

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease that results in the progressive death of motor neurons, leading to paralysis and eventual death. There is presently no cure for ALS, and only two drugs are available, neither of which provide significant extension of life. The wide variation in onset and progression of the disease, both in sporadic and even in strongly penetrant monogenic familial forms of ALS, indicate that in addition to background genetic variation impacting the disease process, environmental exposures are likely contributors. Epidemiological evidence worldwide implicates exposures to bacterial toxins, heavy metals, pesticides, and trauma as probable environmental factors. Here, we review current advances in gene-environment interactions in ALS animal models. We report our recent discoveries in a zebrafish model of ALS in relation to exposure to the cyanobacterial toxin BMAA, and discuss several results from mouse models that show interactions with exposure to mercury and statin drugs, both leading to acceleration of the disease process. The increasing research into this combinatorial gene-environment process is just starting, but shows early promise to uncover the underlying biochemical pathways that instigate the initial motor neuron defects and lead to their rapidly progressive dysfunction.
Keywords: amyotrophic lateral sclerosis, gene-environment, animal models, toxicants, genetic susceptibility, risk factors
Amyotrophic Lateral Sclerosis (ALS)
ALS causes degeneration of motor neurons in the spinal cord, brainstem, and motor cortex. Degeneration of the motor neurons leads to progressive paralysis, with a median survival of less than 5 years. About 10% of ALS is familial (FALS, with ~20% of FALS resulting from mutations in Cu/Zn superoxide dismutase (SOD1) (Siddique et al., 1991). However, the vast majority of ALS (~ 90%) is sporadic (SALS), with epidemiological, clinical, and experimental evidence indicating that early developmental exposures to neurotoxicants can have consequences for neurodegeneration later in life (Fox et al., 2012), and the large variation in onset and progression of FALS and SALS is likely due to developmental exposures to environmental toxicants in genetically susceptible individuals (Trojsi et al., 2013). Pathological late-onset neurodegenerative diseases are increasingly being seen as beginning early in development, and therefore investigating the proteins and pathways disrupted during development and pre-clinically are vital to understanding the eventual pathogenesis (Kovacs et al., 2014). ALS also seems to disrupt spinal motor neuron maturation and aging pathways, supporting the idea that age-related neurodegeneration begins decades before clinical onset (Ho et al., 2016). Evidence points towards an increased risk of ALS due to exposure to a range of environmental toxicants, with the interaction between genetic susceptibility and environmental exposure probably responsible for most neurodegenerative diseases (Papapetropoulos, 2007). The most likely explanation for the development of ALS is that ALS is the result of environmental exposures and time acting together (Al-Chalabi and Hardiman, 2013).
Genetic Factors
At present, there are 29 known genetic risk factors causing or contributing to ALS (Guerreiro et al., 2015), but these genes only account for approximately 5–10% of all ALS cases. Although there are more, presently unknown, genetic factors leading to the development of ALS, exposure to environmental toxicants in combination with underlying genetic risk is the most likely explanation for how ALS develops. In fact, an increasing body of evidence indicates that the development of ALS is a multi-step process where an underlying genetic defect can lead to several outcomes depending on subsequent environmental triggers (Al-Chalabi et al., 2014), similar in etiology to the development of cancer. Perhaps in parallel, it is also thought that environmental exposures may epigenetically modify the genome, and this may be one of the pathways by which ALS develops (Callaghan et al., 2011). ALS, therefore, results after a series of steps in disease progression, where individual genetic and environmental factors serve as steps in disease progress (Figure 1). This need for multiple steps in disease progression would account for the observed adult onset (even among patients with SALS), pleiotropy of some ALS genes, and spread of symptoms from an initial focal point.
Figure 1.
