

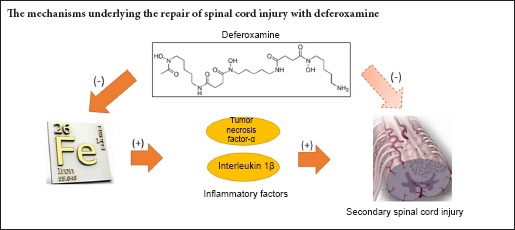
Keywords: nerve regeneration, spinal cord injury, deferoxamine, tumor necrosis factor-α, interleukin-1β; apoptosis, iron, anti-inflammatory, glial scar, proinflammatory, rats, motor function, lipid peroxidation, neural regeneration
Abstract
Deferoxamine, a clinically safe drug used for treating iron overload, also repairs spinal cord injury although the mechanism for this action remains unknown. Here, we determined whether deferoxamine was therapeutic in a rat model of spinal cord injury and explored potential mechanisms for this effect. Spinal cord injury was induced by impacting the spinal cord at the thoracic T10 vertebra level. One group of injured rats received deferoxamine, a second injured group received saline, and a third group was sham operated. Both 2 days and 2 weeks after spinal cord injury, total iron ion levels and protein expression levels of the proinflammatory cytokines tumor necrosis factor-α and interleukin-1β and the pro-apoptotic protein caspase-3 in the spinal cords of the injured deferoxamine-treated rats were significantly lower than those in the injured saline-treated group. The percentage of the area positive for glial fibrillary acidic protein immunoreactivity and the number of terminal deoxynucleotidyl transferase dUTP nick end labeling-positive cells were also significantly decreased both 2 days and 2 weeks post injury, while the number of NeuN-positive cells and the percentage of the area positive for the oligodendrocyte marker CNPase were increased in the injured deferoxamine-treated rats. At 14–56 days post injury, hind limb motor function in the deferoxamine-treated rats was superior to that in the saline-treated rats. These results suggest that deferoxamine decreases total iron ion, tumor necrosis factor-α, interleukin-1β, and caspase-3 expression levels after spinal cord injury and inhibits apoptosis and glial scar formation to promote motor function recovery.
Introduction
Spinal cord injury (SCI) can be divided into two major phases: the primary injury phase and the secondary injury phase. Investigation into the pathophysiology of secondary injury has revealed a number of proposed underlying mechanisms, including ischemia, edema, ion homeostasis changes, biochemical alterations, and metabolic disorders, that result in cell death and tissue damage (Ahn et al., 2006; Genovese et al., 2007; Oyinbo, 2011; Wang et al., 2011). However, the cascade of the inflammatory responses elicited by tumor necrosis factor-α (TNF-α) is considered to be central to secondary injury (Si et al., 2000; Leal-Filho, 2011; Oyinbo, 2011).
The influx of erythrocytes caused by hemorrhage during SCI provides a rich source of iron (Koszyca et al., 2002). Iron plays an important role in glutamate excitotoxicity in spinal cord motor neurons. Free iron can also lead to formation of reactive oxygen species through the Fenton reaction, producing free radicals that contribute to secondary injury (Emerit et al., 2001; Liu et al., 2003; Routhe and Moos, 2015).
Deferoxamine (DFO), an iron chelator, has a high affinity for trivalent iron ions and is used therapeutically for treatment of chronic hyperferremia and acute iron poisoning. Previous studies have demonstrated that treatment with DFO can also promote recovery after SCI in rats (Genovese et al., 2007; Liu et al., 2011). Treatment with DFO reduces lesion volume in vivo and increases regeneration in vitro. Chelation of iron appears to block secondary damage, and experiments aimed at elucidating the underlying mechanism revealed that DFO inhibits lipid peroxidation (Chojnacki and Weiss, 2008).
