytokine expression, implying multiple disease mechanisms (13, 14). Noneosinophilic disease is much more prevalent in Asian countries such as China, Korea, and Japan, although prevalence of eosinophilic disease has risen along with industrialization (see below). In addition to CRSwNP, other eosinophilic forms of disease include polyps associated with AERD, AFS, and Churg-Strauss disease, whereas noneosinophilic forms include cystic fibrosis (CF), antrochoanal polyps (usually single polyps growing out of the maxillary sinus), Young syndrome, and Kartagener syndrome–associated polyps. Early endotyping differentiated between edematous, fibrous, and cystic nasal polyps, though these terms are not used widely at present. Kountakis et al. (15) found that eosinophilic forms of both CRSwNP and CRSsNP, having more edema, were more severe than the corresponding noneosinophilic forms, whereas noneosinophilic forms have more glandular hyperplasia and dense collagen deposition. Independent of eosinophilia, fibrosis and collagen deposition are commonly seen in CRSsNP but not CRSwNP, whereas edema is a prominent feature of the polypoid forms of disease but less so CRSsNP. Van Bruaene & Bachert (16) have reported differences in tissue remodeling and patterns of cytokine expression in Belgium and China. A large histopathologic study of CRS in Wuhan, China, affirmed the link of eosinophilic infiltration with edema and the association of neutrophils with transforming growth factor β (TGF-β) and fibrosis (17). Hyperplasia of submucosal glands is common in nonpolypoid CRS but rare within nasal polyps themselves, especially in the eosinophilic forms. In nasal polyps, glandular density actually decreases significantly—a decrease associated with a stretching of the glandular canals and formation of tubular glands within the polyp tissue. Both forms of CRS have copious secretions, increased numbers of goblet cells, and activation of glands and goblet cells. Pseudocyst formation occurs in CRSwNP but is not generally observed in CRSsNP (18, 19).
Evaluation using trichrome or picrosirius staining shows increased collagen in CRSsNP but minimal expression in nasal polyps (less than control tissue in most cases) (20). These stains do not detect all collagen forms: In addition to fibrillar collagens (I, II, III, V, XI), there are short-chain forms (VIII, X), basement membrane forms (IV), multiplexins (XV, XVIII), fibril-associated forms (IX, XII, XIV, XVI, XIX), and membrane-associated forms (XIII, XVII), among others (20). Dr. Sergejs Berdnikovs at Northwestern has performed a multistudy analysis of nasal polyp microarray findings deposited in the Gene Expression Omnibus (GEO) database and has found significant elevations of mRNA levels in collagen X, VI, and VIII as well as lumican (S. Berdnikovs, personal communication). Several studies have reported increased matrix metalloproteinase (MMP) levels that degrade the extracellular matrix (e.g., collagen, gelatin, fibronectin, laminin, and elastin), regulate many biologically active molecules, and modify tissue structure (Table 1) (19, 21–23). A polymorphism of MMP-9 has been reported to be a risk gene for CRS (24). Elevated levels of endogenous MMP inhibitors, tissue inhibitors of metalloproteinases (TIMPs), have also been described. Degradation by MMPs has been proposed to be important in pseudocyst formation (19).
Relatively few studies have attempted to enumerate the phenotypes of fibroblasts in CRS. One important study evaluated polyp histopathology during the growth process (25). Early stage polyps were collected with the entire stalk and pedicle from the middle turbinate and were compared with mature ethmoid polyps. Early stage polyps had more epithelial loss, whereas mature polyps had greater deficits in junctional proteins E-cadherin, zonula occludens-1 (ZO-1), and occludin. Early polyps had increased TGF-β, more vimentin staining, and elevated activated myofibroblasts that were α-smooth muscle actin (αSMA) positive, suggesting greater epithelial-to-mesenchymal transition (EMT) and ongoing repair. Early polyps also had more M2 macrophages and more fibronectin staining, reflecting more extensive type 2 inflammation. The stalk of the early polyps had extensive deposition of collagen, suggesting greater activation of fibroblasts early during polyp formation (25). Phenotyping by flow cytometry of fibroblasts isolated from CRS and control tissues using fibroblast specific protein 1 (FSP1, also known as S100A4) positive and mucin 1 (MUC1) negative staining phenotype found a hierarchy in fibroblast numbers: AFRS > CRSwNP > CRSsNP = Controls (26). Considering the low levels of typical collagen in polyps, determining whether polyp fibroblasts make atypical collagens is worthwhile. Fibroblasts isolated from nasal polyp tissue showed significantly enhanced expression of CXCL10 but only when the patients also had asthma. Higher expression of this chemokine has been proposed to contribute to virus-induced exacerbation of disease (27).
An alternative hypothesis by Takabayashi et al. (28, 29) explains nasal polyp growth by the deposition of fibrin mesh within the tissue. In this model, vascular leak leads to activation of thrombin via tissue factor and cleavage of plasma fibrinogen. Fibrin is crosslinked by Factor XIIIA derived from M2 macrophages in the tissue. Polyps have reduced fibrinolytic capacity resulting from impaired epithelial expression of tissue plasminogen activator (tPA) (28, 29). Reduced tPA and elevated M2 macrophages containing XIIIA result from increased expression of interleukin 13 (IL-13) in polyp tissue. Deposition of fibrin resulting from strong type 2 inflammation may be of benefit in immobilization and destruction of parasite worms (30, 31).
In the epithelium, one can observe both acanthosis and acantholysis in samples from patients with CRS. Within the tissue, vascular leak contributes to tissue swelling and formation of nasal polyps, as demonstrated by elevated presence of plasma proteins and clear edema. Reports of angiogenesis with increased levels of vascular endothelial cell growth factor (VEGF) and markers of blood vessel growth, along with increased blood vessels in polyp, are variable. The density of vessels within polyps may be similar to that in normal tissue, and expansion of vessels may parallel expansion of the tissue. A view of the mechanism of polyp formation, informed by newer data on cellular responses and based on the action of new generation biological drugs in eosinophilic CRSwNP, suggests that the infiltration, activation, and mediator release of eosinophils, mast cells, and basophils are important in polyp formation.
Epithelial Dysfunction in CRS: Ongoing Epithelial-to-Mesenchymal Transition and the Injury/Repair Cycle
Effective barrier function is essential for survival, and chronic compromise of the epithelial barrier is a characteristic feature of a number of important diseases. Together, the physical barrier, mucociliary escalator, and local innate antimicrobial responses compose what is sometimes called the immune barrier. The main structures that maintain the physical barrier are the tight junctions and adherens junctions that make strong contacts between epithelial cells and the desmosomes/hemidesmosomes that anchor columnar cells to basal cells and basal cells to the basement membrane (32). Tight junctions contain ZO-1, occludin, and claudins as well as junctional adhesion molecule 1 (JAM-1) proteins. These transmembrane proteins adhere to each other by homotypic binding in the space between adjoining cells, and some have cytoplasmic domains attached to the actin cytoskeleton within the cell. Adherens junctions contain the transmembrane proteins E-cadherin and nectin and intracellular proteins α- and β-catenin, which maintain actin binding. Desmosomal attachments consist of heterotypic binding of desmoglein to desmocollin, which binds inside the cell to desmoplakin. Loss of barrier can result from genetic defects or reduced expression of barrier structural proteins resulting from injury, infection, or inflammation. For instance, prolonged loss of barrier in the skin can result from mutations in the tight junction protein claudin-1, leading to loss of barrier, increased susceptibility to sensitization, and atopic dermatitis (33). Defects in claudin expression or function are also important in a variety of gastrointestinal diseases (34). More relevant to airways inflammation and CRS, studies by Holgate and others have convincingly shown a role for defective lung epithelial barrier in asthma pathogenesis (35, 36).
An EMT occurs in mucosal and cutaneous barriers during inflammation and repair, and chronic EMT is observed in CRS, asthma, and cancer. During EMT, differentiated epithelial cells lose their typical morphology, polarity, and junctional attachments to one another, proliferate, and become spindle-shaped mesenchymal cells with migratory function. These mesenchymal cells, sometimes referred to as myofibroblasts, often traverse the lamina reticularis and synthesize collagens and other matrix proteins, expanding the size of the basement membrane. Figure 2 illustrates changes in the expression of molecules that are biomarkers of the EMT process in CRS (35, 37, 38). For example, epithelial cells undergoing EMT lose expression of tight junction and adherens junction proteins, including ZO-1, claudins, occludin, and E-cadherin; the myofibroblasts that they become express matrix proteins, including desmin; fibronectin; collagens I, III, IV; tenascin; laminin; and elastin. These mesenchymal cells also express vimentin, MMP2 and MMP9, αSMA, and transcription factors associated with their differentiation, including Snail, Slug, and Twist. EMT can be induced by TGF-β, members of the EGF family, VEGF and FGF. Prominent signaling pathways inducing EMT include tyrosine kinase receptor pathways, the Notch pathway, SMAD pathways, and Wnt pathways (39). Other pathways initiating EMT include STAT3 as well as gp130-Src-YAP (40). The hormone-processing enzyme proconvertase 1 (PC1/3), usually only expressed in neuroendocrine tissue, is elevated in polyps and is expressed in basal epithelial cells in CRS but not in healthy control tissue. Overexpression of PC1/3 in an epithelial cell line induced profound EMT (41). One of the matricellular proteins expressed by epithelial cells in type 2 inflammation, periostin, has also been shown to be a potent activator of EMT by a TGF-β–dependent pathway, illustrating that diverse stimuli, including cytokines and adhesion substrates, can initiate the process (42). Myofibroblasts produce fibronectin, tenascin, and periostin and form a provisional matrix important in temporary protection from a breach in the epithelial barrier.
Figure 2.
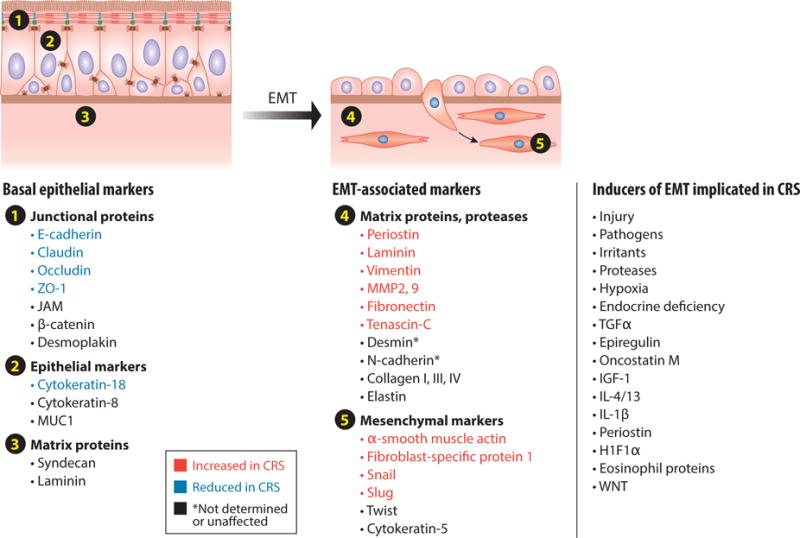
Tabulation of some of the important biomarkers of differentiated healthy epithelium (left) and epithelium undergoing repair-associated epithelial-to-mesenchymal transition (EMT) (right). Markers shown in blue on the left are reduced in chronic rhinosinusitis (CRS), and markers shown in red on the right are increased in CRS. The literature strongly supports the existence of an ongoing, chronic EMT response in CRS. Shown on the far right of the figure are some known EMT inducers that have been implicated in CRS. Abbreviations: H1F1α, hypoxia inducible factor-1-alpha; IGF-1, insulin-like growth factor-1; IL, interleukin; JAM, junctional adhesion molecule; MMP, matrix metalloproteinases; MUC1, mucin 1; TGF-α, transforming growth factor α; ZO-1; zonula occludens.
Several studies have shown a physical or functional defect in barrier in patients with CRS (43, 44). Soyka et al. (45) reported reduced barrier function in tissues from CRS patients. Many studies have confirmed the loss of epithelial markers and gain of mesenchymal markers in CRS (reviewed in 46). Markers shown in Figure 2 in blue are epithelial markers that are lost in CRS, and those shown in red are mesenchymal markers that are gained in CRS. Figure 2 also lists known inducers of EMT, including growth factors, cytokines, proteases, and other stimuli. Jang et al. (47) reported alterations in expression of both ZO-1 and E-cadherin, Shahana et al. (48) described epithelial damage in an EM study and reported shortening of desmosomes in polyp tissue from asthmatic patients, Rogers et al. (49) reported reductions in claudin-1 and occludin in polyps, and Meng et al. (25) found that mature polyps have reduced ZO-1, occludin, and E-cadherin. Epithelial loss was highest in early stage, middle turbinate polyps compared with mature ethmoid polyps, and αSMA was lower in mature polyps (25). Interestingly, more EMT was found in the stalk of early stage polyps than in the body of the early stage polyp or in the body of mature polyps, suggesting that the stalk is an important site for the EMT process, which would agree with the higher deposition of collagen in the stalk (25). The same pattern was observed with staining for vimentin and the TGF-β signaling molecule pSMAD, i.e., it was in the stalk rather than the body of the early stage polyps (25). Hupin and colleagues (50) reported decreased E-cadherin in CRS, along with decreased cytokeratins and vimentin; the vimentin expression correlated with basement membrane thickness and eosinophils in the tissue. They observed increased thickening in basement membrane in both CRSwNP and CRSsNP.