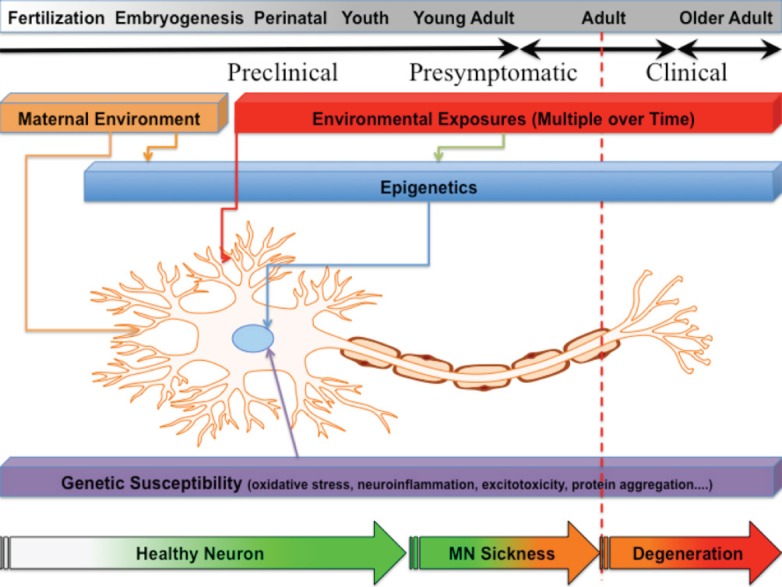
Influences on the development and progression of amyotrophic lateral sclerosis (ALS).
Initiating events may occur through genetic and/or maternal environmental influences immediately after fertilization and throughout fetal development. Multiple insults occur over the lifetime of a person, and these can compound in susceptible motor neurons and surrounding nervous system cells, leading to disease initiation. Additional environmental hits can occur later that impact the progression and eventual fate of the disease process. Motor neurons become stressed and start to develop motor neuron sickness. After many years, compensatory systems are overwhelmed and patients progress into the late pre-clinical stages. Eventually, neurons increasingly fail and ALS becomes both clinically emergent and increasingly progressive. Developing biomarker identifications for preclinical/presymptomatic phases, and understanding the cellular mechanisms underlying the ontology of preclinical gene-environment interactions that lead to initiation and progression of motor neuron sickness may enable the development of preventive, protective, or supportive therapies.
Need for Animal Models
In order to properly assess the risks and pathways affected by the interaction of genetics, epigenetics, and environmental exposures on the initiation and progression of ALS, we need studies that make the best use of the strengths of animal models of neurodegeneration. Animal models allow us to investigate preclinical stages of disease that parallel and represent the human disease process long before clinical diagnosis. Once a patient is diagnosed with ALS, they are already well into decades of disease that has been masked by functional compensation of intact motor systems. Within a newly diagnosed patient's spinal cord, there is a heterogeneous population of motor neurons, some dead, some dying, some diseased but not yet dying, and some healthy. The key to finding interventions that allow for increased survival and function in these patients it to be able to target the healthy and diseased neurons, and to prevent them from becoming sick/dying neurons. With ALS, a long preclinical and presymptomatic period, that may start perinatally, provides the opportunity to target dysfunctional neurons prior to neuronal death, even after disease diagnosis, and studies of this preclinical phase may lead to therapies targeted to compensatory or protective mechanisms (Paratore et al., 2012). Because of the long latency of ALS and the difficulty in studying CNS in living patients, human studies have difficulty studying the mechanisms impacted by these early life exposures. Animals such as mice and zebrafish, on the other hand, have been shown to be durable models for investigating the preclinical and presymptomatic stages of compound exposure.
Exposing an animal to a particular neurotoxicant may result in acute and/or chronic neurological defects. Determining whether these are truly involved in a neurodegenerative disease like ALS, however, is much harder to measure. Using genetic animal models of ALS as a “sensitized” background on which to test the impact of toxicant exposure on known timeframes of disease progression can lead to novel phenotypic or cellular insights into disease progression. Comparing phenotypic outcomes between genetic and genetic/environmental treatments can provide a more direct measure of how toxicants may influence known neurological disease pathways, allowing us to distinguish between acute toxicities that are not involved in the disease course and those that are amplified due to the combination of genes and the environment.
Current dogma is that early developmental defects are an initial manifestation of the cellular and molecular pathways that eventually result in adult neurodegeneration and death, but this idea has never been adequately tested in a vertebrate model of ALS (Krakora et al., 2012). There is evidence for early involvement of neuromuscular junctions in the pathophysiology of ALS, and neuromuscular defects are found in affected muscles of human ALS patients (Liu et al., 2013), but these are of necessity determined AFTER disease diagnosis. The ability to study impacts on early neuromuscular defects (prior to disease onset) in animal models is crucial to finding pathways leading to neurodegeneration, not just reflecting changes in diseased tissues.
