

Abstract
Nitric oxide (NO) exerts its biological function through S-nitrosylation of cellular proteins. Due to the labile nature of this modification under physiological condition, identification of S-nitrosylated residue in enzymes involved in signaling regulation remains technically challenging. The present study investigated whether intrinsic NO produced in endothelium-derived MS-1 cells response to insulin stimulation might target endogenous protein tyrosine phosphatases (PTPs). For this, we have developed an approach using a synthetic reagent that introduces a phenylacetamidyl moiety on S-nitrosylated Cys, followed by detection with anti-phenylacetamidyl Cys (PAC) antibody. Coupling with sequential blocking of free thiols with multiple iodoacetyl-based Cys-reactive chemicals, we employed this PAC-switch method to show that endogenous SHP-2 and PTP1B were S-nitrosylated in MS-1 cells exposed to insulin. The mass spectrometry detected a phenylacetamidyl moiety specifically present on the active-site Cys463 of SHP-2. Focusing on the regulatory role of PTP1B, we showed S-nitrosylation to be the principal Cys reversible redox modification in endothelial insulin signaling. The PAC-switch method in an imaging format illustrated that a pool of S-nitrosylated PTP1B was colocalized with activated insulin receptor to the cell periphery, and that such event was endothelial NO synthase (eNOS)-dependent. Moreover, ectopic expression of the C215S mutant of PTP1B that mimics the active-site Cys215 S-nitrosylated form restored insulin responsiveness in eNOS-ablated cells, which was otherwise insensitive to insulin stimulation. This work not only introduces a new method that explores the role of physiological NO in regulating signal transduction, but also highlights a positive NO effect on promoting insulin responsiveness through S-nitrosylation of PTP1B’s active-site Cys215.
Keywords: Endothelial cell, Insulin signaling, Nitric oxide, New method, S-nitrosylation, SHP-2, PTP1B
1. Introduction
A gaseous form of bioactive molecule functioning as a second messenger, nitric oxide (NO) must target specific molecule in cells response to specific stimulus. To date, a high-affinity binding of NO to protein metal centers, such as in the case of guanylate cyclase and cytochrome c oxidase, has been attributed to the classical mechanism of NO’s biological function [1,2]. Alternatively, NO may regulate cell signaling through post-translational modifications (PTMs). One such well-characterized PTM is S-nitrosylation, which refers to covalent bond formation between a NO moiety and the reduced thiol of a cysteine (Cys) residue, rendering a S-nitrosothiol (SNO) group attached to the target protein [3,4]. Accumulated evidence suggests underlying mechanisms for nonenzymatic formation of S-nitrosylation via few possible routes of biochemical reactions [5,6]. In addition, S-nitrosyation may be generated by enzyme-mediated process of transnitrosylation, which moves a NO moiety from a SNO donor to a thiolate recipient [6–8]. Removal of SNO group involved in signaling events is generally considered an enzymatic process catalyzed by a number of denitrosylases [9–11].
It has been well documented that S-nitrosylation is involved in various physiological processes and development of human diseases [4]. In this context, SNO formation may occur in key enzymes regulating cell metabolism or critical modulators governing signal transduction. Interestingly, the reactive thiol group susceptible to S-nitrosylation is present in the active-site Cys of many enzymes identified so far [12]. The active-site Cys of at least five out of six classes of enzymes categorized by the Enzyme Commission, ranging from oxidoreductases, transferases, hydrolases, isomerases to ligases, is coordinated by a unique secondary structure, promoting deprotonation of the sulfur to form a thiolate (S−) readily reacted with a NO moiety [13]. One example that illustrates the specific structural feature of S-nitrosylation was shown in PTP1B, a Cys-based hydrolase belonging to the superfamily of protein tyrosine phosphatases (PTPs). Data obtained from quantitative mass spectrometry (MS) and X-ray crystallography demonstrated that the active-site Cys215, which bears a unique low pKa character thus being thiolated at neutral pH, was the primary residue of PTP1B susceptible to S-nitrosylation in vitro [14]. Because a large number of enzymes including all members in the PTP superfamily that adopt Cys thiolate essential for catalysis are susceptible to SNO formation, it is not surprising that S-nitrosylation participates in regulation of diverse physiological processes such as mitochondrial function [9], insulin responsiveness [15] and vascular homeostasis [16]. It has been also shown that a high degree of S-nitrosylation may promote the progression of human diseases exampled by development of neuronal degeneration [17,18] and cerebral ischemia [19].
Clearly, identification of S-nitrosylated targets in a given pathophysiological process is the key to define the functional role of NO, which is produced by intrinsic NO synthase (NOS) under this specific condition. To this end, neuronal NOS (nNOS or NOS1) and endothelial NOS (eNOS or NOS3) are important enzymes responsible for extracellular ligands-stimulated NO production, leading to S-nitorsylation of endogenous proteins [20,21]. In the case of neurodegenerative diseases contributed by overproduction of NO, excessive activation of N-methyl-D-aspartic acid (NMDA) receptor acts as a critical driver that empowers nNOS for accumulation of intracellular NO [22,23]. It was subsequently demonstrated that neuronal damage-related proteins including parkin [17,24], GADPH [25], protein disulfide isomerase [26] and dynamin-related protein 1 [27] were S-nitrosylated under disease conditions. Using the biotin switch method, a recent study showed that S-nitrosylation level of SHP-2, a SH2 domain-containing PTP, was increased in ischemic brain or NMDA-stimulated neurons [28]. The inhibition of SHP-2 phosphatase activity via SNO formation was further linked to enhance NMDA receptor-mediated excitotoxic neuronal damage [28]. Contrary to nNOS-dependent accumulation of NO in diseased brain, eNOS produces a transient burst of NO inside endothelium when exposed to physiological stimuli including insulin [29,30], vascular endothelial growth factor (VEGF) [31], cytokines [32] and shear flow [33]. It has been shown that the half-life of NO in vivo is less than 1 s [34]. Therefore, signaling-dependent nascent formation of proteins S-nitrosylation in endothelial cells is likely limited by the short-range NO diffusion and localized near sources of NO [6]. In addition, complicated redox microenvironments and the presence of transnitrosylases or denitrosylases within particular subcellular compartmentations may affect significantly the duration and localization of eNOS-mediated SNO modifications [3]. These findings manifest a labile nature of such PTM, thus rendering in situ measurement of S-nitrosylation on specific endogenous protein of endothelial cells very challenging.
One of the most characterized physiological functions of eNOS is its control in endothelial insulin signaling. Previous studies have demonstrated that genetic deletion of eNOS led to insulin resistance in mice [35–37], suggesting that endothelial insulin responsiveness requires eNOS-produced NO. The treatment of pharmacological inhibitors against eNOS activity also showed that the bioavailability of NO is critical for enhancement of insulin signaling [38,39]. Together, these results highlighted an important role of NO that may promote insulin responsiveness via S-nitrosylation of specific signaling modulators in endothelial cells. In this context, Cys-based PTPs that target insulin receptor (IR) as a substrate are prominent candidates of S-nitrosylated proteins. Using eNOS-overexpressed monkey kidney epithelial cells as a model, we provided preliminary results suggesting that three IR phosphatases, SHP-1, SHP-2 and PTP1B, are possible NO targets in insulin signaling pathway [40]. However, it is not known whether insulin-activated endogenous eNOS may produce sufficient amount of NO capable of modifying IR phosphatases in endothelial cells. Moreover, important issues including site-specific modification and subcellular localization of S-nitrosylated PTPs in insulinstimulated endothelium remain elusive. Clearly, advanced techniques are needed to answer such challenging questions.
This study investigated whether intrinsic NO produced in endothelial MS-1 cells response to insulin stimulation might target endogenous PTPs. For this, we developed a new technique that allows precise detection and visualization of cellular proteins susceptible to S-nitrosylation under physiological conditions. During the course of our study, we discovered that sequential blocking of free thiols with multiple iodoacetyl-based Cys-reactive chemicals is essential for eliminating false-positive results of protein S-nitrosylation. To ensure reliability of signals that register the S-nitrosylated Cys residues specifically, we applied a customer-made antibody to capture ascorbate-reduced nitrosothiols rather than relying on the biotinylated derivative recommended by the biotin switch method [41]. Employing this newly established analytic platform, we confirmed specific SNO modification on the active-site Cys of SHP-2, and monitored the localization of S-nitrosylated PTP1B in cells stimulated with insulin. These findings reveal an essential role that NO plays in regulation of insulin responsiveness through its reversible inactivation of IR phosphatases.
2. Materials and methods
2.1. Reagents
All chemicals were purchased from Sigma unless otherwise indicated. Reagents used in the PAC-switch methods are described in details in the Section 2.4. The 21-nucleotide siRNA duplexes (J-040956-07 andJ-040956-08) against mouse eNOS were purchased from Dharmacon Scientific. The HA-tagged C215S (C/S) mutant form of full-length PTP1B was constructed by site-direct mutagenesis using the wild type (WT) form of human PTP1B as the template and then subcloned into pcDNA3.1 vector. The following antibodies were purchased from various vendors: GAPDH, IR β form, SHP-2, PTP1B for immunoprecipitation (sc-1718-R) and for immunoblotting (sc-1718-G) from Santa Cruz; PTP1B (clone 15 for immunofluorescence staining) from BD; tubulin from Sigma; phospho-specific pYpY1162/1163-IR β from Invitrogen; eNOS and phospho-specific pS1177-eNOS (for immunoblotting) from BD; pS1177-eNOS (for immunofluorescence staining) from Cell Signaling; HA from Millipore. Alexa 647-conjugated anti-mouse IgG, Alexa 488-conjugated anti-rabbit IgG, Alexa 555-conjugated anti-guinea pig IgG, and ProLong Gold antifade reagent were purchased from Invitrogen. Protein A/G-Sepharose beads were from GE Healthcare.