The underlying causes of the EMT process in CRS are not known and could be related to many of the inducers shown in Figure 2. Respiratory allergens containing proteases can disrupt the barrier and initiate EMT (51–53). Protease 1 from Dermatophagoides pteronyssinus disrupts tight junctions and degrades ZO-1 and occludin (53). Decreased levels of serine protease inhibitor Kazal-type 5 can contribute to loss of barrier in skin and have been found in the nasal epithelium in CRS (54, 55). In a mouse asthma model, extended exposure to house dust mite antigen (HDM) caused thickening of smooth muscle along with loss of E-cadherin and occludin and gain in vimentin, αSMA, and procollagen 1 associated with the induction of Snail1, a transcription factor that drives many of these EMT changes (51). Similar changes in HDM-induced allergic rhinitis are observed in patients (52). Stimulation of human bronchial epithelial cells in vitro with HDM increased vimentin and fibronectin and slightly decreased E-cadherin due to EGF signaling (56). Shin et al. (57) demonstrated that hypoxia induces many EMT changes, and hypoxia inducible factor 1 (HIF1α) levels correlated with loss of E-cadherin and gain of αSMA in polyp tissues in vivo. Hypoxia also induces MUC5AC via a HIF-1α–dependent mechanism. Smoking is another factor that worsens CRS in patients and is now known to induce EMT based on studies in patients with chronic obstructive pulmonary disease (58). The type 2 cytokines IL-13 and IL-4, found at high levels within eosinophilic polyps, may act as inducers of chronic EMT in CRS (45). These type 2 cytokines decrease electrical resistance, JAM-A, and E-cadherin of differentiated epithelial cultures (45). As discussed in another section below, treatment of CRSwNP patients with anti-IL-5 to reduce eosinophilia shrinks nasal polyps (59). An important article by Flood-Page et al. (60), who studied anti-IL-5 in asthmatic patients, showed a marked reduction in several EMT markers, including tenascin, lumican, and procollagen III in the reticular basement membrane. This finding suggests that eosinophils and their products are important in barrier dysfunction, damage, and induction of EMT in the airways. An overview of hypothetical dysregulation of the injury–repair cycle in CRS, and its impact on epithelial barrier function, is shown in Figure 3.
Figure 3.
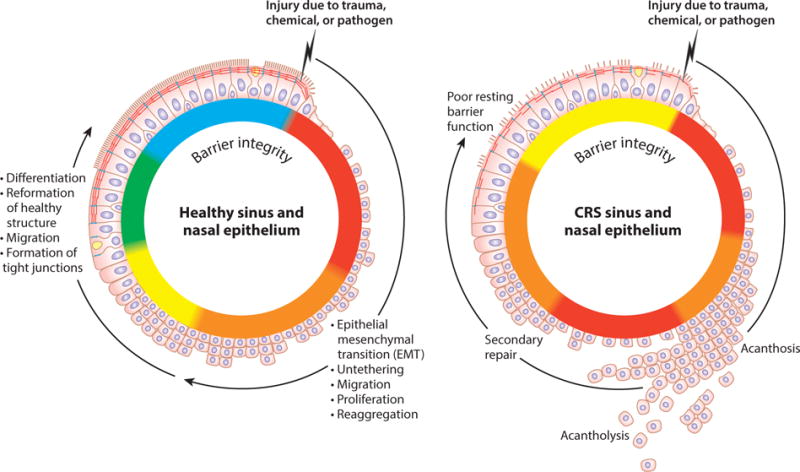
Epithelial dysfunction in chronic rhinosinusitis (CRS). A normal injury–repair cycle is shown on the left, illustrating loss of differentiation and polarity of epithelial cells associated with migration, proliferation, and reformation of junctional structures with neighboring epithelial cells. Epithelial permeability is indicated by the color on the basolateral side of the mucosae, with red indicating significant leak and green indicating a tight barrier. Changes in the injury–repair cycle hypothetically associated with CRS are shown on the right.
Epithelial Dysfunction in CRS: Mucociliary Dysfunction
Proper mucociliary function is essential in nasal physiology and immunity (61). Ciliary motion through the sol phase of the mucus blanket propels mucus and the trapped microbes and particulates out of the sinuses and nasal airways. Mucociliary function in CRS can be tested using the saccharine test, which measures mucociliary movement of saccharine from the sinuses into the mouth where it is tasted. The majority of early studies found that mucociliary function is impaired and that the degree of impairment of clearance is correlated with the severity of CRS (reviewed in 62). Most studies have found that ciliary beat frequency (CBF) is in the normal range in CRS patients. Not surprisingly, studies of the viscoelasticity of mucus yielded variable results, likely depending on the proportions of gel and sol phase, the purulence of the mucus, the hydration of the mucus, etc. Because CRSsNP is often characterized by frequent infections/colonization of the sinonasal cavity with bacteria, it is important to note that several bacteria commonly associated with CRS, including Pseudomonas aeruginosa, Haemophilus influenzae, and Streptococcus pneumoniae, produce toxins that are ciliostatic or ciliotoxic (63). Many studies have reported loss or inactivity of cilia in CRS tissue due to ciliary injury. Defects in mucociliary function promote bacterial growth and formation of biofilms, creating a vicious cycle. Chen et al. (62) found normal resting CBF in CRS explants with a marked defect in the increase of CBF in response to stimulation with ATP, possibly due to desensitization of purinergic receptors or a decrease in signaling molecules such as Ca2+, adenyl cyclase, or protein kinase A. Alterations of CBF-activating factors, including cytokines like tumor necrosis factor (TNF) and IL-1β and autacoids like bradykinin or substance P, and changes in pH and temperature may be relevant (63). Cilia length in tissue sections and cytospin preparations was nearly double in CRS patients compared with normal tissues. The cilia also showed signs of damage and disorganized structure, and their number per cell was increased and therefore overly dense (64). Forkhead box protein J1 (Foxj1), a transcription factor involved in elongation of cilia and cilia density on cells, and centriolar coiled-coil protein of 110kDa (CP110) and TAp73 (a member of the p53 tumor suppressor family), factors involved in initiation and execution of assembly of cilia, were overexpressed in CRS, and levels correlated with cilia length. Thus, both ciliary injury and an abnormal upregulation of ciliogenesis leading to impairment of ciliary architecture and function have been found in CRS (64). The increased cilia length, asymmetry, and disorganization as well as overexpression of CP110, Foxj1, and TAp73 were stable in epithelial stem cells expanded in vitro and matured to air-liquid interface monolayers, suggesting either a genetic or epigenetic intrinsic defect (64).
Epithelial Dysfunction in CRS: Antimicrobial Responses
More than 100 molecules are involved in pathogen recognition and killing, most of which are expressed by the epithelium in the upper airways and sinuses (see Table 2). Not surprisingly, many of these molecules are highly induced in CRS. Although not all have been evaluated, a few have been reported to be dysfunctional or reduced at the mucosal surface in CRS, as indicated in red within Table 2 and discussed here. The reader is also referred to reviews discussing the host–microbe interactions in CRS (55, 65–67).
Table 2.
Pathogen recognition structures that mediate innate immunity. Those shown in red text have at least one member reported to be reduced in CRS
| Type of structure | Protein family | Number of members in the family |
|---|---|---|
| Recognition receptors | Toll-like receptors (TLRs) | 10 |
| Nod-like receptors (NLRs) | 22 | |
| TAS2R | 25 | |
| RLRs | 4 | |
| CD14 | NA | |
| Cytosolic DNA sensors | 12 | |
| Antimicrobial peptides | S100 proteins | 18 |
| α-Defensins | 7 | |
| β-Defensins | 39 | |
| Cathelicidin (hCAP-18)/LL-37 | NA | |
| Collectins | 9 | |
| Pentraxins | 8 | |
| Endotoxin-binding molecules | LBP-BPI-PLUNC | 12 |
| AOAH | NA | |
| Enzymatic mediators | Lysozyme | NA |
| Lactoferrin | NA | |
| Peroxidase/thiocyanate | NA | |
| Complement | NA |
Abbreviations: AOAH, acyloxyacyl hydrolase; BPI, bactericidal/permeability-increasing; CD14, cluster of differentiation 14; LBP, lipopolysaccharide binding protein; LL-37, member of the cathelicidin antimicrobial peptide family; PLUNC, palate lung nasal epithelial clone; NA, not applicable.
Toll-like receptors
Toll-like receptors (TLRs) play fundamental roles in innate immunity, are expressed by epithelial cells and immune cells in the nose and sinuses, and activate expression of inflammatory mediators and host defense molecules (68–70). Many studies have evaluated TLR expression in CRS but without consensus, as some studies have reported increased levels of one or more TLRs (71–73) and some have reported reduced levels or no differences (68, 72, 74, 75). Most studies used PCR and evaluated surgical tissue, which has variable expression, and studies using relatively pure primary epithelial cells collected by scraping or brushing are needed. Two studies of single-nucleotide polymorphism (SNPs) in TLR genes have been done, one of which reported a SNP associated with increased risk of CRS in a Korean population (76).
Bitter taste receptors
Bitter taste receptors such as T2R38 detect quorum sensing molecules produced by gram-negative bacteria in biofilms such as acyl-homoserine lactone compounds (77). These ligands activate T2R38-induced Ca2+ responses in primary nasal epithelial cells and in specialized solitary chemosensory cells (SCC), leading to increased mucociliary function, production of the antimicrobial gas NO, and rapid release of antimicrobial peptides such as β-defensin 2 (77–79). Polymorphisms in T2R38 underlie the lack of ability to taste various bitter compounds in food, and the nonfunctional allele of T2R38 (AVI) is a risk allele for disease severity in CRS and CF (80–82). It is not known whether allelic variants in the 25 other T2Rs influence CRS risk or whether T2R38 is unique (83). The sweet receptor that detects glucose, T1R 2/3, can suppress the protective response of T2R38, and elevated nasal glucose–mediated T2R suppression may explain why patients with diabetes mellitus are 3–4 times more likely to be infected with P. aeruginosa and gram-negative rods (79, 84). Insulin is an essential hormone for growth of epithelial cells, however, and subclinical defects in insulin signaling may be associated with barrier defects and increased risk for CRS as suggested by Berdnikovs and colleagues (H. Abdala-Valencia, L.F. Loffredo, I. Martinez Reyes, K.A. Erickson, M. Browning et al., unpublished manuscript).
Endotoxin-binding molecules
The LBP-BPI-PLUNC family of molecules is important in innate immune responses to lipopolysaccharide (LPS or endotoxin) derived from gram-negative bacteria: LBP binds LPS and collaborates with CD14 for the activation of TLR4, and most BPI-PLUNC proteins bind to bacteria and LPS and mediate opsonization, direct killing, neutralization of LPS, disruption of biofilms, or other antimicrobial actions (85). A proteomic analysis of polyp tissue and normal tissue found reduced levels of PLUNC, confirming results of immunohistochemical analysis (86). Two forms of PLUNC were identified in lavage fluid and were decreased after exposure to irritants such as tobacco smoke (87). Seshadri et al. (88) analyzed PLUNC family members and found decreased levels of LPLUNC2 and SPLUNC1 in nasal polyp tissue attributable to reduced submucosal glands in polyps. Interestingly, two other family members, BPI and LPLUNC4, were reduced in uncinate tissue, suggesting a global defect in expression in CRS (88). Although Wei et al. (89) reported that reduced PLUNC was seen only in eosinophilic polyps and not in noneosinophilic polyps in China, Tsou et al. (90) showed decreased PLUNC associated with colonization of bacteria, especially S. aureus and P. aeruginosa. Knockout of the SPLUNC gene in mice led to significant enhancement of eosinophilic inflammation in an OVA challenge model, and SPLUNC protein blocked LPS induction of eotaxin-2 by alveolar macrophages (91). Another endotoxin-binding protein that is of interest in CRS is acyloxyacyl hydrolase (AOAH), an LPS-catabolizing enzyme shown to be genetically associated with asthma and CRS (92, 93). Considering the importance of gram-negative organisms in CRS, further advancement of our understanding of the role of reductions in BPI-PLUNC members and AOAH is in order.
Antimicrobial peptides
Several S100 proteins are antimicrobial and also promote inflammation. Levels of mRNA for S100A7 and S100A8/S100A9 were found to be lower in nasal epithelial cells from CRS patients, and protein was reduced in nasal lavage fluids (54, 94, 95). S100A7 is also lower in lavage fluid from patients with allergic rhinitis (96). The heterodimer of S100A8/S100A9, known as calprotectin, is also expressed by neutrophils and consequently calprotectin levels are highly elevated within sinonasal tissue and polyps due to neutrophil recruitment (95, 97). Levels of S100A8/S100A9 decrease in nasal lavage fluid of CRS patients with age (98).