Zebrafish as a Key Model for Epigenetic Environmental Toxicology
Early life exposure to neurotoxicants may lead to age-related neurodegeneration either through epigenetic alterations that only emerge in later life, or through alterations in neurological development and maturation that impact late-life neurological stability (Grandjean and Landrigan, 2014). Zebrafish, already a well-established model for toxicology and developmental biology, are rapidly becoming a key in studying ALS (Sakowski et al., 2012). A zebrafish ALS model has been developed carrying transgenes containing the endogenous zebrafish sod1 promoter and sod1 gene (Ramesh et al., 2014), with the 93rd amino acid mutated from glycine to arginine, a highly conserved amino acid often mutated in human ALS. These fish exhibit many features of ALS, including larval and adult neuromuscular junction defects, decreased adult swimming endurance, and eventual age-dependent loss of motor neurons, paralysis, and death.
Evidence for Toxicogenomic Interactions in the SOD1-G93R Zebrafish
Early defects in neural circuitry have been found to be associated with late-onset neurological disorders, including both cognitive and degenerative diseases (Fox et al., 2012). The use of transient, early toxicant exposure provides us with the ability to directly study whether phenotypic changes to early ALS defects actually impact later-stage defects and subsequent expression and epigenetic changes. We have recently reported (Powers et al., 2017) that the SOD1-G93R zebrafish model of ALS is uniquely suited to examining the combinatorial gene-environment effect of developmental toxicant exposure to adult onset ALS phenotypic alterations when compared to exposed non-mutant zebrafish. In fact, this model has been shown to be a strong model for investigating the long pre-clinical phase of the development of ALS (Ramesh et al., 2014). We found that embryonic exposure to the cyanobacterial neurotoxin beta-N-methylamino-l-alanine (BMAA), a non-proteinogenic amino acid produced by cyanobacteria, dinoflaggelates, and diatoms and epidemiologically-linked to ALS clusters worldwide, leads to early neurodevelopmental defects and adult pre-clinical motor dysfunction. Our data indicate that exposure to increasing doses of BMAA during embryonic development results in significant changes in 30 hours post fertilization (hpf) motor neuron growth characteristics in a genotype-dependent manner (G93R-zsod1 show this alteration but WT-zsod1 fish do not) indicating that the genetic defect does sensitize the fish in a complex interaction with the neurotoxin. There is an increased nerve length at low doses (2.5 μg/L) and decreased length at medium/high doses (10 μg/L), indicating that compensatory mechanisms may function at higher doses (biphasic activity or hormesis). In addition, we have found neuromuscular junction colocalization defects as early as 72 hpf when Tg-zsod1-G93R embryos are exposed to higher (> 10 μg/L) BMAA, an effect not seen in Tg-zsod1-WT.
We also reared fish (exposed to 0, 2.5, or 5 μg/L BMAA during the first week of development) to adulthood in water free from BMAA to test the impact of early embryonic exposure on adult motor function. We challenged adult fish to a constant current, and time to failure to maintain position was measured for each fish. We found that at 5 months of age (presymptomatic), BMAA-exposed Tg-zsod1-G93R fish had reduced swimming endurance compared to Tg-zsod1-WT, and that they fatigued more easily with increased BMAA dose. Fatigability is a trait also seen in human ALS patients (Sharma et al., 1995).
Exposure to repeated cyanobacterial blooms in freshwater lakes in New Hampshire and the Northeast has been associated with an increased risk of ALS (10–25 fold increase) (Caller et al., 2013), and epidemiological evidence points to a similar cluster in Southern France (Masseret et al., 2013), and ingestion of even low levels of BMAA may increase the risk of ALS in genetically susceptible individuals (Banack et al., 2007). Critically, a recent experimental study has shown that chronic dietary exposure to BMAA for 140 days led to neurofibrillary tangles (NFT) and beta-amyloid plaques in vervet monkeys (Cox et al., 2016), indicating the potential for widespread neurological outcomes in exposed human populations, and validating the results we found in our zebrafish ALS model.