2.2. Cell culture and transient transfection
Monkey kidney epithelial COS-7 cells (ATCC) were maintained in DMEM with 10% fetal bovine serum (FBS). Mouse endothelial MS-1 cells (ATCC) were maintained in DMEM with 5% FBS. For knockdown of endogenous eNOS or ectopic expression of HA-tagged C/S-PTP1B, MS-1 cells were mixed with siRNA (150pmol with 7 × 105 cells) or plasmid DNA (1.5 μg with 7 × 105 cells) respectively, followed by electroporation using the Neon™ Transfection System (Invitrogen) according to the manufacturer’s instructions.
2.3. Production of anti-phenylacetamidyl cysteine (anti-PAC) antibody
Carrier protein bovine serum albumin (BSA, Sigma A1933, 3 mg/ml) was reduced with 50 mM 1,4-dithioerythritol in buffer containing 20 mM Tris-HCl (pH 8.0), 5 mM EDTA, and 8 M urea at 37 °C for 1 h. An equal volume of 20% trichloroacetic acid was added into the solution with brief mixing. After addition of 10 × volume of acetone, the whole solution was vortexed and kept at −20 °C for overnight. Pellet was collected by centrifugation and washed with acetone. The pellet was then dissolved in 1 ml buffer containing 100 mM sodium bicarbonate (pH 9.4), 1% SDS, and 8 M urea. Iodoacetanilide (IAN, ULM-8131-0 from Cambridge Isotope Laboratories, 3 mg/0.1 ml) was prepared in DMSO and then added into the BSA solution followed by incubation at 37 °C for 2 h, for the alkylation reaction between the Cys thiolate in BSA and the alpha carbon of iodoacetanilide. The molar ratio of iodoacetanilide to BSA was approximately 250:1. After spinning centrifugation-dialysis using Concentrators (Pierce), the BSA protein with its Cys residues already phenylacetamidylated was re-suspended in PBS to form the antigen solution. Before injection, the antigen solution was thoroughly mixed with an equal volume of Freund’s incomplete adjuvant. Approximately 200 μg of antigen BSA protein was injected subcutaneously into the back of a guinea pig during each biweekly immunization. Ten days after the fifth injection, the blood was withdrawn by heart puncture, and serum was stored at 4 °C. The specific features of this anti-phenylacetamidyl Cys (anti-PAC) antibody were characterized by various tests shown in Supplementary Fig. S1.
2.4. Detection of protein S-nitrosylation in immunoprecipitated PTPs using the PAC-switch method
Cells were harvested in lysis buffer containing 25 mM HEPES (pH 7.4, BP310-1 from Thermal Fisher Scientific), 150 mM NaCl (BP358-10 from Thermal Fisher Scientific), 1 mM EDTA (disodium salt dihydrate, adjusted to pH 8, S311-500 from Thermal Fisher Scientific), 1% NP-40 (19628 from USB) and protease inhibitors (04-693-132-001 from Roche). An aliquot of total lysates (800 μg) was reacted with 50 mM IAM (M216-30G from Amresco) in lysis buffer containing 2.5% SDS (L5750 from Sigma) at room temperature (RT) for 30 min. Proteins were precipitated by cold acetone (100%, 179,124-4L from Sigma) and collected by centrifugation at 2000xg for 5 min, subsequently washed with cold acetone (70%) 3 times. The sample was next reacted with 1 mM APIAM (R871273-25G from Sigma) in lysis buffer containing 2.5% SDS and 10% DMSO (BP231-100 from Thermal Fisher Scientific) at 37 °C for 30 min. After precipitation and washing with acetone, protein pellets were resuspended by HENS buffer (100 mM HEPES (pH 8.0), 1 mM EDTA, 0.1 mM neocuproine (N1501-1G from Sigma), 1% SDS) containing 10% DMSO in the presence of 20 mM ascorbate (sodium salt, A7631-25G from Sigma) or 5 mM TCEP (77720 from Thermo Fisher Scientific) in the presence of 0.5 mM IAN (ULM-8131-0 from Cambridge Isotope Laboratories) at RT for 1 h. Proteins were precipitated, washed, and then resuspended in HENS buffer containing 1% SDS. The concentration of SDS in the processed lysates was diluted by PBS to 0.2%. For immunoprecipitation, anti-SHP-2 or anti-PTP1B antibody was first immobilized on protein A/G-Sepharose beads, followed by incubation with an aliquot of processed lysates (0.6 mg) at 4 °C for overnight. Immunoprecipitated SHP-2 or PTP1B was eluted by boiling beads in 1 × Laemmli buffer for 5 min. Equal volumes of protein elutes were subjected to immunoblotting with anti-PAC, SHP-2 and PTP1B antibodies.
2.5. In situ imaging of protein S-nitrosylation detected by the PAC-switch method
Cells were fixed/permeabilized with 4% paraformaldehyde (15 min)/0.1% Triton X-100 (5 min) on coverslips at RT. Reagents used in the imaging format of the PAC-switch method are described in Section 2.4. Free thiols were blocked with 50 mM IAM in HEN buffer (100 mM HEPES (pH 8.0), 1 mM EDTA, 0.1 mM neocuproine) at RT for 30 min. After washes (HEN buffer, 5 min, 3 times), samples were reacted with 2 mM APIAM in HEN buffer containing 10% DMSO at 37 °C for 30 min. After washes again (HEN buffer containing 10% DMSO, 5 min, 3 times), samples were incubated with 20 mM ascorbate and 0.5 mM IAN in HEN buffer containing 10% DMSO at RT for 1 h. After the final wash with HEN buffer containing 10% DMSO, samples were covered by 5% BSA at RT for 1 h, followed by reaction with anti-PAC antibody in 5% BSA at 4 °C for overnight. Samples were subsequently incubated with Alexa conjugated secondary antibodies at RT for 60 min. Once nucleus stained with DAPI (0.5 μg/ml), coverslips were mounted with ProLong Gold antifade reagent. Images were captured by Olympus BX50 fluorescence microscope or Zeiss LSM510 inverted confocal microscope.
2.6. Mass spectrometry-based identification of S-nitrosylated Cys residue
After InstantBlue Coomassie staining, the corresponding immunoprecipitated SHP-2 band on the SDS gel was collected. Gel slices were washed twice with 25 mM ABC buffer, destained in 25 mM ABC/50% acetonitrile (ACN) at 26 °C for 15 min, resuspended in 25 mM ABC buffer containing 0.05% Rapigest (Waters) plus 0.1 μg trypsin (sequencing grade, Promega), and then incubated at 37 °C for overnight. Peptides were extracted from the gels by sonication in 5% trifluoroacetic acid (TFA)/50% ACN, and vacuum-dried for MS analysis. All LC-MS analysis was performed on a Q Exactive HF (Thermo Fisher Scientific) mass spectrometer coupled with Ultimate 3000 RSLCnano System (Dionex). SHP-2 peptides were first trapped on a home-made precolumn (ReproSil-Pur 120 C18-AQ, 5 μm, Dr. Maisch GmbH; 100 μm × 5 cm) at 10 μl/ min of loading buffer (2% acetonitrile, 0.02% TFA in water) and then separated on an analytical column (ReproSil-Pur 120 C18-AQ, 1.9 μm, Dr. Maisch GmbH, 75 μm × 30 cm, self-packed) with a 40-min linear gradient ranging from 5% to 60% of buffer B (80% ACN/ 0.1% formic acid) and corresponding composition of Buffer A (0.1% formic acid in water) at a constant flow rate of 300 nl/min. The MS instrument was operated in a targeted parallel reaction monitoring (PRM) mode comprised of a MS1 scan covering m/z range from 430 to 755 with resolution of 120,000 and several targeted MS/MS scans with resolution of 30,000. Pre-defined precursor ions of interest were isolated by the quadrupole and fragmented in the higher-energy collisional dissociation (HCD) cell. The fragment ions were then analyzed with an Orbitrap mass analyzer. The cycle of MS1 and targeted MS/MS scans were repeated over the whole LC gradient. HCD was performed with an NCE of 28 and an isolation window of 2.0 m/z. Automatic gain control target value and maximum ion injection times for MS and MS/MS were 3 × 106 in 200 ms, and 5 × 105 in 400 ms, respectively. The post-acquisition data analysis was performed manually with the Xcalibur software (Thermo Fisher Scientific). Extracted ion currents of all targeted transitions were generated with the mass tolerance of 20 ppm.
2.7. Statistics
Data are presented as mean +/− SEM and assessed by student t-test. Difference was considered significant at p < 0.05.
3. Results
3.1. Generation of an antibody specifically recognizing phenylacetamidyl Cys
We wished to detect S-nitrosylated proteins with high specificity and accuracy. To do this, we established an antibody-based strategy for biochemical measurement and in situ imaging of endogenous proteins, which undergo S-nitrosylation in response to physiological stimuli. As illustrated in Fig. 1A, proteins with a stimulus-dependent modification at the susceptible Cys would be protected from post-fixing blocking (B). The susceptible Cys was subsequently reversed by ascorbate, tagged by alkylation (A), and then recognized by the antibody. Success of this strategy relies on specificity of the antibody, which only targets the alkylated Cys after ascorbate-mediated reduction but does not react with the fraction of Cys residues being blocked. For this, a synthetic alkylating reagent 2-iodo-phenylacetamide (iodoacetanilide, IAN; compound 1 in Fig. 1C) was selected to introduce a high level of phenylacetamidylation on all 35 Cys residues of recombinant BSA, which had been processed to ensure that it was in its reduced and denatured state (Fig. 1B). The treated BSA was subsequently used for immunization of guinea pig and generation of a strain of polyclonal antibodies, that we termed anti-phenylacetamidyl Cys (PAC) antibody.
Fig. 1.