Enzymatic mediators
Lysozyme and lactoferrin have been reported to be either depleted or increased in various sinonasal tissues in CRS (99, 100). However, both are clearly reduced in the epithelium of nasal polyp tissue, owing to large reductions in submucosal glands in polyps (88). In addition, methacholine challenge studies have shown decreased lysozyme release in CRS patients (101). Studies of CRS with biofilm formation have also reported diminished lactoferrin, and a proteomics study reported diminished lysozyme in lavage (102, 103). Due to the loss of glands, it can be predicted that all glandular host defense molecules will be reduced in nasal polyp tissue (88). A study of complement components and complement activators showed that 19% of 87 CRS patients had a complement deficiency (mostly mannose binding lectin deficiency), whereas only 10% of 150 control patients did (104). It is notable that activation of complement occurs in CRS and is probably important in pathogenesis (see below).
INNATE AND ADAPTIVE INFLAMMATORY RESPONSES UNDERLYING THE PATHOGENESIS OF CRS
Several recent reviews have focused on inflammation, cytokines, T and B lymphocytes, and innate lymphoid cells (ILCs) in CRS pathogenesis (13, 30, 66, 105). The goal of this section is therefore to primarily provide an overview of major findings and conclusions in the literature.
When rapid local innate immune responses are insufficient to prevent growth or entry of pathogens, adaptive immune T cells and B cells are activated and recruited to reinforce the response. Figure 4 describes elements of innate and adaptive immunity that are activated in CRS and speculates as to how they interact temporally in the formation of nasal polyps. The damaged epithelium responding to injury from pathogens, proteases, and irritants is shown. Although the injured epithelium makes at least three T helper cell type 2 (Th2)-promoting cytokines, IL-25, IL-33, and TSLP, among these three TSLP is most highly induced in CRS tissue (106–108). Along with the mast cell products PGD2 and leukotrienes, TSLP can activate type 2 innate lymphoid cells (ILC2s) (109). In the presence of TSLP, ILC2s and mast cells express substantial quantities of type 2 cytokines, especially IL-5 and IL-13, and relatively little IL-4 (110). This pattern is the same as seen in nasal polyps, suggesting a role of ILC2s in the type 2 cytokine production in CRSwNP (111). Unlike T cells, ILC2s do not require antigen for activation, and they rapidly mobilize these cytokines. Elevated ILC2 levels have been reported in polyp tissue (112–114). ILC2s also promote epithelial repair via the production of amphiregulin (115). Acanthosis seen in CRS may be attributable to local production of amphiregulin, oncostatin M, epiregulin, and other inducers of EMT (116, 117).
Figure 4.
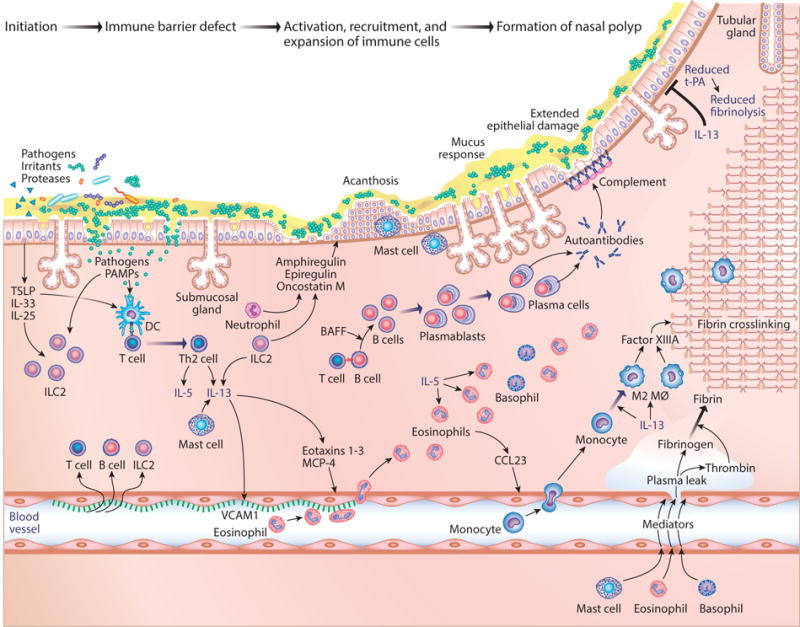
Hypothetical timeline and interactions of events in the formation of nasal polyps. (Left) The damaged epithelium responds to injury from pathogens, proteases, and irritants. Abbreviations: BAFF, B cell–activating factor; CCL23, chemokine (C-C motif) ligand 23; DC, dendritic cell; IL, interleukin; ILC2s, type 2 innate lymphoid cells; MCP-4, chemokine (C-C motif) ligand 13, also known as CCL13; PAMP, pathogen-associated molecular patterns; Th2, T helper cell type 2; tPA, tissue plasminogen activator; TSLP, thymic stromal lymphopoietin; VCAM1, vascular cell adhesion molecule 1.
Epithelial injury may allow pathogen-associated molecular patterns and antigens from mucosal pathogens to cross the barrier and initiate recruitment and activation of antigen-specific T and B cells. Moving left to right in the model, type 2 cytokines from ILC2 are important in recruitment of adaptive immune cells by activation of endothelial adhesion molecules, induction of production of chemokines, and provision of cytokines that shape T cell differentiation. The nature of the ILC that is activated shapes the T cell response; in this case, ILC2s promote Th2 responses (110). Eosinophilic polyps contain type 2 cytokines, whereas noneosinophilic polyps generally do not (Table 2). In parallel with an expansion of T cells in CRSwNP, there is an impressive expansion of B cells, plasmablasts, and plasma cells (118, 119). Local activation, proliferation, class switch recombination, and production of antibodies take place within polyps (118–120). Little is known about the antigen specificity of T or B cells in CRS. IgE antibodies to aeroallergens and staphylococcal enterotoxin have been reported (121). Tan et al. (122) showed that nasal polyps contain autoantibodies that are likely responsible for activation of complement in the tissue. Among the autoantibodies described are antibodies directed against basement membrane, and the complement activation that occurs is localized at the basement membrane region (123, 124). The presence of autoantibodies in polyps may occur as a result of significant levels of the B cell–activating factor (BAFF), a cytokine that drives autoimmunity in mouse models of overexpression (125).
A comprehensive summary of cytokines present in CRS has been compiled by Hulse et al. (30). Whether type 2 cytokines are derived from ILC2, Th2 cells, or mast cells and basophils, the importance of IL-5 and IL-13 in formation of polyps is convincing. Bachert, Gevaert, and colleagues have shown that antibodies against either IL-5 or the IL-4/IL-13 receptor alpha chain shrink polyps (59, 126). As shown in Figure 4, IL-5 and IL-13 drive many features of the inflammation and the effector cell recruitment that is characteristic of the disease. IL-13 activates endothelial expression of VCAM-1 and other adhesion molecules that recruit lymphocytes, eosinophils, and basophils, and IL-13 also induces the expression by the epithelium and other cells of the C-C motif chemokine receptor 3 (CCR3)-specific chemokines that recruit mast cells, eosinophils, and basophils, including C-C motif chemokine ligand 13 (MCP-4, also known as CCL13) and eotaxins-1, 2, and 3, into the tissue. Once eosinophils are recruited to the tissue, IL-5 promotes their survival and activation (127). Activation of mast cells triggers the release of serous and mucous secretions by goblet cells and glands. IL-13 is also important in differentiation of recruited monocytes to become M2 or alternatively activated macrophages. Kato and colleagues (128) showed that chemokine (C-C motif) ligand 23 (CCL23) is highly elevated in polyp tissue and is produced by eosinophils. This chemokine binds CCR1 and recruits monocytes that differentiate to M2 macrophages. IL-13 suppresses expression of tPA and induces Factor XIIIA to promote accumulation of fibrin mesh using thrombin and fibrinogen derived from plasma leak induced by mediators from mast cells, basophils, and eosinophils (28, 29; see also Figure 4). The figure applies to eosinophilic polyps, the type 2–driven primary form of polypoid CRS found in North America, the United Kingdom, and Europe and a secondary form found in Asia (Table 2). Several reports have shown the presence of elevated IL-17 in some polyps from patients in China and Japan, correlating with eosinophilia (129–131). The M2 macrophage response, fibrin deposition, and IgE and autoantibody responses could possibly be restricted to type 2 inflammation in nasal polyps (132).
Whether there are defects in regulatory T cells in CRS is controversial. Although reports have shown reduced levels of the regulatory T cell (Treg) transcription factor Forkhead box P3 (FOXP3) and reduced FOXP3+ staining in polyp tissues, no change in Tregs was detected by flow cytometry in another study (133–135). Treg activation in peripheral blood cells is reported to be reduced in CRS patients compared with control patients, but most studies do not find reduced levels of the suppressive cytokines IL-10 or TGF-β (136–139). Studies in noneosinophilic patients in Asian countries have shown increased presence of IL-17–producing FOXP3+ Tregs (134, 140, 141). On balance, CRS cannot be attributed to defects in Treg responses at present.
RACE, EOSINOPHILIA, AND CRS: WHAT ARE THE ROLES OF GENES AND THE ENVIRONMENT?
To date, no large-scale studies have been performed to identify genetic linkages and polymorphisms that drive CRS pathogenesis. Because the average age of onset of CRS is over 40, building pedigrees and evaluating inheritance has been challenging. Recent studies of the Utah population database (UPDB) analyzed families of more than 25,000 patients with CRS and determined that first-degree relatives (parents, children, and siblings) of CRSwNP patients had a 4.1-fold increased risk of CRS, whereas first-degree relatives of patients with CRSsNP had a 2.4-fold risk (142). Second-degree relatives of CRSwNP and CRSsNP patients had 3.3-fold and 1.4-fold risks, respectively. A similar UPDB study of eosinophilic esophagitis (EE) found a 3.4-fold risk of EE in first-degree relatives (143). Although large studies of primary care CRS patients are lacking, a few family studies and a large number of relatively small studies of surgical patients support a genetic basis of inheritance of CRS (reviewed in 82, 144). Most studies used candidate gene approaches, were underpowered, and still await replication. Findings of genetic associations/linkages worthy of special mention include the identification of the cystic fibrosis gene, CFTR, as a risk gene. CFTR was shown to be present in 7% of CRS patients, versus 2% of the general population, and was confirmed in a linkage study performed in a Hutterite population (145, 146). As discussed above, Cohen and colleagues (77, 81) have identified polymorphisms in the bitter taste receptor T2R38 that exert a strong genetic effect on susceptibility to CRS. Two genome-wide association studies using pooled samples identified associations with matrix proteins laminin α2, laminin β, as well as AOAH (147). Other interesting genes identified were thyroid hormone receptor interactor (TRIPI2) and neuron navigator 3 (NAV3). The association with AOAH was confirmed using a similar approach in a study of Chinese CRS patients and control patients (93). Several studies have shown associations with HLA genes, innate immune genes, and cytokines (82, 144).
The presence of eosinophils in CRS tissues was recognized shortly after the work of Ehrlich identifying dyes that bind to eosinophil granules, and eosinophil granule proteins have been linked with pathology (148, 149). Correlations between disease severity and eosinophils measured in blood or nasal tissues are well established. The Japanese Epidemiological Survey of Refractory Eosinophilic Chronic Rhinosinusitis (JESREC), a 15-center retrospective study in Japan, evaluated eosinophils in the blood and tissue of 1,716 patients who had sinus surgery and concluded that blood eosinophils, ethmoid disease, asthma, and aspirin intolerance were all associated with recurrence of disease and need for further surgical intervention (150). The importance of eosinophils in asthma has been known for decades, and assessment of sputum eosinophil content has significant utility in treatment choice for asthmatic patients (150). Eosinophils have both inflammatory and tissue remodeling effects, and their derived mediators can damage epithelial cells, stimulate EMT, activate or suppress sensory nerves, modulate the activity of stem cells and plasma cells, and alter mechanical responses of airways (151–153). Tan and colleagues (154) took biopsies from the superior meatus, where the olfactory zone resides, and found a strong correlation between eosinophils in that area and anosmia. Indeed, olfactory dysfunction has been shown to be greater in patients with eosinophilic disease and/or asthma (154, 155). Antibodies directed against IL-5 or the IL-4/IL-13 receptor alpha chain reduce eosinophils and shrink polyps in CRSwNP, supporting a role of eosinophils in CRSwNP pathogenesis (59, 126). These drugs also restore olfactory function, supporting the hypothesis that eosinophils mediate anosmia (see below).