Going forward, this new model for gene-environment interactions in ALS can be used to investigate epigenetic, gene-expression, and molecular alterations due to exposure (maternal, embryonically, developmentally) in genetically-susceptible organisms, and to develop novel biomarkers for preclinical disease states. One recent report has shown that exposure of the SOD1-G93A mouse to MeHg starting at 29 days led to acceleration of ALS disease measures, potentially through glutamate-mediated toxicity (Bailey et al., 2017). Another report found that exposure to statins (one of the most commonly prescribed drugs in the human population) in SOD1-G93A mice similarly accelerates the disease process (Su et al., 2016). This opens the way for additional gene-environment studies in ALS that hopefully will lead to new treatment options and earlier treatment windows. We are only at the beginning of this new exploration into the origins of neurodegeneration, and many different pathways may be impacted by specific gene-environment interactions, along with many different cell types such as motor neurons, glia, and astrocytes. There are an enormous number of unknowns in the origins and development of ALS, but progress is being made in unraveling the complexity of this and other neurodegenerative diseases, one experimental model at a time. We do not suggest that any environmental exposure by itself, or any underlying genetic polymorphisms in disease-associated variants by themselves, is solely responsible for ALS, or that all pathways being affected by gene-environment interactions are the same. In fact, strongly penetrant monogenic forms of familial ALS show wide variation in onset and progression, indicating that even these substantial genetic sensitizers are influenced by other factors. To date, the only published reports on gene-environment interactions in ALS are the SOD1 genetic models of zebrafish and mice. There are many other animal models of ALS, including worm, fly, and rat genetic models of TDP43, FUS, and C9orf72, and investigating if/how these genetic defects interact with environmental exposures is an active area of research in our and other laboratories. Whether these genetic models upon toxicant exposure impact the same biochemical pathways that we are presently identifying is unknown, and the results will provide strong data on the commonality or uniqueness of each of these genetic forms of ALS. In addition, these future studies will provide evidence for the cross-species utility of these models in relation to how the pathways identified in different animals converge. At present there is no good information on how well one animal model recapitulates other species in relation to gene-environment interactions in neurodegeneration. We believe that studying “sensitized” or “humanized” animal models of ALS in combination with developmental toxicant exposures can reveal new pathways, new pathologies, and new insights into the underlying mechanisms leading to neurological disease. Understanding how exposures may lead to disease can impact both therapeutic potential as well as minimizing future population exposure risk.
Acknowledgments
We thank Samantha Powers for her tireless work in the original research. We thank Dr. Gregory Cox for a critical reading of this manuscript.
Footnotes
Conflicts of interest: None declared.
Contributor agreement: A statement of “Publishing Agreement” has been signed by an authorized author on behalf of all authors prior to publication.
Plagiarism check: This paper has been checked twice with duplication-checking software iThenticate.
Peer review: A double-blind and stringent peer review process has been performed to ensure the integrity, quality and significance of this paper.
References
- Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A, Calvo A, Chio A, Colville S, Ellis CM, Hardiman O, Heverin M, Howard RS, Huisman MH, Keren N, Leigh PN, Mazzini L, Mora G, Orrell RW, Rooney J, Scott KM, Scotton WJ, Seelen M, Shaw CE, Sidle KS, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 2014;13:1108–1113. doi: 10.1016/S1474-4422(14)70219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JM, Colon-Rodriguez A, Atchison WD. Evaluating a gene-environment interaction in amyotrophic lateral sclerosis: methylmercury exposure and mutated SOD1. Curr Environ Health Rep. 2017;4:200–207. doi: 10.1007/s40572-017-0144-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banack SA, Johnson HE, Cheng R, Cox PA. Production of the neurotoxin BMAA by a marine cyanobacterium. Mar Drugs. 2007;5:180–196. doi: 10.3390/md504180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan B, Feldman D, Gruis K, Feldman E. The association of exposure to lead, mercury, and selenium and the development of amyotrophic lateral sclerosis and the epigenetic implications. Neurodegener Dis. 2011;8:1–8. doi: 10.1159/000315405. [DOI] [PubMed] [Google Scholar]