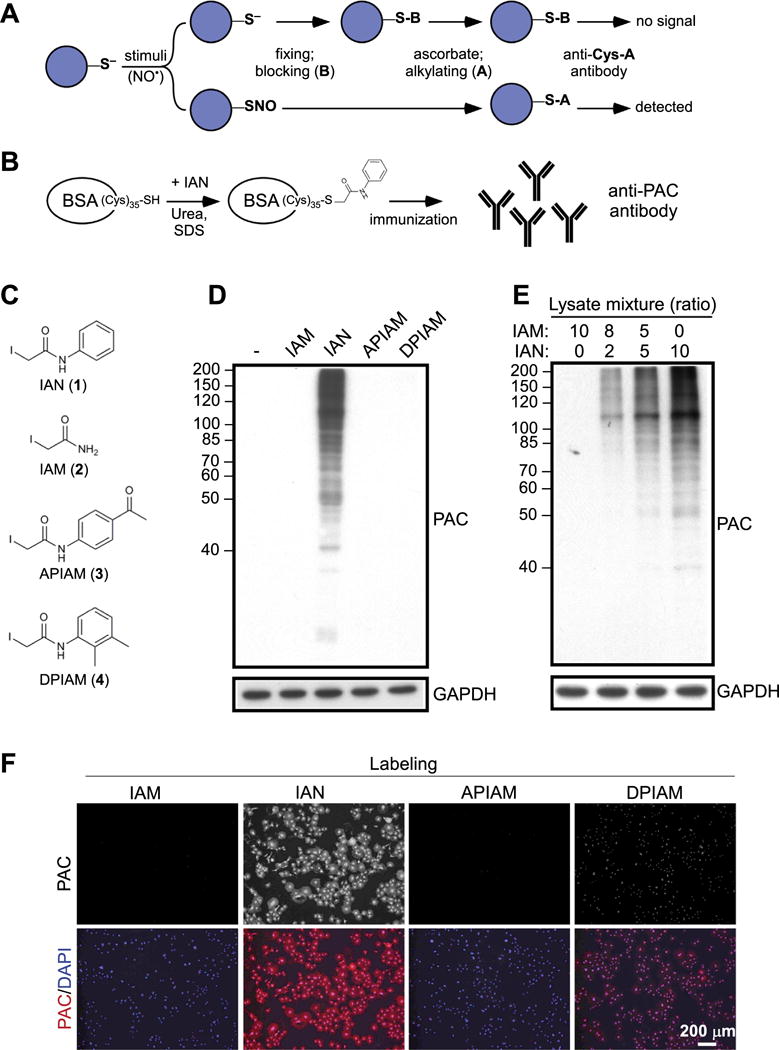
Generation of Anti-PAC Antibody and validation of Its Performance. (A) The workflow was designed to detect S-nitrosylated proteins by a proposed anti-Cys-A antibody. (B) To materialize anti-Cys-A antibody that could be applied in the workflow shown in (A), anti-phenylacetymidyl-cysteine (anti-PAC) antibody was generated using BSA as a carrier protein. (C) Four synthetic alkylating reagents for the specificity test of anti-PAC antibody are shown. (D) An aliquot of total lysates from COS-7 cells was reacted with IAM, IAN, APIAM or DPIAM, respectively, and then subjected to immunoblotting with anti-PAC antibody. (E) Aliquots of IAN- and IAM- reacted total lysates from COS-7 cells were mixed following the ratio shown in the figure. Subsequently samples were subjected to immunoblotting with anti-PAC antibody. (F) COS-7 cells were fixed, permeabilized and then exposed to IAM, IAN, APIAM or DPIAM. After washes, anti-PAC antibody was added for visualization of cellular proteins with Cys tagged byalkylation reagents.
After affinity purification from serum, the specificity of anti-PAC antibody was tested. Aliquots of total lysates from COS-7 cells that do not express a detectable level of endogenous eNOS [42] were incubated with a group of synthetic reagents. These included IAN, iodoacetamide (IAM, compound 2 in Fig. 1C), N-(4-acetylphenyl)-2-iodoacetamide (APIAM, compound 3 in Fig. 1C) and N-(2,3-dimethylphenyl)-2-iodoacetamide (DPIAM, compound 4 in Fig. 1C), which share a common chemical character of Cys alkylation but are structurally diverse. The lysates were subjected to SDS-PAGE, followed by immunoblotting analysis with anti-PAC antibody. As shown in Fig. 1D, the antibody only recognized IAN-treated cellular proteins, but not untreated or other alkylating reagents-reacted lysates. Moreover, analysis of mixtures from IAN-and IAM-treated samples showed a dose-dependent increase of PAC signal proportional to the level of IAN-reacted lysates (Fig. 1E), suggesting that this antibody targeted phenylacetamidyl Cys in total lysates specifically even in the abundant presence of acetamidyl Cys. We next tested the capacity of anti-PAC antibody to recognize cellular proteins with phenylacetamidyl Cys modifications by immunofluorescence staining. Permeabilized COS-7 cells were exposed to IAM, IAN, APIAM or DPIAM and subsequently reacted with anti-PAC antibody for visualization of Cys alkylation. As expected, IAN-treated cells were strongly stained with anti-PAC antibody, whereas cells exposed to IAM or APIAM were resistant to antibody binding (Fig. 1F). We noticed that, to a small degree of cross-reaction, anti-PAC antibody recognized DPIAM-treated cells in this analytic format (Fig. 1F). Thus, we avoided the use of DPIAM in follow-up experiments.
3.2. Development of the PAC-switch method for detecting cellular proteins susceptible to S-nitrosylation
As illustrated in Fig. 1A, precise detection of S-nitrosylated proteins not only requires an antibody specifically targeting the fraction of ascorbate-reduced Cys residues, but also depends on complete blocking of free thiols. Having demonstrated specificity of anti-PAC antibody (Fig. 1), which was reserved for reaction with ascorbate-reduced Cys residues, we next developed a strategy for sufficiently blocking free thiols present in cellular proteins. For this, we selected IAM and APIAM for further tests because their alkylated products did not cross-react with anti-PAC antibody (Fig. 1F). Permeabilized COS-7 cells pre-exposed to IAM or APIAM individually were subsequently incubated with IAN, followed by immunofluorescence staining with anti-PAC antibody. As shown in Fig. 2A, exposure of cells to either reagent markedly decreased, but did not eliminate, the signal detected by anti-PAC antibody. These results indicated that a small fraction of Cys residues in cellular proteins remained in the free thiol form after reaction with IAM or APIAM, and were thereby susceptible to IAN-mediated phenylacetamidylation. Obviously, the blocking condition was not ideal under such circumstances. To alleviate the influence of free thiols as much as possible, we performed sequential treatment of cells with IAM and APIAM before the final addition of IAN. This procedure ablated PAC signal in the cytosol (Fig. 2A), suggesting that complete blocking of free thiols in cellular proteins requires the use of two alkylating reagents with hydrophilic (IAM) and hydrophobic (APIAM) characteristics sequentially. Based on these observations, we established a workflow involving two-step blocking and subsequent PAC modification at the time that ascorbate reduces nitroso-Cys. The concentration of ascorbate was set at 20 mM, which is high enough to react with S-nitrosothiols for the production of thiols [43]. To prevent copper-dependent reaction of ascorbate with S-nitrosothiols that may lead to dis-ulfide formation [43], Cu1+ chelator neocuproine (0.1 mM) and EDTA (1 mM) were added to the sample. For effective reduction of S-nitrosylated proteins in total lysates or in fixed cells, the reaction time of ascorbate was kept for 1 h at room temperature, in line with the experimental conditions being tested in the previous study [44]. We termed this workflow the PAC-switch method, which may be used in the detection of endogenous proteins with S-nitrosylation (Fig. 2B).
Fig. 2.
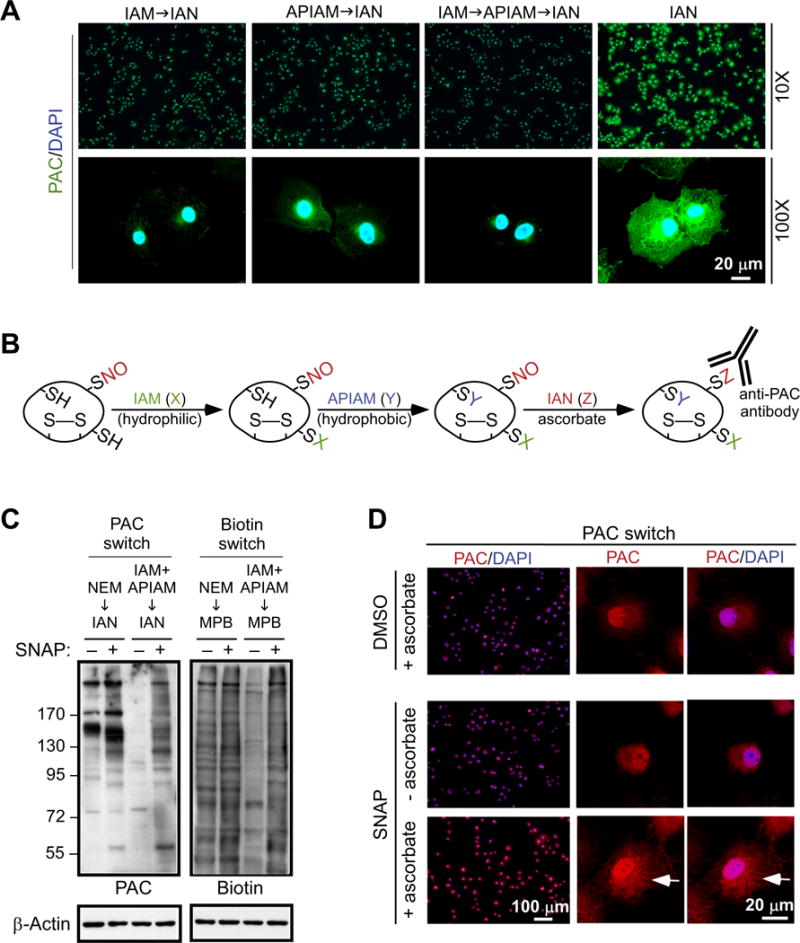
The PAC-switch method for detection of S-nitrosylated proteins. (A) Permeabilized COS-7 cells were treated with one or sequentially two blocking reagents before exposed to IAN, as indicated. Cellular proteins with Cys tagged by IAN were visualized using anti-PAC antibody. (B) The PAC-switch method to probe S-nitrosylated proteins was developed. Free thiols are blocked by sequential addition of IAM (-SX) and APIAM (-SY). S-nitrosylated Cys is reduced by ascorbate, tagged by IAN (-SZ), and then detected by anti-PAC antibody. (C) COS-7 cells were exposed to DMSO vehicle (−) or SNAP (1 mM, mixed with 1 mM L-Cys) for 10 min. Aliquots of total lysates were reacted with NEM, or IAM and APIAM sequentially, for blocking of free thiols. Newly reduced Cys residue by ascorbate was labelled with N-(3-maleimidylpropionyl) biocytin (MPB) or IAN. Processed lysates were subjected to immunoblotting with anti-PAC or anti-Biotin antibody. (D) DMSO or SNAP-treated COS-7 cells were fixed, permeabilized and processed by the PAC-switch method. S-nitrosylated proteins were visualized by anti-PAC antibody. Nucleus was stained with DAPI (A and D). The white arrows in (D) indicate increased PAC signal in paranuclear region. Comparable results shown in (D) were observed in three independent experiments.