Nasal polyp tissues removed from CRS patients during surgery in Asian countries, including China, Korea, and Japan, have fewer eosinophils than tissues from CRS patients collected in Europe and the United States (reviewed in 156). However, Kim et al. (157) studied two similar groups of 230 patients in Korea, one in 1993–1994 and another in 2010–2011, and found that the eosinophil counts rose from 6.8 per high-powered field to 19.3 during this time. In general, less than 50% of polyps in Asian countries are eosinophilic, whereas approximately 75% of polyps in Europe and the United States are eosinophilic (149, 156, 158, 159). In Japan, the JESREC study found a prevalence of eosinophilic polyps similar to that in Western countries, and several other studies suggest that the degree of eosinophilia in polyps in Japan, China, Malaysia, Korea, etc. is increasing (150). Adoption of a Western lifestyle is one possible factor driving this change, suggesting that there are strong environmental influences on eosinophilia (150).
Eosinophilia correlates strongly with type 2 immune and inflammatory responses, and patients with eosinophilic polyp disease, whether in the East or the West, generally have higher levels of IgE, higher prevalence of atopy, and greater prevalence and severity of asthma (156). Periostin is a provisional matrix protein produced during EMT (see above) that is highly induced by IL-4 and IL-13 and therefore has been used increasingly as a marker of Th2 inflammation. In a study of serum levels in 190 asthma patients in Japan, those with high levels of periostin had more aspirin intolerance, nasal polyposis, blood eosinophilia, and reduced lung function, although they did not have increased atopy (160). A study by Cao et al. (132) in China found that polyp tissue from patients with eosinophilic inflammation had higher IgE against aeroallergens and increased mast cell activity, independent of atopy. Local generation of specific IgE in nonatopic subjects, or so-called entopy, was proposed to explain the dissociation of skin test reactivity from presence of local antigen–reactive mast cells in the eosinophilic polyp tissues.
Many findings suggest that genetics plays a strong role in eosinophilia. Eosinophilic polyps compose only 25% of polyp cases in the Asian-American population compared with two-thirds to three-quarters of polyps from African-American, Caucasian, and Hispanic populations, suggesting that genetic or cultural/dietary factors lead to reduced eosinophilia (161). Like CRS, EE has a strong familial association, with nearly 10% of parents of EE patients having a history of esophageal disease. Rothenberg and colleagues (161, 162) reported 26 multiplex families with EE and demonstrated a large familial risk of EE in both dizygotic twins and non-twin siblings. Based on the prevalence of EE in the general population (5/10,000), the risk for siblings was increased approximately 80-fold, compared with a 2-fold increased risk for atopy in the same group, leading them to conclude that genetics has a large role in eosinophilia. Environmental factors that might influence eosinophilia include birth season, breastfeeding, and antibiotic use during infancy; these are considered risk factors that may alter the microbiome (162, 163).
Although asthma studies show very strong influences of race on outcomes, few studies have examined the influence of race on CRS outside of Asian and Caucasian populations (164, 165). A study of three major national surveys in the United States (National Health Interview Survey, National Ambulatory Medical Care Survey, and National Hospital Ambulatory Medical Care Survey) found that 90% of published studies contain little or no information on race. Minority endoscopic sinus surgery patients made up only 18% of the total in the United States, despite an expected percentage of 35%, leading to the conclusion that insurance status influenced the result (166). A retrospective cohort study of CRS cases treated in a large urban center in Chicago found that African-American CRS patients were at higher risk for anosmia, and those requiring functional endoscopic sinus surgery had a higher prevalence of nasal polyposis, AERD, and olfactory loss and had worse CT scores of their sinuses. Those African-American CRS patients with polypoid disease had a history of more hospitalizations for asthma. These findings suggest that African-Americans, who are known to have higher prevalence and worse severity of asthma, also have worse CRSwNP than patients from other races (167). Pinto et al. (168) reported that African-American CRS patients had lower vitamin D levels than control African-American patients, and Bush et al. (169) reported that surgical outcomes were not as durable in African-American patients with polypoid CRS and asthma compared with Caucasian patients. Based on the foregoing, as well as information on asthma, it seems likely that CRSwNP is worse in African-Americans, Puerto Ricans, and Hispanics than in Caucasians and Asians, but clearly more studies are needed (164, 165).
ROLE OF THE NASAL MICROBIOME IN THE DEVELOPMENT OF CRS
Sinusitis is the single-highest indication responsible for antibiotic use, and several hypotheses implicate microorganisms in CRS. The fungal hypothesis proposes that allergic sensitization to fungi by T cells induces response to ambient fungi and drives type 2 disease (170). Although antifungal agents failed to manage CRS in most studies, it is not clear if these drugs would be effective if ambient fungi drive disease (171). Bachert and colleagues (172) have advocated a strong role for colonization with S. aureus in driving CRS, especially severe disease with polyps. Despite associations of disease severity and chronicity with S. aureus colonization, with IgE against S. aureus antigens and enterotoxins and with elevation of Vβ-bearing T cells potentially expanded by enterotoxins supporting this hypothesis, colonization by S. aureus is not observed in all or even most CRS patients. Another hypothesis implicates bacteria that form biofilms in driving CRS disease (173, 174). Yet another hypothesis, which has good support, is that respiratory viruses such as rhinovirus and influenza are responsible for seasonal exacerbations of CRS, which occur largely in the late fall and winter (175). Thus far, microbiome studies have not identified a single causative agent associated with disease; the main findings demonstrate increased abundance and decreased diversity of the nasal microbiome in CRS (176, 177). It is clear that bacteria, fungi, and viruses all can play a role in subsets of patients with CRS and that there is not a single microbial organism that can be implicated.
UNMET NEED FOR EFFECTIVE THERAPEUTIC APPROACHES TO MANAGEMENT OF CRS
In patients who fail medical management, surgery can open the ostia that drain and clear the sinuses and can unblock the nasal passages by removing nasal polyp tissue (3). It can often dramatically improve symptoms, as measured by the Sino-Nasal Outcome Test 22 questionnaire, restore olfaction, and reduce the need for other medications (3). The disease often relapses, and approximately 20% of surgeries performed are revision surgeries. Over a 12-year period, 80% of patients have relapse and 37% undergo revision surgery (178). It is not uncommon for patients with severe nasal polyposis to have had more than 10 prior surgeries, indicating an unmet need for new effective treatments for CRS.
New classes of drugs have been developed for asthma and other diseases that have potential utility in CRS. Bachert, Geveart, and collaborators (59, 126, 179) have performed double-blind clinical trials on omalizumab, mepolizumab, and dupilumab, all of which were able to shrink nasal polyps and improve symptoms. These drugs, which, respectively, block the action of IgE on mast cells and basophils, IL-5 on eosinophils, and IL-4/IL-13 on a host of cells, have great promise for managing intractable type 2 CRSwNP. They also establish the importance of mast cells (and/or basophils) and eosinophils in the pathogenesis of CRSwNP. Other possible strategies to target inflammatory cells in CRS include the following: an anti-Siglec-8 antibody that inhibits mast cell function and induces eosinophil apoptosis; a GATA3 DNAzyme that targets ILC2 and Th2 cells; inhibitors and cytotoxic antibodies against DP2, otherwise known as CRTH2, a PGD receptor important in type 2 skewing; reslizumab and benralizumab, two other antibodies targeting eosinophils; lebrikizumab and other IL-13–targeting antibodies; and many others (180–186). Recently, inhibitors of Jak-STAT signaling, so-called Jakinibs, have had great success in managing inflammatory diseases such as rheumatoid arthritis and are worth evaluation (186). Topical application of inflammatory drugs is challenging due to the difficult access to sinus mucosae, but new advances in topical drug applications, via both implantable devices and new formulations that maintain prolonged elevations of drug levels in the sinonasal cavities, are promising.
CLOSING REMARKS
Important questions remain to challenge CRS researchers: Are nerves involved in producing inflammation or pathogenesis, or are the physical manifestations, such as anosmia, cough, reflexive secretory responses, and headache, all just consequences of the illness? What are the factors that drive polyp formation in noneosinophilic disease? Are submucosal glands passive victims of the process that leads to formation of tubular gland structures, or does dysfunction of glands drive the formation of polyp tissue? What are the activators of ILC2, and is eosinophilic CRS an innate inflammatory disease? What are the antigens that activate T and B cells, and is CRS an adaptive inflammatory disease? Is the barrier defect seen in CRS due to genetic defects in the epithelium? Is the injury–repair cycle associated with EMT, acanthosis, and acantholysis a consequence of inflammation, a cause of inflammation, or both? Will ongoing clinical trials soon inform us of the relative importance of effector cells, including eosinophils, mast cells, basophils, neutrophils, and ILC2s, in driving the inflammatory manifestations? CRS is a heterogeneous illness with high prevalence, cost, and impact on quality of life; it is essential that active investigations answer these and more questions and pursue new strategies for treatment.
Acknowledgments
The author would like to thank Ms. Jacqi Shaffer for outstanding medical illustrations, Dr. Bruce Tan for providing CT and endoscopy images, Dr. Sergejs Berdnikovs for sharing his unpublished data, and Mrs. April Stone for help with the bibliography.
Glossary
- anosmia
loss of the sense of smell
- polyp
an abnormal growth of tissue in the sinuses and nose
- CRSwNP
chronic rhinosinusitis with nasal polyps
- CRSsNP
chronic rhinosinusitis without nasal polyps
- microbiome
the totality of microbial organisms, or their genes, residing in a given niche (e.g., the nose and sinuses)
- allergic fungal sinusitis (AFS)
a form of sinus disease associated with fungal colonization of the nose and sinuses
- antrochoanal polyps
single polyps arising from the maxillary sinus and entering the nasal cavity
- aspirin-exacerbated respiratory disease (AERD)
a syndrome characterized by comorbid asthma, nasal polyps, and sensitivity to aspirin-like drugs
- COX1
a cyclooxygenase enzyme responsible for prostaglandin synthesis
- nasopharynx
the upper pharynx that connects with the nasal cavity
- Eustachian tubes
tubes involved in pressure equalization that link the inner ear with the nasopharynx
- oropharynx
middle pharynx behind the mouth
- M cells
specialized cells in the epithelium of lymphoid follicles that transport solute proteins for immune stimulation
- ostiomeatal complex
channel that links the frontal, ethmoid, and maxillary sinuses to the middle meatus
- nasal vestibule
anteriormost region of the nasal cavity
- Lund-Mackay score
a scoring system to evaluate inflammation in the sinuses using computed tomography
- ethmoid sinuses
sinuses located within the ethmoid bone that are a frequent site of nasal polyp growth
- turbinate tissues
conch-shaped bony structures in the nasal passage
- maxillary sinuses
large sinuses found below the eyes on the cheekbone
- transforming growth factor β (TGF-β)
a member of a family of molecules involved in inflammation, remodeling, and fibrosis
- pseudocyst
a lesion that is cystic in appearance but lacks epithelial and endothelial cells
- lumican
a proteoglycan involved in extracellular matrix formation
- matrix metalloproteinases (MMPs)
proteases involved in degradation and remodeling of the extracellular matrix
- fibronectin
an extracellular matrix protein that mediates cellular binding via integrins
- laminin
an extracellular matrix component that comprises the basal lamina
- elastin
an elastic protein component of connective tissue
- occludin
a tight junction protein
- vimentin
an intracellular filamentous gene expressed by mesenchymal cells
- epithelial-to-mesenchymal transition (EMT)
a process during which differentiated epithelial cells acquire mesenchymal features, such as motility and proliferation, often during the process of epithelial repair
- M2 macrophages
a phenotypic state of macrophages stimulated by IL-4 or IL-13 involved in repair
- acanthosis
hyperplasia of the epithelium
- acantholysis
loss of desmosome intercellular connections causing disruption and shedding of the epithelium
- vascular endothelial cell growth factor (VEGF)
a cytokine that induces vascular leak and growth (angiogenesis)
- mucociliary escalator
the combined action of cilia moving a mucous blanket through the sinonasal spaces and into the nasopharynx
- tight junctions
junctional structures that tightly bind epithelial cells and reduce intercellular permeability
- adherens junctions
intercellular junctions that attach epithelial cells to each other and to the cytoskeleton
- desmosomes
structures involved in intracellular adhesion in epithelia
- hemidesmosomes
structures involved in adhesion of epithelia to the matrix
- zonula occludens (ZO-1)
a protein component of tight junctions
- claudins
proteins that constitute tight junctions in differentiated epithelial cells
- E-cadherin
a structural component of adherens junctions involved in intercellular adhesion in epithelia
- nectin
adhesion molecules involved in adherens junctions
- desmoglein
a structural component of desmosomes that mediates epithelial cell adherence
- desmocollin
a structural component of desmosomes that mediates epithelial cell adherence
- desmoplakin
a structural component of desmosomes that mediates epithelial cell adherence
- lamina reticularis
one of the laminar layers that form the epithelial basement membrane
- tenascin
extracellular matrix glycoprotein involved in provisional matrix
- periostin
multifunctional provisional adhesion molecule that supports integrin binding
- Mucin 5AC (MUC5AC)
a mucus proteoglycan produced by goblet cells and submucosal glands
- Junctional adhesion molecule A (JAM-A)
a protein involved in intercellular epithelial tight junction adhesion
- Toll-like receptors (TLRs)
a family of receptors involved in recognition of pathogen-associated molecular patterns
- bitter taste receptors
a family of seven transmembrane spanning receptors that mediate the sensation of bitter taste and innate immune responses
- defensin
an antimicrobial peptide family produced by inflammatory and structural cells
- LBP
an LPS-binding protein that participates in activation of the LPS receptor
- TLR4
a receptor that mediates inflammatory responses to endotoxin
- PLUNC
palate, lung, and nasal clone family of innate host defense molecules
- BPI
bactericidal/permeability increasing proteins, a family of innate immune receptors
- eotaxins-1, 2, and 3
chemokines that bind to CCR3 and recruit eosinophils and other inflammatory cells
- acyloxyacyl hydrolase (AOAH)
an enzyme that catabolizes and neutralizes bacterial endotoxin (LPS)
- S100 proteins
a family of proteins that exert regulatory, inflammatory, and/or host defense functions
- interleukin 25 (IL-25)
a cytokine that promotes ILC2 cell responses
- interleukin 33 (IL-33)
a cytokine that promotes ILC2 cell responses
- thymic stromal lymphopoietin (TSLP)
a cytokine that induces ILC2 responses
- type 2 innate lymphoid cells (ILC2s)
immune cells that produce type 2 cytokines without direct antigen stimulation
- amphiregulin
a member of the EGF family that stimulates epithelial proliferation and repair
- oncostatin M
member of the IL-6 cytokine family that reduces epithelial barrier
- epiregulin
a member of the EGF family that stimulates epithelial proliferation and repair
- B cell–activating factor (BAFF)
an activating factor of the TNF family, a B cell proliferation and activation factor
- vascular cell adhesion molecule 1 (VCAM-1)
an endothelial adhesion molecule that mediates recruitment of VLA4 positive inflammatory cells
- MCP-4
also known as CCL13, a chemokine that binds to CCR3 and recruits eosinophils and other inflammatory cells
- Treg
regulatory T cell
Footnotes
DISCLOSURE STATEMENT
R.P.S. has consulted over the last two years on topics relevant to CRS with Astra Zeneca, Genentech, GSK, Intersect ENT, Merck, Regeneron, UCB and Sanofi. R.P.S. is a founder and advisor of Allakos, which has an ongoing clinical trial in CRS.
