

Relevance of Alzheimer's disease (AD): AD is one of the greatest health-care challenges of the 21st century. It is known to be the most common neurodegenerative disease and, in the United States, the sixth most common cause of death. It is estimated that 40 million people in the world suffer from dementia, mostly people older than 60 years. With the increase of the survival, this incidence is projected to be doubled every 20 years (Scheltens et al., 2016). Between 1997 and 2050, the elderly population, defined as subjects 65 years of age and older, will increase from 63 to 137 million in the Americas, from 18 to 38 million in Africa, from 113 to 170 million in Europe, and from 172 to 435 million in Asia, so we are looking straight to a public health problem.
There is evidence enough to say that AD pathology starts depositing in the brain in midlife although clinical symptoms remain silent until 65 years and older. The greatest risk factor for the development of AD is old age, but by itself is not enough to cause the disease. Other major risk factors include the presence of one or more apolipoprotein gene E4 alleles, low educational and occupational attainment, family history of AD, moderate or severe traumatic brain injuries, and cardiovascular risk factors (Apostolova, 2016).
A lot of researches have been done since the discovering of amyloid β peptide (Aβ) and Tau, the main components of plaques and tangles respectively, but there is a lack of knowledge about its etiopathogenesis and despite decades of study, there is no disease-modifying therapy so far. We should assume it is time to change the focus towards new therapeutic targets and molecular pathways.
Understanding the pathogenesis: The principal pathological lesions in AD are described by “the amyloid hypothesis of AD”: neuritic plaques (NPs), neurofibrillary tangles (NFTs) and cerebral amyloid angiopathy, which are associated with both neuronal and vascular degeneration. AD is broadly recognized as a proteinopathy-driven disease, essentially by two types of abnormal protein deposition in the human brain: Aβ in neuritic plaques, which is an extracellular deposit that produces neurotoxicity; and intracellular store of hyperphosphorylated forms of a microtubule associated protein Tau, in NFTs (Selkoe and Hardy, 2016) that triggers neuronal death. However, at present, basic research has sentenced that AD is in fact, a multifactorial disease, leading to novel approaches moving from old therapies to multi-target treatments, which allow us to achieve a successful understanding of this condition.
On the basis of previous studies and considering the drugs already used in therapy, the most investigated targets are inhibition of acetylcholinesterase, chelation of copper, iron and/or zinc cations, antioxidant activity, inhibition of Aβ amyloid plaques aggregation, monoamine oxidase enzymes inhibition, and NMDA receptor antagonism. The reduction of chronic inflammation by peroxisome proliferator-activated receptor agonists may represent a strategy to protect neuronal cells that are compromised in these diseases (Lacey and Evans, 2014). However, further studies are needed to find a curative and/or prophylactic treatment.
Heparan sulfate proteoglycans (HSPGs) family: HSPGs constitute a heterogeneous family of complex biological macromolecules composed of heavily glycosylated molecules, in which several heparan sulfate (HS) glycosaminoglycan (GAG) chains are covalently attached to a core protein. Cell surface HSPGs are membrane-spanning syndecans and lipid-anchored glypicans. Secreted HSPGs are agrin, collagen type XVIII, and perlecan (Zhang et al., 2014). The pathological mechanism of these macromolecules is thought to be promoting Aβ fibrillation giving rise NPs and providing resistance against proteolytic breakdown (Figure 1A). In addition, previous data have pointed out that the HS-Aβ interaction contributes to every stage of pathogenesis in AD, including production, clearance, accumulation, aggregation, and toxicity of Aβ (van Horssen et al., 2002; Cui et al., 2013). Moreover, work over the last decade indicates that Taupathology can propagate between cells in a prion-like manner mediated by HSPGs (Holmes and Diamond, 2014).
Figure 1.
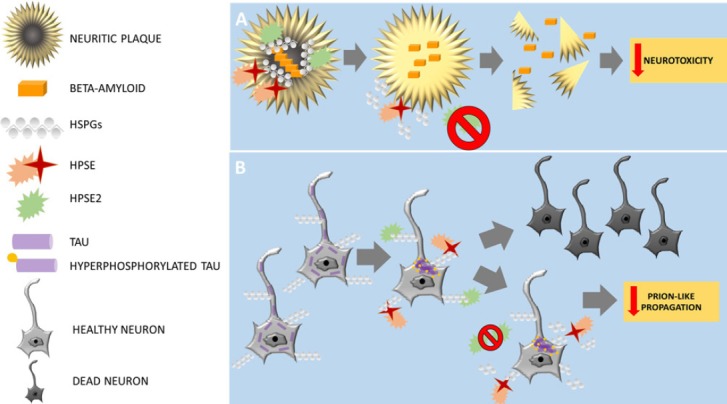
Schematic representation of main Alzheimer's disease pathology and their proposed results after blocking HPSE2 at neuritic plaques (A) and neurons with neurofibrillary tangles (B).
HPSE: Heparanase; HSPGs: Heparan sulfate proteoglycans.
Heparanase (HPSE) and HPSE2: HPSE is an endo-D-glucuronidase that cleaves specific linkages in the structure of HS, yielding biologically active fragments. By altering HSPGs on the cell surface, HPSE is thought to regulate the cellular response to external stimuli. HPSE takes part in multiple functions in cancer, inflammation, infections, diabetes, and atherosclerosis, and in terms of neurodegeneration (Garcia et al., 2016). In fact, recent studies have shown that overexpression of HPSE lowers the amyloid burden in transgenic mouse models of AD (Jendresen et al., 2015). HPSE2 is a homologue of HPSE that lacks HS-degrading activity, although it is able to interact with HS with high affinity (Levy-Adam et al., 2010). HPSE2 may act as a natural competitive inhibitor of HPSE due to its ability to associate with HPSE. Moreover, it seems to modulate its enzymatic activity and signaling properties.