To validate the PAC-switch method, we collected aliquots of total lysates from untreated and NO donor S-nitroso-N-acetylpenicillamine (SNAP, 1 mM, mixed with 1 mM L-Cys in culture medium)-treated COS-7 cells, processed them following the protocol shown in Fig. 2B, and then subjected them to immunoblotting with anti-PAC antibody. Importantly, in the absence of SNAP, the PAC signal was barely detected in total lysates (Fig. 2C, left panel), indicating successful blocking of cellular proteins by the two-step alkylating reaction. On the other hand, treatment with SNAP led to significant increase of the PAC signal in a broad range of cellular proteins (Fig. 2C, left panel). These results suggested that stimulus-induced S-nitrosylation of endogenous proteins can be detected reliably by this method. We also tested the effect of N-ethylmaleimide (NEM), which was recommended in the published biotin switch method [41] to prevent false-positive signal, on blocking of free thiols in the context of SNAP-induced S-nitrosylation of cellular proteins. Interestingly, as shown in Fig. 2C (left panel), NEM was unable to eliminate the PAC signal derived from untreated cells. Even though stimulation of cells with SNAP led to an increase in PAC signals (Fig. 2C, left panel), incomplete blocking of free thiols by NEM rendered visualization of S-nitrosylated proteins questionable under this circumstance.
Having demonstrated the drawback of single alkylating reagent to reduce background while identifying S-nitrosylated proteins, we then examined the potential problem revealed by the widely used biotin switch method, which is entirely dependent upon NEM-mediated blocking of free thiols [41]. As shown in Fig. 2C (right panel), following the published protocol to process total lysates, we were unable to differentiate the biotin signals of control and the SNAP-treated COS-7 cells, suggesting that a large fraction of biotin-tagged proteins would be false-positive rather than actually being S-nitrosylated. On the other hand, when we used sequential addition of IAM and APIAM instead of NEM alone as the blocking procedure, we observed a significant reduction of biotin signal in untreated sample (Fig. 2C, right panel). Under this condition, SNAP-promoted S-nitrosylation of cellular proteins was readily detected (Fig. 2C, right panel).
We next applied the PAC-switch method in the format of immunofluorescence staining to visualize S-nitrosylated proteins in COS-7 cells exposed to SNAP. As shown in Fig. 2D, in the presence of ascorbate, a robust signal registered by anti-PAC antibody throughout the entire cell was detected. This result indicated that proteins located at various subcellular compartments might be susceptible to S-nitrosylation as long as bioavailable NO is nearby. In an exploration of this possibility, cells stained with anti-PAC antibody were co-stained with specific markers for subcellular organelles. In response to SNAP treatment, fractions of proteins associated with endoplasmic reticulum (ER), Golgi and plasma membrane were likely S-nitrosylated (Supplementary Fig. S2).
3.3. S-nitrosylation of endogenous PTP1B and SHP-2 in MS-1 endothelial cells stimulated with insulin
In the next phase of study, we employed the PAC-switch method to explore whether and where physiologically produced NO might target endogenous PTPs. For this, we used insulin-stimulated MS-1 endothelium as a model because insulin signaling in this cell line has been characterized to be NO-dependent [40]. As shown in Fig. 3A, endogenous eNOS was phosphorylated and activated after 2 min of insulin treatment, concomitant with an initial increase of phosphorylation on the tandem tyrosine motif (Y1162/Y1163) at the activation loop of IR. The Y1162/Y1163 motif was robustly phosphorylated at 5 min, peaked at 10 min and significantly dephosphorylated 30 min after insulin stimulation (Fig. 3A). Consistent with our previous report [40], upon ablation of eNOS by RNAi, insulin-boosted phosphorylation of IR in the Y1162/Y1163 motif was significantly diminished in MS-1 cells (Supplementary Fig. S3). Based on these observations, we hypothesized that NO-dependent S-nitrosylation and inactivation of PTPs might be required within the initial phase (5–10 min), thus allowing IR to be fully activated. To test this hypothesis, MS-1 cells exposed to insulin for 5 min or 30 min were harvested and total lysates were subsequently processed following the PAC-switch method. Aliquots of total lysates and immunoprecipitated PTP1B or SHP-2, both function as IR phosphatases [45–47], were then subjected to immunoblotting with anti-PAC antibody. Our data demonstrated that, although PAC signals in total lysates were indistinguishable between control and treated samples (Fig. 3B), stimulation of cells with insulin for 5 min induced increased levels of S-nitrosylation in SHP-2 (Fig. 3C) and PTP1B (Fig. 3D). Interestingly, whereas NO-mediated modification on SHP-2 was substantially prolonged as indicated by similar PAC levels between samples with 5 min and 30 min of insulin treatment (Fig. 3C), S-nitrosylation of PTP1B was transiently increased at 5 min and then markedly reduced after 30 min of stimulation (Fig. 3D).
Fig. 3.
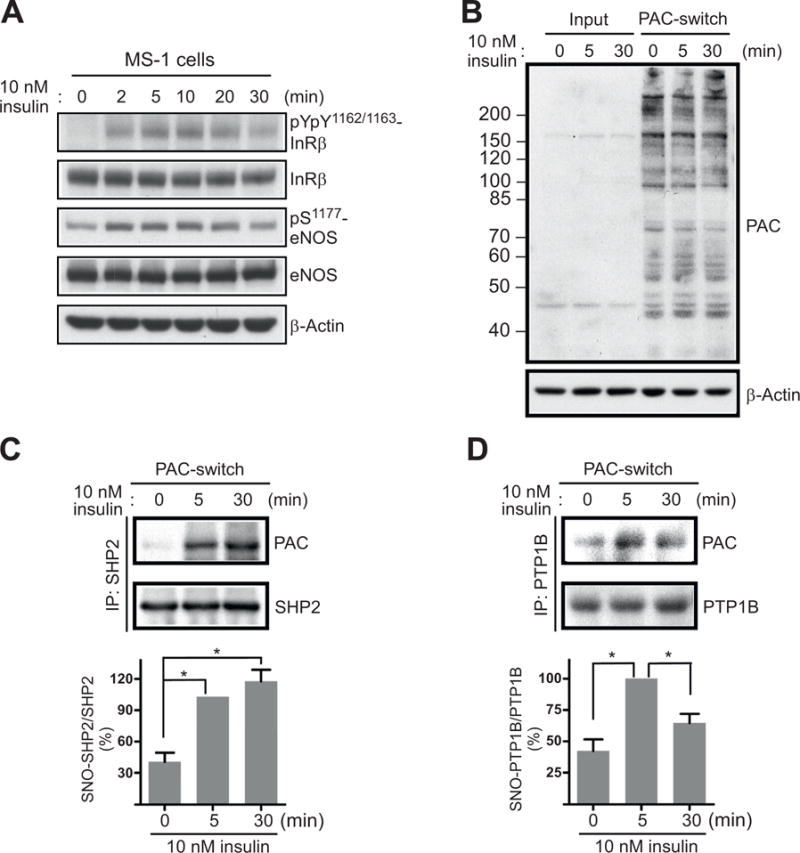
Insulin stimulation-induced S-nitrosylation of SHP-2 and PTP1B. MS-1 cells were exposed to 10 nM insulin for indicated times. (A) Aliquots of total lysates were subjected to immunoblotting with anti-pYpY1162/1163-IR, anti-IR, anti-pS1177 -eNOS, and anti-eNOS antibodies. (B) Aliquots of total lysates (Input) or processed lysates (PAC-switch) were subjected to immunoblotting with anti-PAC antibody. (C-D) An aliquot of processed lysates was subjected to immunoprecipitation with anti-SHP-2 (C) or anti-PTP1B (D) antibody. After washes, immunocomplexes were analyzed by immunoblotting with anti-PAC, anti-SHP-2 or anti-PTP1B antibody (upper panel). Results of densitometric analysis of the gel images as a ratio of PAC intensity relative to total SHP-2 (C) or total PTP1B (D) from three independent experiments were shown in the lower panel (*, p < 0.05).