Errata
An online log of corrections to Annual Review of Pathology: Mechanisms of Disease articles may be found at http://www.annualreviews.org/errata/pathol
LITERATURE CITED
- 1.Meltzer EO, Hamilos DL, Hadley JA, Lanza DC, Marple BF, et al. Rhinosinusitis: establishing definitions for clinical research and patient care. J Allergy Clin Immunol. 2004;114(6 Suppl):155–212. doi: 10.1016/j.jaci.2004.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fokkens WJ, Lund VJ, Mullol J, Bachert C, Alobid I, et al. EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology. 2012;50(1):1–12. doi: 10.4193/Rhino12.000. [DOI] [PubMed] [Google Scholar]
- 3.Senior BA, Kennedy DW, Tanabodee J, Kroger H, Hassab M, et al. Long-term results of functional endoscopic sinus surgery. Laryngoscope. 1998;108(2):151–57. doi: 10.1097/00005537-199802000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Laidlaw TM, Boyce JA. Aspirin-exacerbated respiratory disease—new prime suspects. N Engl J Med. 2016;374(5):484–88. doi: 10.1056/NEJMcibr1514013. [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharyya N, Orlandi RR, Grebner J, Martinson M. Cost burden of chronic rhinosinusitis: a claims-based study. Otolaryngol Head Neck Surg. 2011;144(3):440–45. doi: 10.1177/0194599810391852. [DOI] [PubMed] [Google Scholar]
- 6.Tan BK, Kern RC, Schleimer RP, Schwartz BS. Chronic rhinosinusitis: the unrecognized epidemic. Am J Respir Crit Care Med. 2013;188(11):1275–77. doi: 10.1164/rccm.201308-1500ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevens WW, Peters AT, Suh L, Norton JE, Kern RC, et al. A retrospective, cross-sectional study reveals that women with CRSwNP have more severe disease than men. Immun Inflamm Dis. 2015;3(1):14–22. doi: 10.1002/iid3.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahin-Yilmaz A, Naclerio RM. Anatomy and physiology of the upper airway. Proc Am Thorac Soc. 2011;8(1):31–39. doi: 10.1513/pats.201007-050RN. [DOI] [PubMed] [Google Scholar]
- 9.Kato A, Hulse KE, Tan BK, Schleimer RP. B-lymphocyte lineage cells and the respiratory system. J Allergy Clin Immunol. 2013;131(4):933–57. doi: 10.1016/j.jaci.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avila PC, Schleimer RP. Airway epithelium. In: Kay AB, Kaplan AP, Bousquet J, Holt PG, editors. Allergy and Allergic Diseases. 2nd Vol. 1. Hoboken, NJ: Wiley-Blackwell; 2008. pp. 366–97. [Google Scholar]
- 11.Hopkins C, Browne JP, Slack R, Lund V, Brown P. The Lund-Mackay staging system for chronic rhinosinusitis: How is it used and what does it predict? Otolaryngol Head Neck Surg. 2007;137(4):555–61. doi: 10.1016/j.otohns.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Tos M, Larsen PL, Larsen K, Caye-Thomasen P. Pathogenesis and pathophysiology of nasal polyps. In: Önerci TM, Ferguson BJ, editors. Nasal Polyposis. Berlin: Springer-Verlag; 2010. pp. 53–63. [Google Scholar]
- 13.Akdis CA, Bachert C, Cingi C, Dykewicz MS, Hellings PW, et al. Endotypes and phenotypes of chronic rhinosinusitis: a PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol. 2013;131(6):1479–90. doi: 10.1016/j.jaci.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomassen P, Vandeplas G, Van Zele T, Cardell LO, Arebro J, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. 2016;137(5):1449–56. doi: 10.1016/j.jaci.2015.12.1324. [DOI] [PubMed] [Google Scholar]
- 15.Kountakis SE, Arango P, Bradley D, Wade ZK, Borish L. Molecular and cellular staging for the severity of chronic rhinosinusitis. Laryngoscope. 2004;114(11):1895–905. doi: 10.1097/01.mlg.0000147917.43615.c0. [DOI] [PubMed] [Google Scholar]
- 16.Van Bruaene N, Bachert C. Tissue remodeling in chronic rhinosinusitis. Curr Opin Allergy Clin Immunol. 2011;11(1):8–11. doi: 10.1097/ACI.0b013e32834233ef. [DOI] [PubMed] [Google Scholar]
- 17.Shi LL, Xiong P, Zhang L, Cao PP, Liao B, et al. Features of airway remodeling in different types of Chinese chronic rhinosinusitis are associated with inflammation patterns. Allergy. 2013;68(1):101–9. doi: 10.1111/all.12064. [DOI] [PubMed] [Google Scholar]
- 18.Kim DK, Jin HR, Eun KM, Mutusamy S, Cho SH, et al. Non-eosinophilic nasal polyps shows increased epithelial proliferation and localized disease pattern in the early stage. PLOS ONE. 2015;10(10):e0139945. doi: 10.1371/journal.pone.0139945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watelet JB, Bachert C, Claeys C, Van Cauwenberge P. Matrix metalloproteinases MMP-7, MMP-9 and their tissue inhibitor TIMP-1: expression in chronic sinusitis versus nasal polyposis. Allergy. 2004;59(1):54–60. doi: 10.1046/j.1398-9995.2003.00364.x. [DOI] [PubMed] [Google Scholar]
- 20.Nielsen LF, Moe D, Kirkeby S, Garbarsch C. Sirius red and acid fuchsin staining mechanisms. Biotech Histochem. 1998;73(2):71–77. doi: 10.3109/10520299809140509. [DOI] [PubMed] [Google Scholar]
- 21.Eyibilen A, Cayli S, Aladag I, Koc S, Gurbuzler L, et al. Distribution of matrix metalloproteinases MMP-1, MMP-2, MMP-8 and tissue inhibitor of matrix metalloproteinases-2 in nasal polyposis and chronic rhinosinusitis. Histol Histopathol. 2011;26(5):615–21. doi: 10.14670/HH-26.615. [DOI] [PubMed] [Google Scholar]
- 22.Kostamo K, Tervahartiala T, Sorsa T, Richardson M, Toskala E. Metalloproteinase function in chronic rhinosinusitis with nasal polyposis. Laryngoscope. 2007;117(4):638–43. doi: 10.1097/MLG.0b013e318030aca6. [DOI] [PubMed] [Google Scholar]
- 23.Lechapt-Zalcman E, Coste A, d’Ortho MP, Frisdal E, Harf A, et al. Increased expression of matrix metalloproteinase-9 in nasal polyps. J Pathol. 2001;193(2):233–41. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH771>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 24.Wang LF, Chien CY, Tai CF, Kuo WR, Hsi E, et al. Matrix metalloproteinase-9 gene polymorphisms in nasal polyposis. BMC Med Genet. 2010;11:85. doi: 10.1186/1471-2350-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng J, Zhou P, Liu Y, Liu F, Yi X, et al. The development of nasal polyp disease involves early nasal mucosal inflammation and remodelling. PLOS ONE. 2013;8(12):e82373. doi: 10.1371/journal.pone.0082373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carroll WW, O’Connell BP, Schlosser RJ, Gudis DA, Karnezis TT, et al. Fibroblast levels are increased in chronic rhinosinusitis with nasal polyps and are associated with worse subjective disease severity. Int Forum Allergy Rhinol. 2016;6(2):162–68. doi: 10.1002/alr.21636. [DOI] [PubMed] [Google Scholar]
- 27.Yoshikawa M, Wada K, Yoshimura T, Asaka D, Okada N, et al. Increased CXCL10 expression in nasal fibroblasts from patients with refractory chronic rhinosinusitis and asthma. Allergol Int. 2013;62(4):495–502. doi: 10.2332/allergolint.13-OA-0572. [DOI] [PubMed] [Google Scholar]
- 28.Takabayashi T, Kato A, Peters AT, Hulse KE, Suh LA, et al. Excessive fibrin deposition in nasal polyps caused by fibrinolytic impairment through reduction of tissue plasminogen activator expression. Am J Respir Crit Care Med. 2013;187(1):49–57. doi: 10.1164/rccm.201207-1292OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takabayashi T, Kato A, Peters AT, Hulse KE, Suh LA, et al. Increased expression of factor XIII-A in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2013;132(3):584–592. doi: 10.1016/j.jaci.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hulse KE, Stevens WW, Tan BK, Schleimer RP. Pathogenesis of nasal polyposis. Clin Exp Allergy. 2015;45(2):328–46. doi: 10.1111/cea.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee H, Kim SR, Oh Y, Cho SH, Schleimer RP, et al. Targeting insulin-like growth factor-I and insulin-like growth factor-binding protein-3 signaling pathways. A novel therapeutic approach for asthma. Am J Respir Cell Mol Biol. 2014;50(4):667–77. doi: 10.1165/rcmb.2013-0397TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Investig Dermatol. 2007;127(11):2525–32. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- 33.De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127(3):773–86. doi: 10.1016/j.jaci.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barmeyer C, Schulzke JD, Fromm M. Claudin-related intestinal diseases. Semin Cell Dev Biol. 2015;42:30–38. doi: 10.1016/j.semcdb.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 35.Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol. 2012;12(1):53–59. doi: 10.1097/ACI.0b013e32834ec6eb. [DOI] [PubMed] [Google Scholar]
- 36.Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev. 2011;242(1):205–19. doi: 10.1111/j.1600-065X.2011.01030.x. [DOI] [PubMed] [Google Scholar]
- 37.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Investig. 2009;119(6):1420–28. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pain M, Bermudez O, Lacoste P, Royer PJ, Botturi K, et al. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur Respir Rev. 2014;23(131):118–30. doi: 10.1183/09059180.00004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 40.Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, et al. A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature. 2015;519(7541):57–62. doi: 10.1038/nature14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SN, Lee DH, Sohn MH, Yoon JH. Overexpressed proprotein convertase 1/3 induces an epithelial-mesenchymal transition in airway epithelium. Eur Respir J. 2013;42(5):1379–90. doi: 10.1183/09031936.00100412. [DOI] [PubMed] [Google Scholar]
- 42.Sidhu SS, Yuan S, Innes AL, Kerr S, Woodruff PG, et al. Roles of epithelial cell-derived periostin in TGF-β activation, collagen production, and collagen gel elasticity in asthma. PNAS. 2010;107(32):14170–75. doi: 10.1073/pnas.1009426107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernstein JM, Gorfien J, Noble B, Yankaskas JR. Nasal polyposis: immunohistochemistry and bioelectrical findings (a hypothesis for the development of nasal polyps) J Allergy Clin Immunol. 1997;99(2):165–75. doi: 10.1016/s0091-6749(97)70091-5. [DOI] [PubMed] [Google Scholar]
- 44.Dejima K, Randell SH, Stutts MJ, Senior BA, Boucher RC. Potential role of abnormal ion transport in the pathogenesis of chronic sinusitis. Arch Otolaryngol Head Neck Surg. 2006;132(12):1352–62. doi: 10.1001/archotol.132.12.1352. [DOI] [PubMed] [Google Scholar]
- 45.Soyka MB, Wawrzyniak P, Eiwegger T, Holzmann D, Treis A, et al. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-γ and IL-4. J Allergy Clin Immunol. 2012;130(5):1087–96. doi: 10.1016/j.jaci.2012.05.052. [DOI] [PubMed] [Google Scholar]
- 46.Zhang N, Van Crombruggen K, Gevaert E, Bachert C. Barrier function of the nasal mucosa in health and type-2 biased airway diseases. Allergy. 2016;71(3):295–307. doi: 10.1111/all.12809. [DOI] [PubMed] [Google Scholar]
- 47.Jang YJ, Kim HG, Koo TW, Chung PS. Localization of ZO-1 and E-cadherin in the nasal polyp epithelium. Eur Arch Otorhinolaryngol. 2002;259(9):465–69. doi: 10.1007/s00405-002-0500-z. [DOI] [PubMed] [Google Scholar]
- 48.Shahana S, Jaunmuktane Z, Asplund MS, Roomans GM. Ultrastructural investigation of epithelial damage in asthmatic and non-asthmatic nasal polyps. Respir Med. 2006;100(11):2018–28. doi: 10.1016/j.rmed.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 49.Rogers GA, Den Beste K, Parkos CA, Nusrat A, Delgaudio JM, et al. Epithelial tight junction alterations in nasal polyposis. Int Forum Allergy Rhinol. 2011;1(1):50–54. doi: 10.1002/alr.20014. [DOI] [PubMed] [Google Scholar]