HPSE2 as a potential therapeutic target: A recent work by Garcia et al. (2017) studied the expression of HPSE and HPSE2 in AD brains. The results from HPSE showed overexpression in neurodegenerated areas of AD brains and the amount of expression was well correlated with Braak & Braak stage. As we know that HS-Aβ joint may be useful for impeding Aβ degradation, this increase of expression may be a response from brain cells to reduce the existing AD pathogenesis by HS chains degradation. According to this hypothesis, the HPSE overexpression in this entity may be a consequence of the disease itself, rather than part of the cause. However, concerning HPSE2, the results were similar to HPSE. In addition, HPSE2 was also overexpressed in AD brains, mostly AD-stage-dependent. Histologically, both HPSE and HPSE2 were found to be associated with NPs and degenerated neurons with neurofibrillary tangles. Accordingly, HPSE2 may inhibit HPSE activity and, in consequence, may favour the progression of AD pathogenesis (Figure 1B). Consequently, blocking at different levels HPSE2 using target drugs should increase HPSE activity facilitating NPs degradation and thus decreasing neurotoxicity; and reducing prion-like propagation of hyperphosphorilated Tau in AD (Figure 1A and B).
Conclusions: HSPGs and HPSEs play a crucial role in AD pathogenesis. While HPSE activity may help to cease the progression of the disease by breaking down Aβ-HS deposits or blocking Tau intracellular fibril formation and propagation, HPSE2 appears to act as an inhibitor of HPSE. Thus, the blockade of HPSE by HPSE2 may interfere with its potential beneficial role in stopping AD pathogenesis. Furthermore, the lack of progress in AD investigation may be a consequence of the underestimation of the structural and molecular roles of HS molecules in AD pathogenesis. Preliminary studies are currently ongoing in our laboratory concerning HSPGs in order to better understand their roles in AD pathogenesis. Remarkably, no other molecules are capable of performing the same function, so both enzymes (HPSE and HPSE2) may become a potential target for therapeutical research. Although further research is needed in this field, the questions exposed before may open novel insights to AD pathogenesis and may lead to the development of innovative therapeutic strategies.
Footnotes
Contributor agreement: A statement of “Publishing Agreement” has been signed by an authorized author on behalf of all authors prior to publication.
Plagiarism check: This paper has been checked twice with duplication-checking software iThenticate.
Peer review: A double-blind and stringent peer review process has been performed to ensure the integrity, quality and significance of this paper.
References
- Apostolova LG. Alzheimer disease. Continuum (Minneap Minn) 2016;22:419–434. doi: 10.1212/CON.0000000000000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Freeman C, Jacobson GA, Small DH. Proteoglycans in the central nervous system: role in development, neural repair, and Alzheimer's disease. IUBMB Life. 2013;65:108–120. doi: 10.1002/iub.1118. [DOI] [PubMed] [Google Scholar]
- Garcia B, Garcia-Suarez O, Fernandez-Vega I, Vallina A, Astudillo A, Quiros LM. Heparanase and heparanase 2 display differently deregulation in neuroendocrine tumors, depending on their differentiation grade. Histol Histopathol. 2016;31:73–81. doi: 10.14670/HH-11-650. [DOI] [PubMed] [Google Scholar]
- Garcia B, Martin C, Garcia-Suarez O, Muniz-Alonso B, Ordiales H, Fernandez-Menendez S, Santos-Juanes J, Lorente-Gea L, Castanon S, Vicente-Etxenausia I, Pina Batista KM, Ruiz-Diaz I, Caballero-Martinez MC, Merayo-Lloves J, Guerra-Merino I, Quiros LM, Fernandez-Vega I. Upregulated expression of heparanase and heparanase 2 in the brains of Alzheimer's disease. J Alzheimers Dis. 2017;58:185–192. doi: 10.3233/JAD-161298. [DOI] [PubMed] [Google Scholar]
- Holmes BB, Diamond MI. Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J Biol Chem. 2014;289:19855–19861. doi: 10.1074/jbc.R114.549295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendresen CB, Cui H, Zhang X, Vlodavsky I, Nilsson LN, Li JP. Overexpression of heparanase lowers the amyloid burden in amyloid-beta precursor protein transgenic mice. J Biol Chem. 2015;290:5053–5064. doi: 10.1074/jbc.M114.600569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey R, Evans A. An unusual cause of jaundice in a patient with breast cancer. BMJ Case Rep 2014. 2014:bcr2014205764. doi: 10.1136/bcr-2014-205764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Adam F, Feld S, Cohen-Kaplan V, Shteingauz A, Gross M, Arvatz G, Naroditsky I, Ilan N, Doweck I, Vlodavsky I. Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. J Biol Chem. 2010;285:28010–28019. doi: 10.1074/jbc.M110.116384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer's disease. Lancet. 2016;388:505–517. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Horssen J, Kleinnijenhuis J, Maass CN, Rensink AA, Otte-Holler I, David G, van den Heuvel LP, Wesseling P, de Waal RM, Verbeek MM. Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol Aging. 2002;23:537–545. doi: 10.1016/s0197-4580(02)00010-6. [DOI] [PubMed] [Google Scholar]
- Zhang GL, Zhang X, Wang XM, Li JP. Towards understanding the roles of heparan sulfate proteoglycans in Alzheimer's disease. BioMed Res Int 2014. 2014:516028. doi: 10.1155/2014/516028. [DOI] [PMC free article] [PubMed] [Google Scholar]