Recently, modulation of inflammatory responses in SCI has come under investigation (Li et al., 2016). Vigorous immune responses induced by SCI have been shown to contribute to secondary cell death (Zoppi et al., 2011). Production of TNF-α, primarily by macrophages and microglial cells, enhances the inflammatory response, upregulates intercellular adhesion molecules, and promotes adhesion and recruitment of leukocytes to the site of injury. These leukocytes release free radicals, proteases, and toxic oxidative metabolites, triggering a damage cascade that worsens inflammation.
Here, we explored the effect of DFO on levels of the major proinflammatory cytokines, TNF-α and interleukin-1β (IL-1β), to further investigate the understanding of the mechanisms underlying DFO-induced neural repair in SCI. TNF-α and IL-1β have been shown to induce neuronal apoptosis after SCI (Wang et al., 2005; de Rivero Vaccari et al., 2008). We detected whether DFO treatment reduced apoptosis in this rat model and whether DFO prevented glial scar formation.
Materials and Methods
Animals
Female Wistar rats (n = 54) weighing 230–250 g were provided by the Institute of Radiation Medicine, Chinese Academy of Medical Sciences, Tianjin, China. Rats were housed and maintained on a 12-hour light/dark cycle, and allowed free access to food and water. The rats were kept in standard cages, with five animals per cage.
The study protocol was approved by the Ethics Committee of Tianjin Medical University (Approval No. TMUAMEC 20170004). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animal (NIH Publication No. 85-23, revised 1986), and “Consensus Author Guidelines on Animal Ethics and Welfare” produced by the international Association for Veterinary Editors (IAVE).
The 54 rats were randomly allocated to three different groups containing 18 animals per group: sham group (sham-operated), injury group (SCI + saline-treated), and injury + DFO group (SCI + DFO-treated). Within each group, 3 rats were used for iron detection at 2 days and 3 more rats were used at 2 weeks post-SCI; 3 rats were used for hematoxylin and eosin staining and glial fibrillary acidic protein (GFAP), NeuN, 2’,3’-cyclic-nucleotide 3’-phosphodiesterase (CNPase), and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) detection assays at 2 days post-SCI, and 3 more rats for the same assays at 8 weeks post-SCI; and 3 rats were used for western blot assays at 2 days and 3 more rats at 2 weeks post-SCI.
SCI and intervention
Rats were anesthetized by intraperitoneal injection with 10% chloral hydrate (0.3 mL/100 g body weight). SCI models were established using a modification of Allen's method (Koozekanani et al., 1976). Briefly, a laminectomy was performed at the T9–10 thoracic vertebrae level to expose the spinal cord at T10. SCI was induced by New York University Impactor device (NYU, New York, NY, USA) using a 10 g-rod and a 25-mm drop height. The hind limbs of the rats exhibited involuntary spasms and the tails wriggled, indicating that the injury was consistent with the criteria of SCI in this model. After surgery, each rat was placed in a constant-temperature cage until fully recovered from the anesthesia. During the postoperative period, the bladder of each rat was manually voided twice daily until the bladder control reflex was restored.
The sham group was subjected only to laminectomy. The injury + DFO group was subjected to SCI and then treated with intraperitoneal injections from day 0 to day 7 with DFO (100 mg/kg per day; Novartis, Basel, Switzerland; 500 mg dissolved in 5 mL of 0.9% normal saline). Another control group (injury group) was also subjected to SCI, but was treated with intraperitoneal injections of saline (1 mL/kg).
Histological analysis
Rats were sacrificed at 2 days or 56 days post-SCI. Two rats randomly selected from each group were intracardially perfused with 4% paraformaldehyde under anesthesia. The spinal cord, including the entire T9–10 segment, was dissected and fixed in 4% paraformaldehyde solution. The specimens were embedded in paraffin and cut into 5-μm cross sections using a cryostat. The sections were stained with hematoxylin and eosin. The histopathological analysis was performed by a pathologist who was blinded to the experimental design. The number of nuclei was calculated to show the pathological differences among the groups.