- Caller TA, Chipman JW, Field NC, Stommel EW. Spatial analysis of amyotrophic lateral sclerosis in Northern New England, USA, 1997-2009. Muscle Nerve. 2013;48:235–241. doi: 10.1002/mus.23761. [DOI] [PubMed] [Google Scholar]
- Cox PA, Davis DA, Mash DC, Metcalf JS, Banack SA. Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. Proc Biol Sci. 2016;283:20152397. doi: 10.1098/rspb.2015.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DA, Grandjean P, de Groot D, Paule MG. Developmental origins of adult diseases and neurotoxicity: epidemiological and experimental studies. Neurotoxicology. 2012;33:810–816. doi: 10.1016/j.neuro.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean P, Landrigan PJ. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014;13:330–338. doi: 10.1016/S1474-4422(13)70278-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Bras J, Hardy J. SnapShot: Genetics of ALS and FTD. Cell. 2015;160:798–e791. doi: 10.1016/j.cell.2015.01.052. [DOI] [PubMed] [Google Scholar]
- Ho R, Sances S, Gowing G, Amoroso MW, O’Rourke JG, Sahabian A, Wichterle H, Baloh RH, Sareen D, Svendsen CN. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat Neurosci. 2016;19:1256–1267. doi: 10.1038/nn.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Adle-Biassette H, Milenkovic I, Cipriani S, van Scheppingen J, Aronica E. Linking pathways in the developing and aging brain with neurodegeneration. Neuroscience. 2014;269:152–172. doi: 10.1016/j.neuroscience.2014.03.045. [DOI] [PubMed] [Google Scholar]
- Krakora D, Macrander C, Suzuki M. Neuromuscular junction protection for the potential treatment of amyotrophic lateral sclerosis. Neurol Res Int 2012. 2012:379657. doi: 10.1155/2012/379657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Brannstrom T, Andersen PM, Pedrosa-Domellof F. Distinct changes in synaptic protein composition at neuromuscular junctions of extraocular muscles versus limb muscles of ALS donors. PLoS One. 2013;8:e57473. doi: 10.1371/journal.pone.0057473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masseret E, Banack S, Boumediene F, Abadie E, Brient L, Pernet F, Juntas-Morales R, Pageot N, Metcalf J, Cox P, Camu W. French Network on ALSCD, Investigation (2013) Dietary BMAA exposure in an amyotrophic lateral sclerosis cluster from southern France. PLoS One. 8:e83406. doi: 10.1371/journal.pone.0083406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos S. Is there a role for naturally occurring cyanobacterial toxins in neurodegeneration? The beta-N-methylamino-L-alanine (BMAA) paradigm. Neurochem Int. 2007;50:998–1003. doi: 10.1016/j.neuint.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Paratore S, Pezzino S, Cavallaro S. Identification of pharmacological targets in amyotrophic lateral sclerosis through genomic analysis of deregulated genes and pathways. Curr Genomics. 2012;13:321–333. doi: 10.2174/138920212800793366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers S, Kwok S, Lovejoy E, Lavin T, Sher R. Embryonic exposure to the environmental neurotoxin BMAA negatively impacts early neuronal development and progression of neurodegeneration in the Sod1-G93R Zebrafish model of amyotrophic lateral sclerosis. Toxicol Sci. 2017;157:129–140. doi: 10.1093/toxsci/kfx020. [DOI] [PubMed] [Google Scholar]
- Ramesh TM, Shaw PJ, McDearmid J. A zebrafish model exemplifies the long preclinical period of motor neuron disease. J Neurol Neurosurg Psychiatry. 2014;85:1288–1289. doi: 10.1136/jnnp-2014-308288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakowski SA, Lunn JS, Busta AS, Oh SS, Zamora-Berridi G, Palmer M, Rosenberg AA, Philip SG, Dowling JJ, Feldman EL. Neuromuscular effects of G93A-SOD1 expression in zebrafish. Mol Neurodegener. 2012;7:44. doi: 10.1186/1750-1326-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma KR, Kent-Braun JA, Majumdar S, Huang Y, Mynhier M, Weiner MW, Miller RG. Physiology of fatigue in amyotrophic lateral sclerosis. Neurology. 1995;45:733–740. doi: 10.1212/wnl.45.4.733. [DOI] [PubMed] [Google Scholar]
- Siddique T, Figlewicz DA, Pericak-Vance MA, Haines JL, Rouleau G, Jeffers AJ, Sapp P, Hung WY, Bebout J, McKenna-Yasek D, Deng G, Horvitz HR, Gusella JS, Brown RH, Roses AD. Collaborators (1991) Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med. 324:1381–1384. doi: 10.1056/NEJM199105163242001. [DOI] [PubMed] [Google Scholar]
- Su XW, Nandar W, Neely EB, Simmons Z, Connor JR. Statins accelerate disease progression and shorten survival in SOD1(G93A) mice. Muscle Nerve. 2016;54:284–291. doi: 10.1002/mus.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojsi F, Monsurro MR, Tedeschi G. Exposure to environmental toxicants and pathogenesis of amyotrophic lateral sclerosis: state of the art and research perspectives. Int J Mol Sci. 2013;14:15286–15311. doi: 10.3390/ijms140815286. [DOI] [PMC free article] [PubMed] [Google Scholar]