- 50.Hupin C, Gohy S, Bouzin C, Lecocq M, Polette M, et al. Features of mesenchymal transition in the airway epithelium from chronic rhinosinusitis. Allergy. 2014;69(11):1540–49. doi: 10.1111/all.12503. [DOI] [PubMed] [Google Scholar]
- 51.Johnson JR, Roos A, Berg T, Nord M, Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLOS ONE. 2011;6(1):e16175. doi: 10.1371/journal.pone.0016175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steelant B, Farre R, Wawrzyniak P, Belmans J, Dekimpe E, et al. Impaired barrier function in patients with house dust mite–induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1 expression. J Allergy Clin Immunol. 2016;137(4):1043–53. doi: 10.1016/j.jaci.2015.10.050. [DOI] [PubMed] [Google Scholar]
- 53.Wan H, Winton HL, Soeller C, Tovey ER, Gruenert DC, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Investig. 1999;104(1):123–33. doi: 10.1172/JCI5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richer SL, Truong-Tran AQ, Conley DB, Carter R, Vermylen D, et al. Epithelial genes in chronic rhinosinusitis with and without nasal polyps. Am J Rhinol. 2008;22(3):228–34. doi: 10.2500/ajr.2008.22.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tieu DD, Kern RC, Schleimer RP. Alterations in epithelial barrier function and host defense responses in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124(1):37–42. doi: 10.1016/j.jaci.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heijink IH, Postma DS, Noordhoek JA, Broekema M, Kapus A. House dust mite–promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol. 2010;42(1):69–79. doi: 10.1165/rcmb.2008-0449OC. [DOI] [PubMed] [Google Scholar]
- 57.Shin HW, Cho K, Kim DW, Han DH, Khalmuratova R, et al. Hypoxia-inducible factor 1 mediates nasal polypogenesis by inducing epithelial-to-mesenchymal transition. Am J Respir Crit Care Med. 2012;185(9):944–54. doi: 10.1164/rccm.201109-1706OC. [DOI] [PubMed] [Google Scholar]
- 58.Shaykhiev R, Crystal RG. Early events in the pathogenesis of chronic obstructive pulmonary disease. Smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann Am Thorac Soc. 2014;11(Suppl 5):S252–8. doi: 10.1513/AnnalsATS.201402-049AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gevaert P, Van Bruaene N, Cattaert T, Van Steen K, Van Zele T, et al. Mepolizumab, a humanized anti-IL-5 mAb, as a treatment option for severe nasal polyposis. J Allergy Clin Immunol. 2011;128(5):989–95. doi: 10.1016/j.jaci.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 60.Flood-Page P, Menzies-Gow A, Phipps S, Ying S, Wangoo A, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Investig. 2003;112(7):1029–36. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee RCN. Physiology of the nose and paranasal sinuses. In: deSouza C, et al., editors. Otolaryngology Head and Neck Surgery. 2nd New Delhi: Jaypee Publishers; 2016. In press. [Google Scholar]
- 62.Chen B, Shaari J, Claire SE, Palmer JN, Chiu AG, et al. Altered sinonasal ciliary dynamics in chronic rhinosinusitis. Am J Rhinol. 2006;20(3):325–29. doi: 10.2500/ajr.2006.20.2870. [DOI] [PubMed] [Google Scholar]
- 63.Gudis D, Zhao KQ, Cohen NA. Acquired cilia dysfunction in chronic rhinosinusitis. Am J Rhinol Allergy. 2012;26(1):1–6. doi: 10.2500/ajra.2012.26.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li YY, Li CW, Chao SS, Yu FG, Yu XM, et al. Impairment of cilia architecture and ciliogenesis in hyperplastic nasal epithelium from nasal polyps. J Allergy Clin Immunol. 2014;134(6):1282–92. doi: 10.1016/j.jaci.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 65.Hamilos DL. Host-microbial interactions in patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2014;133(3):640–53. doi: 10.1016/j.jaci.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stevens WW, Lee RJ, Schleimer RP, Cohen NA. Chronic rhinosinusitis pathogenesis. J Allergy Clin Immunol. 2015;136(6):1442–53. doi: 10.1016/j.jaci.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Crombruggen K, Jacob F, Zhang N, Bachert C. Damage-associated molecular patterns and their receptors in upper airway pathologies. Cell Mol Life Sci. 2013;70(22):4307–21. doi: 10.1007/s00018-013-1356-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Claeys S, de Belder T, Holtappels G, Gevaert P, Verhasselt B, et al. Human β-defensins and toll-like receptors in the upper airway. Allergy. 2003;58(8):748–53. doi: 10.1034/j.1398-9995.2003.00180.x. [DOI] [PubMed] [Google Scholar]
- 69.Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31(3):358–64. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- 70.Vandermeer J, Sha Q, Lane AP, Schleimer RP. Innate immunity of the sinonasal cavity: expression of messenger RNA for complement cascade components and toll-like receptors. Arch Otolaryngol Head Neck Surg. 2004;130(12):1374–80. doi: 10.1001/archotol.130.12.1374. [DOI] [PubMed] [Google Scholar]
- 71.Hirschberg A, Kiss M, Kadocsa E, Polyanka H, Szabo K, et al. Different activations of toll-like receptors and antimicrobial peptides in chronic rhinosinusitis with or without nasal polyposis. Eur Arch Otorhinolaryngol. 2015;273:1779–88. doi: 10.1007/s00405-015-3816-1. [DOI] [PubMed] [Google Scholar]
- 72.Lane AP, Truong-Tran QA, Myers A, Bickel C, Schleimer RP. Serum amyloid A, properdin, complement 3, and toll-like receptors are expressed locally in human sinonasal tissue. Am J Rhinol. 2006;20(1):117–23. [PMC free article] [PubMed] [Google Scholar]
- 73.Sun Y, Zhou B, Wang C, Huang Q, Zhang Q, et al. Biofilm formation and Toll-like receptor 2, Toll-like receptor 4, and NF-κB expression in sinus tissues of patients with chronic rhinosinusitis. Am J Rhinol Allergy. 2012;26(2):104–9. doi: 10.2500/ajra.2012.26.3718. [DOI] [PubMed] [Google Scholar]
- 74.Ramanathan M, Jr, Lee WK, Dubin MG, Lin S, Spannhake EW, et al. Sinonasal epithelial cell expression of Toll-like receptor 9 is decreased in chronic rhinosinusitis with polyps. Am J Rhinol. 2007;21(1):110–16. doi: 10.2500/ajr.2007.21.2997. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Q, Wang CS, Han DM, Sy C, Huang Q, et al. Differential expression of Toll-like receptor pathway genes in chronic rhinosinusitis with or without nasal polyps. Acta Otolaryngol. 2013;133(2):165–73. doi: 10.3109/00016489.2012.717713. [DOI] [PubMed] [Google Scholar]
- 76.Park CS, Cho JH, Park YJ. Toll-like receptor 2 gene polymorphisms in a Korean population: association with chronic rhinosinusitis. Otolaryngol Head Neck Surg. 2011;144(1):96–100. doi: 10.1177/0194599810390881. [DOI] [PubMed] [Google Scholar]
- 77.Lee RJ, Xiong G, Kofonow JM, Chen B, Lysenko A, et al. T2R38 taste receptor polymorphisms underlie susceptibility to upper respiratory infection. J Clin Investig. 2012;122(11):4145–59. doi: 10.1172/JCI64240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee RJ, Cohen NA. Role of the bitter taste receptor T2R38 in upper respiratory infection and chronic rhinosinusitis. Curr Opin Allergy Clin Immunol. 2015;15(1):14–20. doi: 10.1097/ACI.0000000000000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee RJ, Kofonow JM, Rosen PL, Siebert AP, Chen B, et al. Bitter and sweet taste receptors regulate human upper respiratory innate immunity. J Clin Investig. 2014;124(3):1393–405. doi: 10.1172/JCI72094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adappa ND, Workman AD, Hadjiliadis D, Dorgan DJ, Frame D, et al. T2R38 genotype is correlated with sinonasal quality of life in homozygous ΔF508 cystic fibrosis patients. Int Forum Allergy Rhinol. 2016;6(4):356–61. doi: 10.1002/alr.21675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adappa ND, Zhang Z, Palmer JN, Kennedy DW, Doghramji L, et al. The bitter taste receptor T2R38 is an independent risk factor for chronic rhinosinusitis requiring sinus surgery. Int Forum Allergy Rhinol. 2014;4(1):3–7. doi: 10.1002/alr.21253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mfuna-Endam L, Zhang Y, Desrosiers MY. Genetics of rhinosinusitis. Curr Allergy Asthma Rep. 2011;11(3):236–46. doi: 10.1007/s11882-011-0189-4. [DOI] [PubMed] [Google Scholar]
- 83.Wiener A, Shudler M, Levit A, Niv MY. BitterDB: a database of bitter compounds. Nucleic Acids Res. 2012;40:D413–19. doi: 10.1093/nar/gkr755. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Z, Adappa ND, Lautenbach E, Chiu AG, Doghramji L, et al. The effect of diabetes mellitus on chronic rhinosinusitis and sinus surgery outcome. Int Forum Allergy Rhinol. 2014;4(4):315–20. doi: 10.1002/alr.21269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bingle CD, Craven CJ. PLUNC: a novel family of candidate host defence proteins expressed in the upper airways and nasopharynx. Hum Mol Genet. 2002;11(8):937–43. doi: 10.1093/hmg/11.8.937. [DOI] [PubMed] [Google Scholar]
- 86.Min-man W, Hong S, Zhi-qiang X, Xue-ping F, Chang-qi L, et al. Differential proteomic analysis of nasal polyps, chronic sinusitis, and normal nasal mucosa tissues. Otolaryngol Head Neck Surg. 2009;141(3):364–68. doi: 10.1016/j.otohns.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 87.Ghafouri B, Kihlstrom E, Stahlbom B, Tagesson C, Lindahl M. PLUNC (palate, lung and nasal epithelial clone) proteins in human nasal lavage fluid. Biochem Soc Trans. 2003;31(Pt 4):810–14. doi: 10.1042/bst0310810. [DOI] [PubMed] [Google Scholar]
- 88.Seshadri S, Lin DC, Rosati M, Carter RG, Norton JE, et al. Reduced expression of antimicrobial PLUNC proteins in nasal polyp tissues of patients with chronic rhinosinusitis. Allergy. 2012;67(7):920–28. doi: 10.1111/j.1398-9995.2012.02848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wei Y, Xia W, Ye X, Fan Y, Shi J, et al. The antimicrobial protein short palate, lung, and nasal epithelium clone 1 (SPLUNC1) is differentially modulated in eosinophilic and noneosinophilic chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2014;133(2):420–28. doi: 10.1016/j.jaci.2013.09.052. [DOI] [PubMed] [Google Scholar]
- 90.Tsou YA, Peng MT, Wu YF, Lai CH, Lin CD, et al. Decreased PLUNC expression in nasal polyps is associated with multibacterial colonization in chronic rhinosinusitis patients. Eur Arch Otorhinolaryngol. 2014;271(2):299–304. doi: 10.1007/s00405-013-2535-8. [DOI] [PubMed] [Google Scholar]
- 91.Thaikoottathil JV, Martin RJ, Di PY, Minor M, Case S, et al. SPLUNC1 deficiency enhances airway eosinophilic inflammation in mice. Am J Respir Cell Mol Biol. 2012;47(2):253–60. doi: 10.1165/rcmb.2012-0064OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barnes KC, Grant A, Gao P, Baltadjieva D, Berg T, et al. Polymorphisms in the novel gene acyloxyacyl hydroxylase (AOAH) are associated with asthma and associated phenotypes. J Allergy Clin Immunol. 2006;118(1):70–77. doi: 10.1016/j.jaci.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 93.Zhang Y, Endam LM, Filali-Mouhim A, Zhao L, Desrosiers M, et al. Polymorphisms in RYBP and AOAH genes are associated with chronic rhinosinusitis in a Chinese population: a replication study. PLOS ONE. 2012;7(6):e39247. doi: 10.1371/journal.pone.0039247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bryborn M, Adner M, Cardell LO. Psoriasin, one of several new proteins identified in nasal lavage fluid from allergic and non-allergic individuals using 2-dimensional gel electrophoresis and mass spectrometry. Respir Res. 2005;6:118. doi: 10.1186/1465-9921-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tieu DD, Peters AT, Carter RG, Suh L, Conley DB, et al. Evidence for diminished levels of epithelial psoriasin and calprotectin in chronic rhinosinusitis. J Allergy Clin Immunol. 2010;125(3):667–75. doi: 10.1016/j.jaci.2009.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kvarnhammar AM, Rydberg C, Jarnkrants M, Eriksson M, Uddman R, et al. Diminished levels of nasal S100A7 (psoriasin) in seasonal allergic rhinitis: an effect mediated by Th2 cytokines. Respir Res. 2012;13:2. doi: 10.1186/1465-9921-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Van Crombruggen K, Vogl T, Perez-Novo C, Holtappels G, Bachert C. Differential release and deposition of S100A8/A9 proteins in inflamed upper airway tissue. Eur Respir J. 2016;47(1):264–74. doi: 10.1183/13993003.00159-2015. [DOI] [PubMed] [Google Scholar]