Immunohistochemistry and immunofluorescence
Rats were sacrificed 2 days or 8 weeks post-SCI. For immunohistochemical staining, paraffin sections (5 μm) from each specimen were deparaffinized with xylene (two times for 30 minutes each), rehydrated in graded concentrations of ethanol (twice exposures to each of the following concentrations: 100%, 95%, 85%, and 75%), and washed in phosphate-buffered saline (PBS) for 5 minutes. The sections were then incubated with a rabbit anti-rat NeuN monoclonal antibody (1:500; Abcam, Branford, CT, USA) to detect neurons or a rabbit anti-rat CNPase monoclonal antibody (1:100; Proteintech Rosemont, IL, USA) to detect oligodendrocytes overnight at 4°C. After being washed in PBS for 5 minutes, the sections were incubated with goat anti-rabbit IgG (1:200; Beyotime, Haimen, Jiangsu Province, China) for 30 minutes at 37°C. Subsequently, the sections were incubated with avidin-biotin-peroxidase (Beyotime) for 45 minutes at 37°C and then transferred to a solution containing 30% hydrogen peroxide in 0.05 M PBS containing 1 drop of 3,3’-diaminobenzidine (0.0125 g/25 mL) for 10 minutes at 37°C. After being washed in PBS, the sections were dehydrated in series of increasing concentrations of ethanol (25%, 50%, 80%, 100%; twice at each concentration), washed twice with xylene, and then mounted onto glass slides for microscopic analysis. Image J software (version 2.1.4.7; National Institutes of Health, Bethesda, MD, USA) was used to assess the percentages of the areas in sections that were immunopositive.
For immunofluorescence, the sections were rehydrated as described above and incubated with a primary antibody (polyclonal goat anti-rat GFAP, 1:400; Abcam) overnight at 4°C. The next day, the sections were washed twice for 5 minutes each in PBS and incubated with a Cy3-labeled donkey anti-goat IgG secondary antibody (1:200; Beyotime) for 60 minutes at room temperature. PBS was used to wash the sections, and nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI, 1 g/mL; Biosharp, Hefei, Anhui Province, China) for 5 minutes. The sections were washed, and one drop of anti-fade mounting medium (Solarbio, Beijing, China) was placed on each slide, and a coverslip was placed on top of each sample. All sections were photographed using an inverted fluorescence microscope (IX71, Olympus, Tokyo, Japan).
Western blot assay
Rats were sacrificed 2 days or 2 weeks post-SCI. After being anesthetized, the rats were intracardially perfused with 9% saline (300 mL per rat; 4°C). Control and injured spinal cord tissue (0.5 cm above and below the site of injury) was dissected, frozen in liquid nitrogen, and stored at −80°C. After being homogenized, tissue samples were centrifuged at 12,000 × g for 15 minutes at 4°C. The supernatants were collected, and total protein was extracted from tissues and quantified using a Bicinchoninic Acid Protein Assay kit according to the manufacturer's instructions (Wanleibio; Shenyang, Liaoning Province, China). Proteins were resolved on 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% fat-free milk in PBS for 60 minutes and then incubated overnight at 4°C with the following primary antibodies: anti-IL-1β (1:500; Wanleibio), anti-TNF-α (1:500; Wanleibio), anti-caspase-3 (1:500; Wanleibio), and anti-β-actin (1:10,000; Wanleibio). The membranes were then washed with PBS and subsequently incubated at 37°C for 45 minutes with goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibodies (1:10,000; Wanleibio). Bands were visualized using chemiluminescence on X-ray film (Kodak, Rochester, NY, USA). Band grayscale values were quantified using Gel-Pro Analyzer software (Media Cybernetics, Rockville, MD, USA), and all grayscale values were normalized to that of β-actin. All assays were performed in triplicate.
Total iron detection
Spinal cord samples were collected as described above and homogenized under ice-cold conditions. Iron levels of spinal cords at 2 days and 2 weeks post-SCI were detected by following the tissue iron assay kit instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu Province, China). All assays were performed in triplicate.