3.4. Specific SNO modification on the active-site Cys463 of endogenous SHP-2 in response to insulin stimulation
The sustained S-nitrosylation level of SHP-2 (Fig. 3C) suggested that a relatively stable SNO adduct might be present in this phosphatase. Therefore, we examined whether a specific Cys residue of SHP-2 is susceptible to insulin-induced S-nitrosylation. To do so, lysates harvested from MS-1 cells exposed to insulin for 5 min were processed using the PAC-switch method, and then subjected to immunoprecipitation with anti-SHP-2 antibodies. For detecting S-nitrosylated Cys residue by MS, tryptic peptides of SHP-2 recovered from SDS-gel were analyzed by a highly selective and sensitive method of parallel reaction monitoring [48]. The mass spectrometer instrument was operated full-time on the mode of pre-defined precursor ions only (see Materials and Methods for details). Selected targets included two proteotypic peptides (INAAEIESR and VGQALLQGNTER) and two additional tryptic peptides containing either Cys104 (YPLNCADPTSER) or the active-site Cys463 (QESIVDAGPVVVHCSAGIGR) in both IAM- and IAN-labelled forms (summarized in Supplementary Table S1). Extracted ion currents (XICs) of four most abundant fragment ions per targeted peptide were then generated and examined after intensity normalization. This workflow was first validated by analyzing SHP-2 which was immunoprecipitated from an aliquot of lysates exposed to IAN in vitro. As expected, only IAN-labelled peptides containing Cys104 or Cys463 of SHP-2 were detected in the control sample (upper panel of Fig. 4A). In the insulin-treated sample processed by the PAC-switch method, however, both peptides labelled with IAM showed relatively high intensities (lower panel of Fig. 4A), indicating that a significant portion of Cys residues in endogenous SHP-2 stayed in free thiol form. Importantly, XICs of the fragment ions unraveled an IAN label on the Cys463 peptide but did not show a detectable IAN signal in the Cys104 peptide (lower panel of Fig. 4A), suggesting specific SNO modification on the active-site of SHP-2. The nature of IAM-(Fig. 4B) and IAN-tagged Cys463 (Fig. 4C) was ultimately demonstrated by the MS/MS spectra. It should be noted that in an attempt to monitor IAN labeling on other non-catalytic Cys residues, we were unable to detect any of them in SHP-2 isolated from insulin-treated cells (data not shown). Collectively, our results confirmed that, while some SHP-2 remained in its active form, a fraction of endogenous SHP-2 in MS-1 cells was targeted by NO on the active-site Cys463, leading to catalytic inactivation in response to insulin stimulation.
Fig. 4.
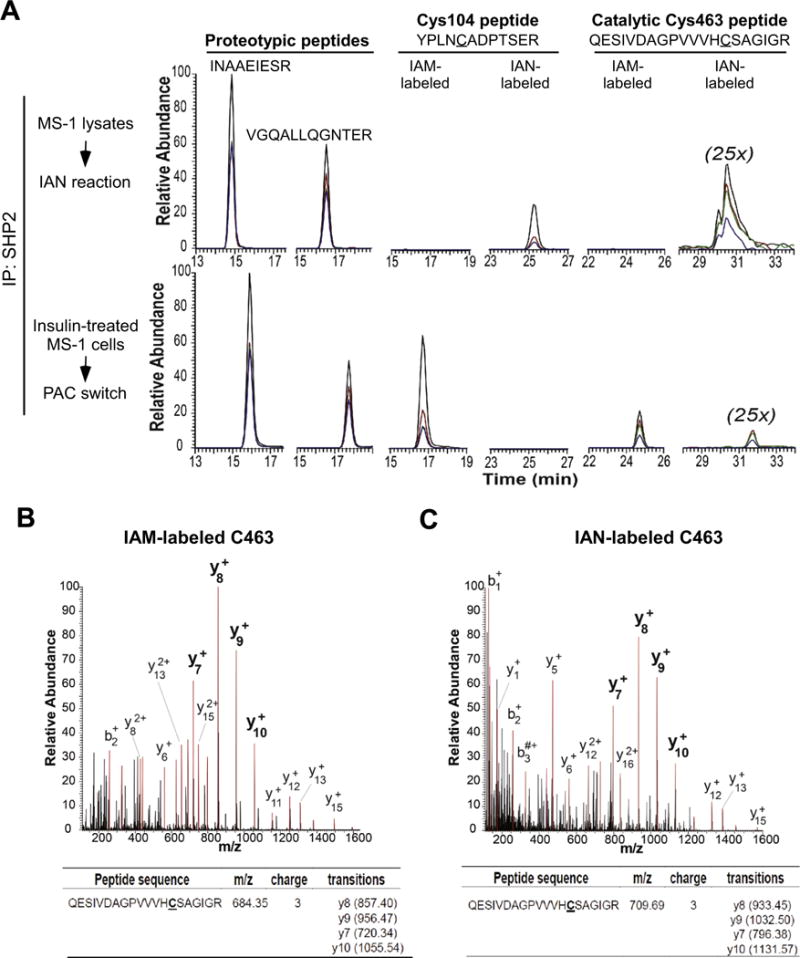
Targeted mass spectrometric determination of S-nitrosylated Cys in endogenous SHP-2. SHP-2 immunoprecipitated from IAN-reacted MS-1 lysates, or from an aliquot of PAC-switch processed lysates of insulin (10 nM, 5 min)-treated MS-1 cells, was tryptically digested, and subsequently analyzed on the Q Exactive HF MS instrument operated in a parallel-reaction monitoring mode. Two proteotypic peptides and two tryptic peptides bearing Cys104 and the active-site Cys463 in both IAM- and IAN-labelled forms were selected as targets. (A) Extract ion currents (XICs) of the four most abundant fragment ions from individual targets were color-coded and overlapped after normalizing to the proteotypic peptide INAAEIESR. From the most intense to the fourth intense, fragment ions were colored with black, red, green and blue, respectively. The intensities of XICs from the IAN-labelled C463-containing peptide were magnified by 25 folds. (B–C) Annotated MS/MS spectrum of IAM- (B) or IAN-labelled (C) C463-containing peptide was acquired from immunoprecipitated endogenous SHP-2 of insulin-treated MS-1 cells processed by the PAC-switch method. Peaks highlighted in red were fragment ions assigned by MS/MS. The list of selected fragment ions for generating XICs of each peptide precursor is shown in the lower panel.
3.5. eNOS-dependent S-nitrosylation of endogenous PTP1B in MS-1 cells exposed to insulin
The transient nature of PTP1B S-nitrosylation (Fig. 3D) suggested that such modification is involved in the regulation of insulin signaling. It has been documented that the active form of IR is a well-characterized substrate of PTP1B [49]. Thus, we proposed that a significant increase of IR phosphorylation within the first 5 min of insulin treatment (Fig. 3A) requires rapid inactivation of PTP1B, likely achieved by NO-driven S-nitrosylation of its active-site Cys residue [14]. Nonetheless, insulin-induced production of hydrogen peroxide (H2O2) may also target the active-site Cys of PTP1B, rendering the formation of sulfenic acid (S-OH) that leads to inhibition of the phosphatase activity [50,51]. To examine the specific role of NO in endothelial insulin signaling, we processed the total lysates from insulin-stimulated MS-1 cells following the PAC-switch method. Upon the completion of alkylating steps to block free thiols, either ascorbate or TCEP, which reduces S-nitrosylated Cys or all forms of reversibly modified Cys (including S-NO and S-OH) respectively [52], was added to the lysates at the time of IAN labeling. Endogenous PTP1B was then immunoprecipitated and subjected to immunoblotting with anti-PAC antibody. Consistent with our previous observation (Fig. 3D), insulin stimulation promoted a significant increase of S-nitrosylation on PTP1B (Fig. 5A). Interestingly, the degree of S-nitrosylation was indistinguishable from that of all reversible Cys modifications as indicated by similar levels of the PAC signal between ascorbate-and TCEP-reduced PTP1B (Fig. 5A). These results clearly demonstrated that insulin-induced production of NO plays a key role in rapid S-nitrosylation and inactivation of PTP1B in endothelial cells. Although H2O2-mediated reversible Cys oxidation of PTP1B might promote insulin responsiveness of fibroblasts or cancer cells [51], apparently this regulatory mechanism is not involved directly in endothelial insulin signaling. We next explored whether eNOS-dependent NO production is responsible for insulin-induced S-nitrosylation of PTP1B. For this, MS-1 cells were transfected with specific siRNA oligonucleotides which suppress the expression of endogenous eNOS. After insulin treatment, cells were lysed and processed by the PAC-switch method using ascorbate to reduce S-nitrosylated Cys residues. After immunoprecipitation, PTP1B was then probed with anti-PAC antibody. As shown in Fig. 5B, upon ablation of endogenous eNOS by RNA interference, insulin-stimulated S-nitrosylation of PTP1B was completely lost. Taken together, these results unravel an essential function of eNOS that promotes NO-dependent inactivation of PTP1B, consequently leading to the enhancement of endothelial insulin signaling.
Fig. 5.