- 98.Cho SH, Hong SJ, Han B, Lee SH, Suh L, et al. Age-related differences in the pathogenesis of chronic rhinosinusitis. J Allergy Clin Immunol. 2012;129(3):858–60. doi: 10.1016/j.jaci.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dong D, Yulin Z, Yan X, Hongyan Z, Shitao Z, et al. Enhanced expressions of lysozyme, SLPI and glycoprotein 340 in biofilm-associated chronic rhinosinusitis. Eur Arch Otorhinolaryngol. 2014;271(6):1563–71. doi: 10.1007/s00405-013-2758-8. [DOI] [PubMed] [Google Scholar]
- 100.Ishida M, Matsunaga T, Uda H. An immunohistological study of nasal and paranasal mucosa of patients with relapsing chronic sinusitis. Rhinology. 1984;22(2):115–18. [PubMed] [Google Scholar]
- 101.Jeney EV, Raphael GD, Meredith SD, Kaliner MA. Abnormal nasal glandular secretion in recurrent sinusitis. J Allergy Clin Immunol. 1990;86(1):10–18. doi: 10.1016/s0091-6749(05)80117-4. [DOI] [PubMed] [Google Scholar]
- 102.Psaltis AJ, Wormald PJ, Ha KR, Tan LW. Reduced levels of lactoferrin in biofilm-associated chronic rhinosinusitis. Laryngoscope. 2008;118(5):895–901. doi: 10.1097/MLG.0b013e31816381d4. [DOI] [PubMed] [Google Scholar]
- 103.Tewfik MA, Latterich M, DiFalco MR, Samaha M. Proteomics of nasal mucus in chronic rhinosinusitis. Am J Rhinol. 2007;21(6):680–85. doi: 10.2500/ajr.2007.21.3103. [DOI] [PubMed] [Google Scholar]
- 104.Gaunsbaek MQ, Lange B, Kjeldsen AD, Svane-Knudsen V, Skjoedt K, et al. Complement defects in patients with chronic rhinosinusitis. PLOS ONE. 2012;7(11):e47383. doi: 10.1371/journal.pone.0047383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hamilos DL. Drivers of chronic rhinosinusitis: inflammation versus infection. J Allergy Clin Immunol. 2015;136(6):1454–59. doi: 10.1016/j.jaci.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 106.Liu T, Li TL, Zhao F, Xie C, Liu AM, et al. Role of thymic stromal lymphopoietin in the pathogenesis of nasal polyposis. Am J Med Sci. 2011;341(1):40–47. doi: 10.1097/MAJ.0b013e3181f20489. [DOI] [PubMed] [Google Scholar]
- 107.Miljkovic D, Bassiouni A, Cooksley C, Ou J, Hauben E, et al. Association between group 2 innate lymphoid cells enrichment, nasal polyps and allergy in chronic rhinosinusitis. Allergy. 2014;69(9):1154–61. doi: 10.1111/all.12440. [DOI] [PubMed] [Google Scholar]
- 108.Nagarkar DR, Poposki JA, Tan BK, Comeau MR, Peters AT, et al. Thymic stromal lymphopoietin activity is increased in nasal polyps of patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2013;132(3):593–600. doi: 10.1016/j.jaci.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lund S, Walford HH, Doherty TA. Type 2 innate lymphoid cells in allergic disease. Curr Immunol Rev. 2013;9(4):214–21. doi: 10.2174/1573395510666140304235916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McKenzie AN, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity. 2014;41(3):366–74. doi: 10.1016/j.immuni.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 111.Stevens WW, Ocampo CJ, Berdnikovs S, Sakashita M, Mahdavinia M, et al. Cytokines in chronic rhinosinusitis. Role in eosinophilia and aspirin-exacerbated respiratory disease. Am J Respir Crit Care Med. 2015;192(6):682–94. doi: 10.1164/rccm.201412-2278OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ho J, Bailey M, Zaunders J, Mrad N, Sacks R, et al. Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin Exp Allergy. 2015;45(2):394–403. doi: 10.1111/cea.12462. [DOI] [PubMed] [Google Scholar]
- 113.Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 114.Shaw JL, Fakhri S, Citardi MJ, Porter PC, Corry DB, et al. IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am J Respir Crit Care Med. 2013;188(4):432–39. doi: 10.1164/rccm.201212-2227OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zaiss DM, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. 2015;42(2):216–26. doi: 10.1016/j.immuni.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pothoven KL, Norton JE, Hulse KE, Suh LA, Carter RG, et al. Oncostatin M promotes mucosal epithelial barrier dysfunction, and its expression is increased in patients with eosinophilic mucosal disease. J Allergy Clin Immunol. 2015;136(3):737–46. doi: 10.1016/j.jaci.2015.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Homma T, Kato A, Masafumi S, Norton J, Suh L, et al. Involvement of EGFR ligands and MMP-1 in pathogenesis of CRS. Am J Respir Crit Care Med. 2015;191:A1377. (Abstr.) [Google Scholar]
- 118.Gevaert P, Holtappels G, Johansson SG, Cuvelier C, Cauwenberge P, et al. Organization of secondary lymphoid tissue and local IgE formation to Staphylococcus aureus enterotoxins in nasal polyp tissue. Allergy. 2005;60(1):71–79. doi: 10.1111/j.1398-9995.2004.00621.x. [DOI] [PubMed] [Google Scholar]
- 119.Hulse KE, Norton JE, Suh L, Zhong Q, Mahdavinia M, et al. Chronic rhinosinusitis with nasal polyps is characterized by B-cell inflammation and EBV-induced protein 2 expression. J Allergy Clin Immunol. 2013;131(4):1075–83. doi: 10.1016/j.jaci.2013.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mechtcheriakova D, Sobanov Y, Holtappels G, Bajna E, Svoboda M, et al. Activation-induced cytidine deaminase (AID)-associated multigene signature to assess impact of AID in etiology of diseases with inflammatory component. PLOS ONE. 2011;6(10):e25611. doi: 10.1371/journal.pone.0025611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bachert C, Zhang N, Holtappels G, De Lobel L, van Cauwenberge P, et al. Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. J Allergy Clin Immunol. 2010;126(5):962–68. doi: 10.1016/j.jaci.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 122.Tan BK, Li QZ, Suh L, Kato A, Conley DB, et al. Evidence for intranasal antinuclear autoantibodies in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2011;128(6):1198–206. doi: 10.1016/j.jaci.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jeffe JS, Seshadri S, Hamill KJ, Huang JH, Carter R, et al. A role for anti-BP180 autoantibodies in chronic rhinosinusitis. Laryngoscope. 2013;123(9):2104–11. doi: 10.1002/lary.24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Van Roey GA, Vanison C, Huang H, Kern R, Chandra R, et al. Complement activation in nasal tissue of patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2015;135(2):AB237. doi: 10.1016/j.jaci.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kato A, Peters A, Suh L, Carter R, Harris KE, et al. Evidence of a role for B cell-activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2008;121(6):1385–92. doi: 10.1016/j.jaci.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bachert C, Mannent L, Naclerio RM, Mullol J, Ferguson BJ, et al. Effect of subcutaneous dupilumab on nasal polyp burden in patients with chronic sinusitis and nasal polyposis: a randomized clinical trial. JAMA. 2016;315(5):469–79. doi: 10.1001/jama.2015.19330. [DOI] [PubMed] [Google Scholar]
- 127.Bochner BS, Schleimer RP. Mast cells, basophils, and eosinophils: distinct but overlapping pathways for recruitment. Immunol Rev. 2001;179:5–15. doi: 10.1034/j.1600-065x.2001.790101.x. [DOI] [PubMed] [Google Scholar]
- 128.Poposki JA, Uzzaman A, Nagarkar DR, Chustz RT, Peters AT, et al. Increased expression of the chemokine CCL23 in eosinophilic chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2011;128(1):73–81. doi: 10.1016/j.jaci.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cao PP, Li HB, Wang BF, Wang SB, You XJ, et al. Distinct immunopathologic characteristics of various types of chronic rhinosinusitis in adult Chinese. J Allergy Clin Immunol. 2009;124(3):478–84. doi: 10.1016/j.jaci.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 130.Saitoh T, Kusunoki T, Yao T, Kawano K, Kojima Y, et al. Role of interleukin-17A in the eosinophil accumulation and mucosal remodeling in chronic rhinosinusitis with nasal polyps associated with asthma. Int Arch Allergy Immunol. 2010;151(1):8–16. doi: 10.1159/000232566. [DOI] [PubMed] [Google Scholar]
- 131.Shen Y, Pan CK, Tang XY, Yang YC, Ke X, et al. Significance of interleukin-17A in patients with nasal polyposis. Asian Pac J Allergy Immunol. 2011;29(2):169–75. [PubMed] [Google Scholar]
- 132.Cao PP, Zhang YN, Liao B, Ma J, Wang BF, et al. Increased local IgE production induced by common aeroallergens and phenotypic alteration of mast cells in Chinese eosinophilic, but not non-eosinophilic, chronic rhinosinusitis with nasal polyps. Clin Exp Allergy. 2014;44(5):690–700. doi: 10.1111/cea.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ho J, Bailey M, Zaunders J, Mrad N, Sacks R, et al. Cellular comparison of sinus mucosa versus polyp tissue from a single sinus cavity in chronic rhinosinusitis. Int Forum Allergy Rhinol. 2015;5(1):14–27. doi: 10.1002/alr.21417. [DOI] [PubMed] [Google Scholar]
- 134.Jin J, Chang DY, Kim SH, Rha KS, Mo JH, et al. Role of hypoxia-inducible factor-1α expression in regulatory T cells on nasal polypogenesis. Laryngoscope. 2014;124(5):E151–59. doi: 10.1002/lary.24472. [DOI] [PubMed] [Google Scholar]
- 135.Van Bruaene N, Perez-Novo CA, Basinski TM, Van Zele T, Holtappels G, et al. T-cell regulation in chronic paranasal sinus disease. J Allergy Clin Immunol. 2008;121(6):1435–41. doi: 10.1016/j.jaci.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 136.Bachert C, Wagenmann M, Hauser U, Rudack C. IL-5 synthesis is upregulated in human nasal polyp tissue. J Allergy Clin Immunol. 1997;99(6):837–42. doi: 10.1016/s0091-6749(97)80019-x. [DOI] [PubMed] [Google Scholar]
- 137.Bolard F, Gosset P, Lamblin C, Bergoin C, Tonnel AB, et al. Cell and cytokine profiles in nasal secretions from patients with nasal polyposis: effects of topical steroids and surgical treatment. Allergy. 2001;56(4):333–38. doi: 10.1034/j.1398-9995.2001.00835.x. [DOI] [PubMed] [Google Scholar]
- 138.Kim YM, Munoz A, Hwang PH, Nadeau KC. Migration of regulatory T cells toward airway epithelial cells is impaired in chronic rhinosinusitis with nasal polyposis. Clin Immunol. 2010;137(1):111–21. doi: 10.1016/j.clim.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Riechelmann H, Deutschle T, Rozsasi A, Keck T, Polzehl D, et al. Nasal biomarker profiles in acute and chronic rhinosinusitis. Clin Exp Allergy. 2005;35(9):1186–91. doi: 10.1111/j.1365-2222.2005.02316.x. [DOI] [PubMed] [Google Scholar]