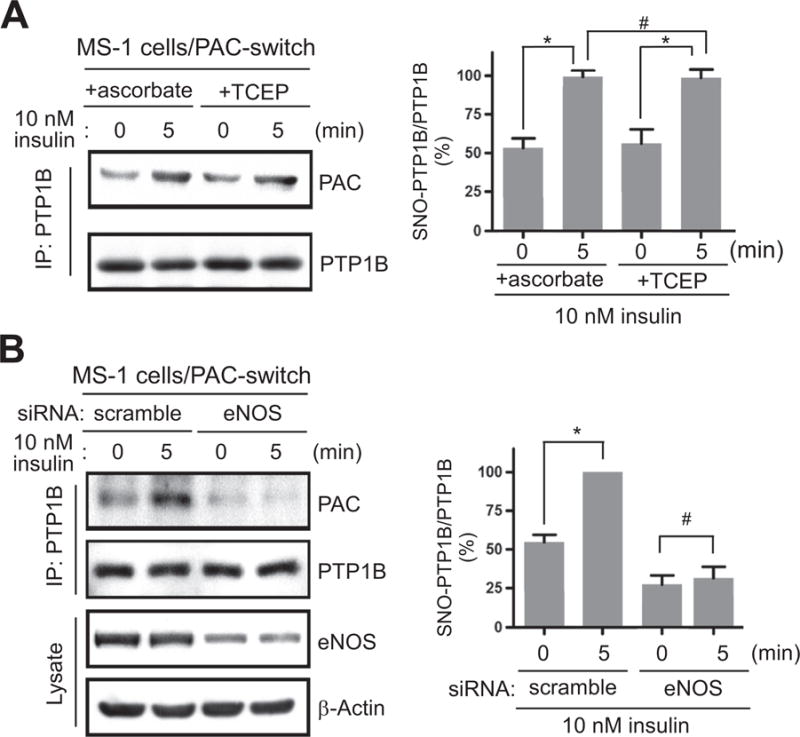
The SNO modification of endogenous PTP1B in endothelial insulin signaling. (A) MS-1 cells were treated with insulin for 5 min, subsequently lysed and processed by the PAC-switch method. After blocking of free thiols, an aliquot of total lysates was reacted with ascorbate or TCEP in the presence of IAN. PTP1B was then immunoprecipitated and analyzed by immunoblotting with anti-PAC antibody. (B) MS-1 cells electroporated with scramble or eNOS-specific siRNA oligonucleotides were treated with insulin for 5 min. After harvesting, total lysates were processed by the PAC-switch method using ascorbate to reduce S-nitrosylated Cys. The immunoprecipitated PTP1B was subsequently probed with anti-PAC antibody. Aliquots of total lysates were analyzed by immunoblotting with anti-eNOS antibody. Results of densitometric analysis of the gel images as a ratio of PAC intensity relative to total PTP1B from three independent experiments were shown in the right panels of (A) and (B) (*, p < 0.05; #, no significant difference).
3.6. Colocalization of S-nitrosylated PTP1B and activated insulin receptor in insulin-stimulated cells
We further examined the respective localization of S-nitrosylated PTP1B and active IR in cells under stimulation of insulin signaling. For this, the PAC-switch method was used in the format of in situ imagining, which allows visualization of PTP1B undergoing S-nitrosylation. Our pilot study showed that, no matter ectopically (Supplementary Fig. S4) or endogenously expressed (Supplementary Fig. S5), PTP1B was positively stained with anti-PAC antibody in COS-7 cells treated with SNAP. These results highlighted the feasibility that the PAC-switch method may be used to capture images of S-nitrosylated PTP1B induced by physiological ligands. Employing this method, we showed that a pool of PTP1B translocated to the boundary of MS-1 cells was clearly co-stained with anti-PAC antibody in response to insulin stimulation for 5 min, whereas the association between PTP1B and PAC signals was significantly diminished after 30 min of treatment (Fig. 6A). Apparently, S-nitrosylation of PTP1B localized to the cell periphery was transient and reversible in insulin signaling. We investigated whether this fraction of S-nitrosylated PTP1B might be co-localized with the activated form of IR. For this, MS-1 control cells or eNOS knockdown cells were exposed to insulin for 5 min, processed by the PAC-switch method, and subsequently examined by immunofluorescence. In insulin-stimulated cells where endogenous eNOS was expressed, PTP1B localized to the cell periphery was co-stained with anti-PAC and anti-pYpY1162/1163-IR antibodies (Fig. 6B). Importantly, co-localization of PTP1B, PAC and pIR was abrogated upon ablation of eNOS by specific siRNA (Fig. 6B), suggesting that the complex formation between S-nitrosylated PTP1B and activated IR was NO production-dependent.
Fig. 6.
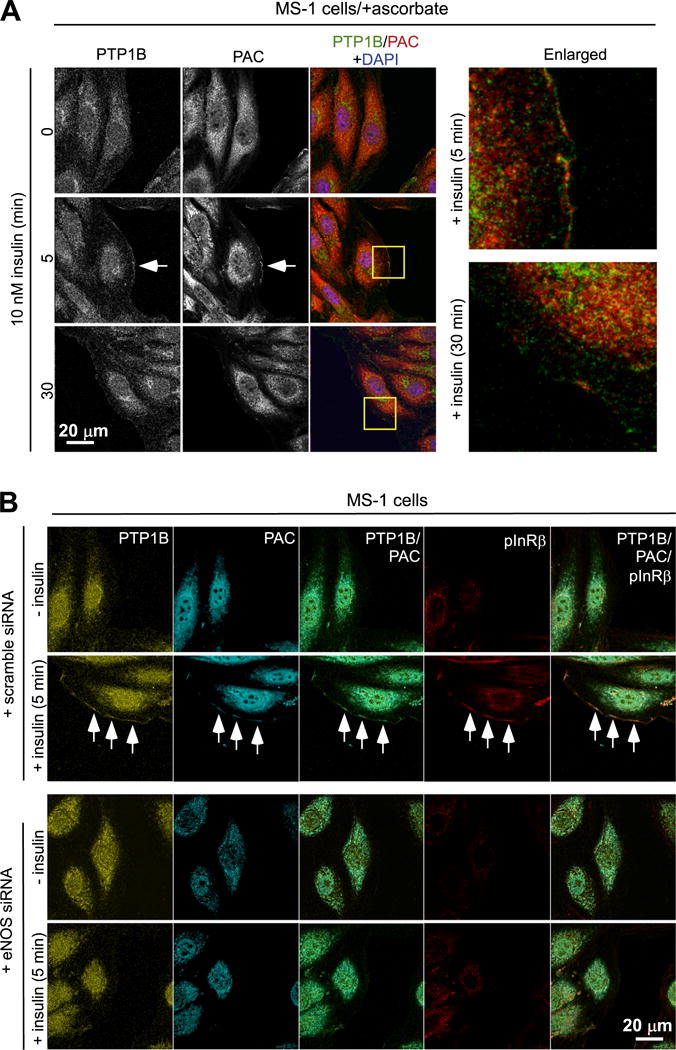
Insulin-induced colocalization of S-nitrosylated PTP1B and activated insulin receptor at cell periphery. (A) MS-1 cells treated with insulin were fixed and processed by the PAC-switch method. Subsequently cells were stained with anti-PTP1B and anti-PAC antibodies. The white arrow indicates that a pool of PTP1B transloctaed to the cell periphery was co-stained with anti-PAC antibody in cells stimulated with insulin for 5 min. The enlarged view (images extracted from yellow boxes) shows clear co-staining between PTP1B and PAC. (B) MS-1 cells electroporated with scramble or eNOS-specific siRAN oligonucleotides were exposed to insulin for 5 min. After fixing, cells were processed by the PAC-switch method, and then stained with anti-PTP1B, PAC and pYpY1162/1163- IR (pIR) antibodies. A pool of PTP1B transloctaed to the cell periphery was co-stained with anti-PAC and anti-pIR antibodies in cells exposed to insulin (white arrows). This event was abolished when eNOS was ablated by RNAi. Comparable results in (A) and (B) were observed in three independent experiments.
3.7. Enhanced insulin responsiveness via PTP1B S-nitrosylation on the active-site Cys215
Having demonstrated ligand-stimulated and eNOS-dependent association of S-nitrosylated PTP1B and activated IR, we proposed that the NO moiety attached on the active-site Cys215 of PTP1B in this particular pool of IR complex is critical to promote insulin signaling. To test this hypothesis, the C215S mutant of PTP1B (PTP1B-C215S), which mimics the active-site Cys215 S-nitrosylated, hence the inactivated form of PTP1B, was ectopically expressed in eNOS-ablated MS-1 cells. This PTP1B-C215S might function as a dominant negative mutant to compete with endogenous PTP1B, which would be in the reduced and active form upon eNOS knockdown, for IR complex formation. We then examined the role of PTP1B-C215S in regulating insulin responsiveness without the influence of intrinsic NO. As expected, IR in its pYpY1162/1163 form was barely detected in eNOS-ablated control cells (Fig. 7A). Interestingly, even in the absence of endogenous eNOS, ectopic expression of PTP1B-C215S promoted the accumulation of activated IR at the cell periphery (Fig. 7A). The intensity of insulin signaling was then monitored by IR pYpY1162/1163 phosphorylation in eNOS-ablated cells with mock transfection or ectopic expression of PTP1B-C215S. We showed that eNOS ablation-caused defect of insulin responsiveness was significantly restored by expression of PTP1B-C215S (Fig. 7B). Collectively, these results indicate that PTP1B S-nitrosylation of the active-site Cys215 plays a key role in NO-promoted insulin signaling. Our data suggest that NO-dependent modification inhibits PTP1B, thus allowing rapid IR phosphorylation for enhancement of insulin responsiveness.
Fig. 7.
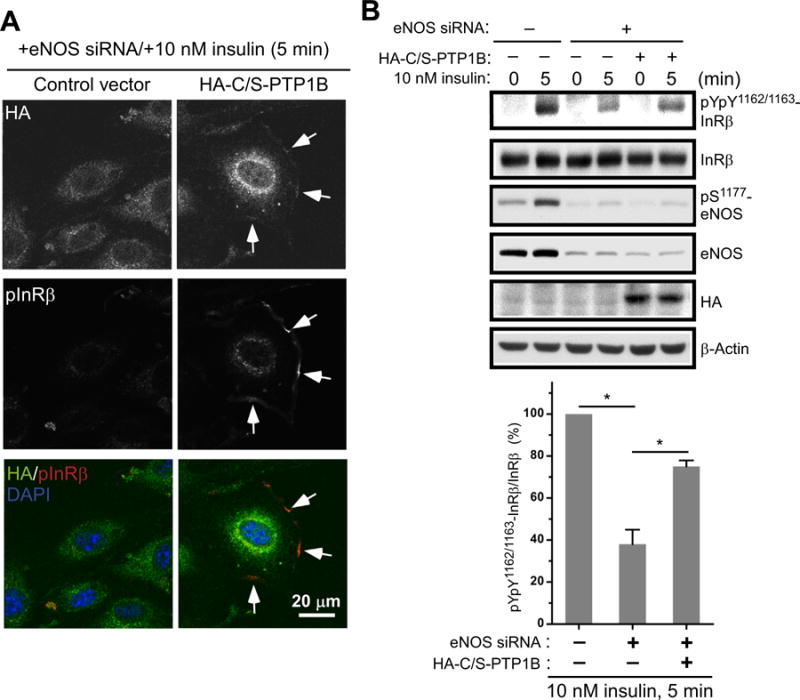
Restoration of insulin responsiveness in eNOS-ablated endothelium by ectopic expression of C215S mutant of PTP1B. eNOS-ablated MS-1 cells were transfected with a control vector or a vector expressing HA-tagged C215S mutant of PTP1B (HA-C/S-PTP1B). (A) Transfectants treated with insulin for 5 min were fixed, permeabilized and then stained with anti-HA and pYpY1162/1163-IR (pIR) antibodies. A pool of activated pYpY1162/1163-IR located to the cell periphery was co-stained with anti-HA antibody in HA-C/S-PTP1B transfectants exposed to insulin (white arrows). Comparable results were observed in three independent experiments. (B) MS-1 control cells (-siRAN/-HA-C/S-PTP1B) and transfectants as described in (A) were treated with insulin for 5 min. Aliquots of total lysates were subjected to immunoblotting with indicated antibodies (upper panel). Results of densitometric analysis of the gel image from three independent experiments as a ratio of pIR relative to total IR were shown in the lower panel (*, p < 0.05).
4. Discussion
In the present study, we employed the newly established PAC-switch method to show that endogenous SHP-2 and PTP1B were targeted directly by eNOS-produced NO in MS-1 cells shortly after insulin stimulation. In conjunction with MS analysis, SNO modification was detected preferentially on the active-site Cys463 of SHP-2, indicating that insulin-induced NO plays a critical role in regulating phosphatase activity in vivo. Applying the imaging approach in situ, we demonstrated that S-nitrosylated PTP1B, a phosphatase long regarded to antagonize IR activity [45,46,49], colocalized with the active form of IR following an insulin stimulation-dependent manner. The site-specific regulation of NO on target PTP1B was supported by the finding that ectopically expressed phosphatase-inactive C215S mutant of PTP1B restored insulin signaling, which was otherwise alleviated upon ablation of endogenous eNOS via RNAi (summarized in Supplementary Fig. S6).
Development of the PAC-switch method allowed us to capture the labile SNO modification that occurs on SHP-2 and PTP1B, both which function as signaling modulators thus appearing to be low abundant in cells. As illustrated by experimental evidences shown in Fig. 2, sequential blocking of free thiols present in cellular proteins by the two-step alkylating reaction instead of single reagent was a key advantage of our method. In addition, we propose that the use of anti-PAC antibody but not avidin- or streptavidin-based detection system for tagging S-nitrosothiols was another critical approach to enhance accuracy of our method. Expression of endogenous biotin in various tissues including kidney, liver and brain has been well documented in the literature [53,54]. It was also shown that mitochondrial matrix contains a significant level of biotinylated proteins [55]. The presence of endogenous biotin and biotinylated proteins may cause unexpected background signals in any application of biotin-avidin or biotin-streptavidin technique, including the widely used biotin-switch method [41]. The combined problems of incomplete blocking of NEM together with the influence of endogenous biotin and biotinylated proteins suggest that many false-positive results might have been generated by the biotin switch method since its introduction 15 years ago. We propose that published studies using biotin-avidin or biotin-streptavidin-based detection for protein S-nitrosylation should be critically re-evaluated by the PAC-switch method.
The sustained S-nitrosylation of SHP-2 in insulin signaling suggested that this phosphatase stays inert 30 min after stimulation (Fig. 3). A unique SNO-dependent conformational change might prevent the access of denitrosylase, thus accounting for the slow process of SHP-2 reduction. However, the biological significant of SHP-2 S-nitrosylation and the exact reason of retarded SHP-2 denitrosylation in insulin-treated cells remain elusive. In contract to what being observed in SHP-2, insulin-induced S-nitrosylation of PTP1B was transient (Fig. 3), suggesting that a speedy process of denitrosylation may remove SNO moiety rapidly from PTP1B after the peak activation of insulin signaling. This finding illustrated that the activity of PTP1B was temporarily inhibited by insulin-induced S-nitrosylation. Interestingly, previous studies have suggested that eNOS-mediated NO production is essential for physiological homeostasis of insulin signaling [35–37]. Nevertheless, the mechanism underlying such NO-promoted insulin responsiveness was unknown. With the new evidence provided herein, it is clear that eNOS-produced NO inhibits PTP1B temporarily via S-nitrosylation (Figs. 5–7), allowing transient activation of IR to govern an optimal degree and duration of insulin signaling.
Our observation that activated IR and S-nitrosylated PTP1B colocalized during the initial phase of insulin response (Fig. 6) is important. Because of this finding, we are able to explain how to prevent the rapid “switch-off” of insulin signaling from immediate association between PTP1B and IR in cells under such stimulation. It was observed a decade ago that PTP1B interacts with IR in a diverse array of insulin responsive cells soon after insulin stimulation. The effect of insulin on the dynamic interaction of IR with a trapping mutant form of PTP1B was documented as early as 30 s in living human embryonic kidney cells [56], suggesting that the tyrosine kinase activity of IR may drive the recruitment of PTP1B in this context. It was subsequently shown that association between an inactive mutant form of PTP1B and IR peaked at 5 min in adipocytes stimulated with insulin, and that their interaction was independent of IR internalization [57]. Bimolecular Fluorescence Complementation (BiFC) demonstrate that not only a trapping mutant or catalytically inactivate mutant but also WT PTP1B can target IR in human embryonic kidney cells within 5–10 min post insulin stimulation [58]. Using proximity ligation assay (PLA), which generates signals only if neighboring proteins are within 40 nm [59], we further demonstrated the colocalization of endogenous PTP1B and pYpY1162/1163-IR (pIR) at the periphery of MS-1 cells soon after exposure to insulin (Supplementary Fig. S7). Together with the new evidence shown in Fig. 7, we postulate that the pool of PTP1B in the complex with pIR is S-nitrosylated and inactivated, thus protecting IR from immediate dephosphorylation by PTP1B already present in close vicinity. Our data suggest that, even though PTP1B and pIR are complexed shortly after ligand stimulation, SNO-dependent inhibition in this particular pool of PTP1B may permit efficient insulin responsiveness not only shown in this study but also documented in the literature [56–58].
We showed that endogenous PTP1B, which is anchored to the cytosolic surface of the ER membrane, can interact with pIR at the cell periphery in MS-1 cells exposed to insulin (Figs. 6 and 7). The extension of ER membrane plays a key role in targeting PTP1B to plasma membrane-proximal regions upon insulin stimulation. Recent studies demonstrated that ER membranes are positioned close to the plasma membrane by microtubules [60]. It was subsequently shown that dynamic ER network might carry PTP1B to its regulatory pTyr site of substrates located at the plasma membrane [61]. However, if contacts between ER membranes and plasma membranes occur randomly, the process would be insufficient to establish the specific interaction of PTP1B with pIR. Our recent study revealed an important role of the adaptor protein Nck, which binds to PTP1B constitutively through the N-terminal SH3 domain, in recruiting PTP1B to the activated IR in signaling response to insulin stimulation [62]. Together with new results presented in the current work, we hypothesize that SH2 domains of Nck interact with pTyr residues of IR, leading to the inducible recruitment of Nck/S-nitrosylated PTP1B complex to specific sites where activated IR is located. This pool of S-nitrosylated PTP1B may subsequently undergo Cys reduction, leading to rapid rebound of its phosphatase activity for down-regulating pIR before dissociation of Nck/PTP1B complex from dephosphorylated IR. Further investigation is required to examine the regulatory role of Nck in the control of degree and duration of insulin responsiveness. In conclusion, with the application of new reagents and methodology in detection of endogenous proteins undergoing S-nitrosylation, we demonstrate that intrinsic NO targets the active-site Cys residue of PTPs in endothelium exposed to insulin. It should be pointed out that the PAC-switch method developed in this work relies on specific reducing reagent such as ascorbate or TCEP to probe reversibly modified Cys residues. Additional studies are required to confirm the specificity of the reactions carried out by these reducing reagents in situ. Nonetheless, our findings open a new avenue for understanding previously unexplored role of physiological NO in regulating reversible S-nitrosylation involved in a diverse array of signaling networks.
Supplementary Material
Acknowledgments
We thank Chin-Chun Hung for the confocal microscopy assistance, Chun-Lin Chiang for the graphic assistance. This work was supported by National Taiwan University Grants NTU-CESRP-102R7602B1 and NTU-CESRP-101R7602B1 to G.D.C.; Taiwan’s Ministry of Science and Technology Grant MoST103-2325-B-001-002 to T.C.M.; Academia Sinica Grant AS-100-TP-003 to T.C.M.; and US National Institutes of Health (NIH) Grants RO1DK090492 and RO1DK095359 to F.G.H.
Abbreviations
- ACN
acetonitrile
- APIAM
N-(4-acetylphenyl)-2-iodoacetamide
- BiFC
Bimolecular Fluorescence Complementation
- BSA
bovine serum albumin
- Cys
cysteine
- DAPI
(4′,6-diamidino-2-phenylindole)
- DPIAM
N-(2,3-dimethylphenyl)-2-iodoacetamide
- DMEM
Dulbecco’s modified Eagle’s medium
- EGFP
enhanced green fluorescence protein
- ER
endoplasmic reticulum
- eNOS
endothelial NO synthase
- GADPH
glyceraldehyde-3-phosphate dehydrogenase
- IAM
iodoacetamide
- IAN
iodoacetanilide
- IR
insulin receptor
- MPB
N-(3-maleimidylpropionyl) biocytin
- MS
mass spectrometry
- NEM
N-ethylmaleimide
- NMDA
N-methyl-D-aspartic acid
- nNOS
neuronal NO synthase
- NO
nitric oxide
- PAC
phenylacetamidyl Cys
- pIR
phosphorylated insulin receptor
- PTM
post-translational modification
- PTP
protein tyrosine phosphatase
- SNAP
S-nitroso-N-acetylpenicillamine
- SNO
S-nitrosothiol
- TCEP
Tris(2-carboxyethyl)phosphine
- VEGF
vascular endothelial growth factor
- XIC
extracted ion currents
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2016.08.012.