- 140.Lan F, Zhang N, Zhang J, Krysko O, Zhang Q, et al. Forkhead box protein 3 in human nasal polyp regulatory T cells is regulated by the protein suppressor of cytokine signaling 3. J Allergy Clin Immunol. 2013;132(6):1314–21. doi: 10.1016/j.jaci.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 141.Liu T, Song CH, Liu AM, Xie C, Zhao F, et al. Forkhead box P3+ T cells express interleukin-17 in nasal mucosa of patients with both allergic rhinitis and polyposis. Clin Exp Immunol. 2011;163(1):59–64. doi: 10.1111/j.1365-2249.2010.04278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Oakley GM, Curtin K, Orb Q, Schaefer C, Orlandi RR, et al. Familial risk of chronic rhinosinusitis with and without nasal polyposis: genetics or environment. Int Forum Allergy Rhinol. 2015;5(4):276–82. doi: 10.1002/alr.21469. [DOI] [PubMed] [Google Scholar]
- 143.Padia R, Curtin K, Peterson K, Orlandi RR, Alt J. Eosinophilic esophagitis strongly linked to chronic rhinosinusitis. Laryngoscope. 2016;126(6):1279–83. doi: 10.1002/lary.25798. [DOI] [PubMed] [Google Scholar]
- 144.Hsu J, Avila PC, Kern RC, Hayes MG, Schleimer RP, et al. Genetics of chronic rhinosinusitis: state of the field and directions forward. J Allergy Clin Immunol. 2013;131(4):977–93. doi: 10.1016/j.jaci.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pinto JM, Hayes MG, Schneider D, Naclerio RM, Ober C. A genomewide screen for chronic rhinosinusitis genes identifies a locus on chromosome 7q. Laryngoscope. 2008;118(11):2067–72. doi: 10.1097/MLG.0b013e3181805147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang X, Moylan B, Leopold DA, Kim J, Rubenstein RC, et al. Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. JAMA. 2000;284(14):1814–19. doi: 10.1001/jama.284.14.1814. [DOI] [PubMed] [Google Scholar]
- 147.Bosse Y, Bacot F, Montpetit A, Rung J, Qu HQ, et al. Identification of susceptibility genes for complex diseases using pooling-based genome-wide association scans. Hum Genet. 2009;125(3):305–18. doi: 10.1007/s00439-009-0626-9. [DOI] [PubMed] [Google Scholar]
- 148.Mygind N, Dahl R, Bachert C. Nasal polyposis, eosinophil dominated inflammation, and allergy. Thorax. 2000;55(Suppl 2):S79–83. doi: 10.1136/thorax.55.suppl_2.S79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schleimer R, Kato A, Kern RC. Eosinophils and chronic rhinosinusitis. In: Lee JJ, Rosenberg H, editors. Eosinophils in Health and Disease. London: Elsevier; 2012. pp. 508–519. [Google Scholar]
- 150.Tokunaga T, Sakashita M, Haruna T, Asaka D, Takeno S, et al. Novel scoring system and algorithm for classifying chronic rhinosinusitis: the JESREC Study. Allergy. 2015;70(8):995–1003. doi: 10.1111/all.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.George L, Brightling CE. Eosinophilic airway inflammation: role in asthma and chronic obstructive pulmonary disease. Ther Adv Chronic Dis. 2016;7(1):34–51. doi: 10.1177/2040622315609251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee JJ, Jacobsen EA, McGarry MP, Schleimer RP, Lee NA. Eosinophils in health and disease: the LIAR hypothesis. Clin Exp Allergy. 2010;40(4):563–75. doi: 10.1111/j.1365-2222.2010.03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Postma DS, Rabe KF. The asthma-COPD overlap syndrome. N Engl J Med. 2015;373(13):1241–49. doi: 10.1056/NEJMra1411863. [DOI] [PubMed] [Google Scholar]
- 154.Lavin J, Meen E, Lam K, Kato A, Huang H, et al. The impact and nature of inflammation in the olfactory cleft on olfaction in patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2013;131(2):AB59. [Google Scholar]
- 155.Yoshimura K, Kawata R, Haruna S, Moriyama H, Hirakawa K, et al. Clinical epidemiological study of 553 patients with chronic rhinosinusitis in Japan. Allergol Int. 2011;60(4):491–96. doi: 10.2332/allergolint.10-OA-0234. [DOI] [PubMed] [Google Scholar]
- 156.Wang ET, Zheng Y, Liu PF, Guo LJ. Eosinophilic chronic rhinosinusitis in East Asians. World J Clin Cases. 2014;2(12):873–82. doi: 10.12998/wjcc.v2.i12.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim SJ, Lee KH, Kim SW, Cho JS, Park YK, et al. Changes in histological features of nasal polyps in a Korean population over a 17-year period. Otolaryngol Head Neck Surg. 2013;149(3):431–37. doi: 10.1177/0194599813495363. [DOI] [PubMed] [Google Scholar]
- 158.Hu Y, Cao PP, Liang GT, Cui YH, Liu Z. Diagnostic significance of blood eosinophil count in eosinophilic chronic rhinosinusitis with nasal polyps in Chinese adults. Laryngoscope. 2012;122(3):498–503. doi: 10.1002/lary.22507. [DOI] [PubMed] [Google Scholar]
- 159.Katotomichelakis M, Tantilipikorn P, Holtappels G, De Ruyck N, Feng L, et al. Inflammatory patterns in upper airway disease in the same geographical area may change over time. Am J Rhinol Allergy. 2013;27(5):354–60. doi: 10.2500/ajra.2013.27.3922. [DOI] [PubMed] [Google Scholar]
- 160.Matsusaka M, Kabata H, Fukunaga K, Suzuki Y, Masaki K, et al. Phenotype of asthma related with high serum periostin levels. Allergol Int. 2015;64(2):175–80. doi: 10.1016/j.alit.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 161.Mahdavinia M, Suh LA, Carter RG, Stevens WW, Norton JE, et al. Increased noneosinophilic nasal polyps in chronic rhinosinusitis in US second-generation Asians suggest genetic regulation of eosinophilia. J Allergy Clin Immunol. 2015;135(2):576–79. doi: 10.1016/j.jaci.2014.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Alexander ES, Martin LJ, Collins MH, Kottyan LC, Sucharew H, et al. Twin and family studies reveal strong environmental and weaker genetic cues explaining heritability of eosinophilic esophagitis. J Allergy Clin Immunol. 2014;134(5):1084–92. doi: 10.1016/j.jaci.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rothenberg ME. Biology and treatment of eosinophilic esophagitis. Gastroenterology. 2009;137(4):1238–49. doi: 10.1053/j.gastro.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Leong AB, Ramsey CD, Celedon JC. The challenge of asthma in minority populations. Clin Rev Allergy Immunol. 2012;43(1–2):156–83. doi: 10.1007/s12016-011-8263-1. [DOI] [PubMed] [Google Scholar]
- 165.Pino-Yanes M, Thakur N, Gignoux CR, Galanter JM, Roth LA, et al. Genetic ancestry influences asthma susceptibility and lung function among Latinos. J Allergy Clin Immunol. 2015;135(1):228–35. doi: 10.1016/j.jaci.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Soler ZM, Mace JC, Litvack JR, Smith TL. Chronic rhinosinusitis, race, and ethnicity. Am J Rhinol Allergy. 2012;26(2):110–16. doi: 10.2500/ajra.2012.26.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mahdavinia M, Benhammuda M, Codispoti CD, Tobin MC, Losavio PS, et al. African American patients with chronic rhinosinusitis have a distinct phenotype of polyposis associated with increased asthma hospitalization. J Allergy Clin Immunol Pract. 2016;4(4):658–64. doi: 10.1016/j.jaip.2015.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pinto JM, Schneider J, Perez R, DeTineo M, Baroody FM, et al. Serum 25-hydroxyvitamin D levels are lower in urban African American subjects with chronic rhinosinusitis. J Allergy Clin Immunol. 2008;122(2):415–17. doi: 10.1016/j.jaci.2008.05.038. [DOI] [PubMed] [Google Scholar]
- 169.Bush CM, Jang DW, Champagne JP, Kountakis SE. Epidemiologic factors and surgical outcomes in patients with nasal polyposis and asthma. ORL J Otorhinolaryngol Relat Spec. 2013;75(6):320–24. doi: 10.1159/000354804. [DOI] [PubMed] [Google Scholar]
- 170.Ponikau JU, Sherris DA, Kephart GM, Adolphson C, Kita H. The role of ubiquitous airborne fungi in chronic rhinosinusitis. Clin Rev Allergy Immunol. 2006;30(3):187–94. doi: 10.1385/CRIAI:30:3:187. [DOI] [PubMed] [Google Scholar]
- 171.Fokkens WJ, van Drunen C, Georgalas C, Ebbens F. Role of fungi in pathogenesis of chronic rhinosinusitis: the hypothesis rejected. Curr Opin Otolaryngol Head Neck Surg. 2012;20(1):19–23. doi: 10.1097/MOO.0b013e32834e9084. [DOI] [PubMed] [Google Scholar]
- 172.Bachert C, Gevaert P, van Cauwenberge P. Staphylococcus aureus enterotoxins: A key in airway disease? Allergy. 2002;57(6):480–87. doi: 10.1034/j.1398-9995.2002.02156.x. [DOI] [PubMed] [Google Scholar]
- 173.Ramadan HH, Sanclement JA, Thomas JG. Chronic rhinosinusitis and biofilms. Otolaryngol Head Neck Surg. 2005;132(3):414–17. doi: 10.1016/j.otohns.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 174.Sanderson AR, Leid JG, Hunsaker D. Bacterial biofilms on the sinus mucosa of human subjects with chronic rhinosinusitis. Laryngoscope. 2006;116(7):1121–26. doi: 10.1097/01.mlg.0000221954.05467.54. [DOI] [PubMed] [Google Scholar]
- 175.Rank MA, Wollan P, Kita H, Yawn BP. Acute exacerbations of chronic rhinosinusitis occur in a distinct seasonal pattern. J Allergy Clin Immunol. 2010;126(1):168–69. doi: 10.1016/j.jaci.2010.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chalermwatanachai T, Velasquez LC, Bachert C. The microbiome of the upper airways: focus on chronic rhinosinusitis. World Allergy Organ J. 2015;8(1):3. doi: 10.1186/s40413-014-0048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mahdavinia M, Keshavarzian A, Tobin MC, Landay AL, Schleimer RP. A comprehensive review of the nasal microbiome in chronic rhinosinusitis (CRS) Clin Exp Allergy. 2016;46(1):21–41. doi: 10.1111/cea.12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bachert C, Holtappels G. Pathophysiology of chronic rhinosinusitis, pharmaceutical therapy options. GMS Curr Top Otorhinolaryngol Head Neck Surg. 2015;14:Doc09. doi: 10.3205/cto000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gevaert P, Calus L, Van Zele T, Blomme K, De Ruyck N, et al. Omalizumab is effective in allergic and nonallergic patients with nasal polyps and asthma. J Allergy Clin Immunol. 2013;131(1):110–16. doi: 10.1016/j.jaci.2012.07.047. [DOI] [PubMed] [Google Scholar]
- 180.Kiwamoto T, Kawasaki N, Paulson JC, Bochner BS. Siglec-8 as a drugable target to treat eosinophil and mast cell-associated conditions. Pharmacol Ther. 2012;135(3):327–36. doi: 10.1016/j.pharmthera.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Krug N, Hohlfeld JM, Kirsten AM, Kornmann O, Beeh KM, et al. Allergen-induced asthmatic responses modified by a GATA3-specific DNAzyme. N Engl J Med. 2015;372(21):1987–95. doi: 10.1056/NEJMoa1411776. [DOI] [PubMed] [Google Scholar]
- 182.Ly TW, Bacon KB. Small-molecule CRTH2 antagonists for the treatment of allergic inflammation: an overview. Expert Opin Investig Drugs. 2005;14(7):769–73. doi: 10.1517/13543784.14.7.769. [DOI] [PubMed] [Google Scholar]
- 183.Markham A. Reslizumab: first global approval. Drugs. 2016;76(8):907–11. doi: 10.1007/s40265-016-0583-2. Erratum. 2016. Drugs 76(11):1159. [DOI] [PubMed] [Google Scholar]
- 184.Castro M, Wenzel SE, Bleecker ER, Pizzichini E, Kuna P, et al. Benralizumab, an anti-interleukin 5 receptor α monoclonal antibody, versus placebo for uncontrolled eosinophilic asthma: a phase 2b randomised dose-ranging study. Lancet Respir Med. 2014;2(11):879–90. doi: 10.1016/S2213-2600(14)70201-2. [DOI] [PubMed] [Google Scholar]
- 185.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365(12):1088–98. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 186.Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. 2016;12(1):25–36. doi: 10.1038/nrrheum.2015.167. [DOI] [PMC free article] [PubMed] [Google Scholar]