Footnotes
Conflict of interest
The authors declare that they have no competing interests.
References
- 1.Cary SP, Winger JA, Derbyshire ER, Marletta MA. Nitric oxide signaling: no longer simply on or off. Trends Biochem Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Martinez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms, Free Radic. Biol Med. 2011;51:17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Evangelista AM, Kohr MJ, Murphy E. S-nitrosylation: specificity, occupancy, and interaction with other post-translational modifications, Antioxid. Redox Signal. 2013;19:1209–1219. doi: 10.1089/ars.2012.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 5.Hogg N. The kinetics of S-transnitrosation—a reversible second-order reaction. Anal Biochem. 1999;272:257–262. doi: 10.1006/abio.1999.4199. [DOI] [PubMed] [Google Scholar]
- 6.Martinez-Ruiz A, Araujo IM, Izquierdo-Alvarez A, Hernansanz-Agustin P, Lamas S, Serrador JM. Specificity in S-nitrosylation: a short-range mechanism for NO signaling? Antioxid Redox Signal. 2013;19:1220–1235. doi: 10.1089/ars.2012.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12:1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 9.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 11.Hashemy SI, Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J Biol Chem. 2008;283:21890–21898. doi: 10.1074/jbc.M801047200. [DOI] [PubMed] [Google Scholar]
- 12.Nagahara N. Catalytic site cysteines of thiol enzyme: sulfurtransferases. J Amino Acids 2011. 2011:709404. doi: 10.4061/2011/709404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Go YM, Chandler JD, Jones DP. The cysteine proteome, Free Radic. Biol Med. 2015;84:227–245. doi: 10.1016/j.freeradbiomed.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen YY, Chu HM, Pan KT, Teng CH, Wang DL, Wang AH, Khoo KH, Meng TC. Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem. 2008;283:35265–35272. doi: 10.1074/jbc.M805287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Wang AX, Aylor K, Barrett EJ. Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes. 2013;62:4030–4042. doi: 10.2337/db13-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 17.Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 18.Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 19.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 20.Kiss JP, Vizi ES. Nitric oxide: a novel link between synaptic and nonsynaptic transmission. Trends Neurosci. 2001;24:211–215. doi: 10.1016/s0166-2236(00)01745-8. [DOI] [PubMed] [Google Scholar]
- 21.Tomasian D, Keaney JF, Vita JA. Antioxidants and the bioactivity of endothelium-derived nitric oxide. Cardiovasc Res. 2000;47:426–435. doi: 10.1016/s0008-6363(00)00103-6. [DOI] [PubMed] [Google Scholar]
- 22.Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura T, Lipton SA. S-Nitrosylation and uncompetitive/fast off-rate (UFO) drug therapy in neurodegenerative disorders of protein misfolding. Cell Death Differ. 2007;14:1305–1314. doi: 10.1038/sj.cdd.4402138. [DOI] [PubMed] [Google Scholar]
- 24.Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 26.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 27.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi ZQ, Sunico CR, McKercher SR, Cui J, Feng GS, Nakamura T, Lipton SA. S-nitrosylated SHP-2 contributes to NMDA receptor-mediated excitotoxicity in acute ischemic stroke. Proc Natl Acad Sci USA. 2013;110:3137–3142. doi: 10.1073/pnas.1215501110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000;101:1539–1545. doi: 10.1161/01.cir.101.13.1539. [DOI] [PubMed] [Google Scholar]
- 30.Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Investig. 1996;98:894–898. doi: 10.1172/JCI118871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, Gratton JP. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell. 2010;39:468–476. doi: 10.1016/j.molcel.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 32.Pi X, Wu Y, Ferguson JE, 3rd, Portbury AL, Patterson C. SDF-1alpha stimulates JNK3 activity via eNOS-dependent nitrosylation of MKP7 to enhance endothelial migration. Proc Natl Acad Sci USA. 2009;106:5675–5680. doi: 10.1073/pnas.0809568106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol. 2003;285:C499–C508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- 34.Crow JP, Spruell C, Chen J, Gunn C, Ischiropoulos H, Tsai M, Smith CD, Radi R, Koppenol WH, Beckman JS. On the pH-dependent yield of hydroxyl radical products from peroxynitrite. Free Radic Biol Med. 1994;16:331–338. doi: 10.1016/0891-5849(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 35.Cook S, Hugli O, Egli M, Menard B, Thalmann S, Sartori C, Perrin C, Nicod P, Thorens B, Vollenweider P, Scherrer U, Burcelin R. Partial gene deletion of endothelial nitric oxide synthase predisposes to exaggerated high-fat diet-induced insulin resistance and arterial hypertension. Diabetes. 2004;53:2067–2072. doi: 10.2337/diabetes.53.8.2067. [DOI] [PubMed] [Google Scholar]
- 36.Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- 37.Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes. 2000;49:684–687. doi: 10.2337/diabetes.49.5.684. [DOI] [PubMed] [Google Scholar]
- 38.Baron AD, Zhu JS, Marshall S, Irsula O, Brechtel G, Keech C. Insulin resistance after hypertension induced by the nitric oxide synthesis inhibitor l-NMMA in rats. Am J Physiol. 1995;269:E709–15. doi: 10.1152/ajpendo.1995.269.4.E709. [DOI] [PubMed] [Google Scholar]
- 39.Roy D, Perreault M, Marette A. Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. Am J Physiol. 1998;274:E692–9. doi: 10.1152/ajpendo.1998.274.4.E692. [DOI] [PubMed] [Google Scholar]
- 40.Hsu MF, Meng TC. Enhancement of insulin responsiveness by nitric oxidemediated inactivation of protein-tyrosine phosphatases. J Biol Chem. 2010;285:7919–7928. doi: 10.1074/jbc.M109.057513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 42.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann RJ, Shah VH, Sessa WC. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci USA. 2006;103:19777–19782. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmes AJ, Williams DLH. Reaction of ascorbic acid with S-nitrosothiols: clear evidence for two distinct reaction pathways. J Chem Soc Perkin Trans. 2000;2:1639–1644. [Google Scholar]
- 44.Zhang Y, Keszler A, Broniowska KA, Hogg N. Characterization and application of the biotin-switch assay for the identification of S-nitrosated proteins. Free Radic Biol Med. 2005;38:874–881. doi: 10.1016/j.freeradbiomed.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 46.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ouwens DM, van der Zon GC, Maassen JA. Modulation of insulin-stimulated glycogen synthesis by Src Homology Phosphatase 2. Mol Cell Endocrinol. 2001;175:131–140. doi: 10.1016/s0303-7207(01)00389-6. [DOI] [PubMed] [Google Scholar]
- 48.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteom. 2012;11:1475–1488. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 50.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005;7:1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 52.Wojdyla K, Rogowska-Wrzesinska A. Differential alkylation-based redox proteomics—Lessons learnt. Redox Biol. 2015;6:240–252. doi: 10.1016/j.redox.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKay BE, Molineux ML, Turner RW. Biotin is endogenously expressed in select regions of the rat central nervous system. J Comp Neurol. 2004;473:86–96. doi: 10.1002/cne.20109. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Pevsner J. Detection of endogenous biotin in various tissues: novel functions in the hippocampus and implications for its use in avidin-biotin technology. Cell Tissue Res. 1999;296:511–516. doi: 10.1007/s004410051311. [DOI] [PubMed] [Google Scholar]
- 55.Hollinshead M, Sanderson J, Vaux DJ. Anti-biotin antibodies offer superior organelle-specific labeling of mitochondria over avidin or streptavidin. J Histochem Cytochem. 1997;45:1053–1057. doi: 10.1177/002215549704500803. [DOI] [PubMed] [Google Scholar]
- 56.Boute N, Boubekeur S, Lacasa D, Issad T. Dynamics of the interaction between the insulin receptor and protein tyrosine-phosphatase 1B in living cells. EMBO Rep. 2003;4:313–319. doi: 10.1038/sj.embor.embor767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi K, Egawa K, Maegawa H, Nakamura T, Ugi S, Nishio Y, Kashiwagi A. Protein-tyrosine phosphatase 1B associates with insulin receptor and negatively regulates insulin signaling without receptor internalization. J Biochem. 2004;136:89–96. doi: 10.1093/jb/mvh094. [DOI] [PubMed] [Google Scholar]
- 58.Anderie I, Schulz I, Schmid A. Direct interaction between ER membrane-bound PTP1B and its plasma membrane-anchored targets. Cell Signal. 2011;19:582–592. doi: 10.1016/j.cellsig.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 59.Salmon M, Gomez D, Greene E, Shankman L, Owens GK. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22alpha promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res. 2012;111:685–696. doi: 10.1161/CIRCRESAHA.112.269811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Monteleone MC, Gonzalez AE, Wusener JE, Burdisso C, Conde A, Caceres C, Arregui O. ER-bound protein tyrosine phosphatase PTP1B interacts with Src at the plasma membrane/substrate interface. PLoS One. 2012;7:e38948. doi: 10.1371/journal.pone.0038948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haj FG, Sabet O, Kinkhabwala A, Wimmer-Kleikamp S, Roukos V, Han HM, Grabenbauer M, Bierbaum M, Antony C, Neel BG, Bastiaens PI. Regulation of signaling at regions of cell-cell contact by endoplasmic reticulum-bound protein-tyrosine phosphatase 1B. PLoS One. 2012;7:e36633. doi: 10.1371/journal.pone.0036633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu CL, Buszard B, Teng CH, Chen WL, Warr CG, Tiganis T, Meng TC. Dock/Nck facilitates PTP61F/PTP1B regulation of insulin signalling. Biochem J. 2011;439:151–159. doi: 10.1042/BJ20110799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.