TUNEL assay
Rats were sacrificed 2 days or 2 weeks post-SCI. The TUNEL assay was performed using an In Situ Cell Death Detection kit (Roche, USA) according to the manufacturer's recommended instructions. Briefly, spinal cord sections were rehydrated and incubated for 15–30 minutes at 37°C with the Proteinase K working solution. Subsequently, the slides were rinsed twice with PBS. The Converter-POD solution (50 μL) was added to the sections, which were then incubated in a humidified chamber for 30 minutes at 37°C and washed three times with PBS. The 3,3’-diaminobenzidine substrate (50–100 μL) was added to each sample, which was then incubated for 10 minutes at 25°C and washed three times. The samples were mounted under glass coverslips using glycerol.
Behavioral evaluation
Recovery of locomotor function post-SCI was evaluated using the Basso, Beattie, Bresnahan (BBB) locomotor rating scale (Barros Filho and Molina, 2008). This scale ranges from 0 to 21, with complete paralysis scored 0 and normal locomotor function scored 21. Each rat was evaluated weekly post-SCI by two independent observers blinded to the experimental design. The rats were allowed to move freely in an open field apparatus (1 m × 1 m), and their movements were recorded for 5 minutes. The BBB scores were determined 0, 7, 14, 21, 28, 35, 42, 49, and 56 days post-SCI to assess locomotor recovery.
Statistical analysis
All data are presented as the mean ± SEM. All statistical analyses were conducted using SPSS 19.0 software (IBM Corporation, NY, USA). Significant differences among groups were assessed by one-way analysis of variance followed by Tukey's post hoc test. P-values less than 0.05 were considered statistically significant.
Results
DFO treatment blocked the SCI-induced increase in iron levels
Iron overload in injured spinal cord tissue plays a detrimental role in secondary injury. To analyze changes in iron concentrations post-SCI, iron levels were analyzed in rats from the sham, injury, and injury + DFO groups at 2 days and 2 weeks after injury (Figure 1). Compared with those in the sham group, iron levels significantly increased in the injury group 2 and 14 days post-SCI (P < 0.01, P < 0.001). By contrast, iron levels in the injured rats treated with DFO were not significantly different from those in the sham group, but were significantly lower than those in saline-treated SCI rats 2 days and 2 weeks post-SCI (P < 0.01 and P < 0.001, respectively). Thus, DFO exhibited a strong efficacy to maintain the iron level of the injured spinal cord at a level similar to that of uninjured tissue.
Figure 1.
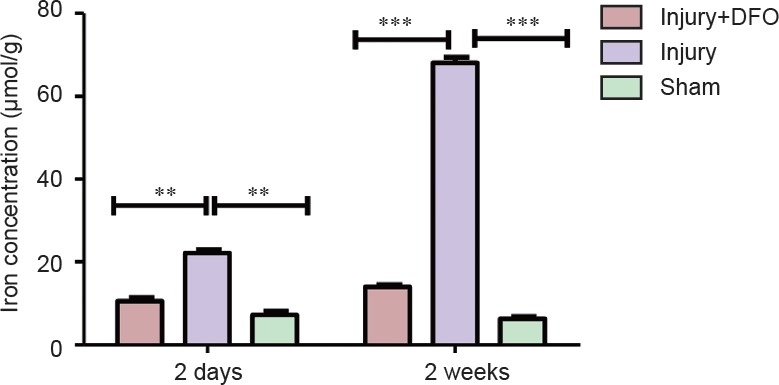
Deferoxamine (DFO) effects on total iron levels in rats with spinal cord injury.
Compared with that in sham-operated rats, the iron level at the injury site is significantly higher 2 days after the damage (**P < 0.01). DFO-treated rats show iron levels markedly lower than those in injured saline-treated rats and similar to those in the sham-operated rats, especially 2 weeks post injury (***P < 0.001). Data are expressed as the mean ± SEM (n = 3 per group, one-way analysis of variance followed by Tukey's post hoc test). DFO: Deferoxamine.
DFO treatment blocked the SCI-induced increase in IL-1β and TNF-α protein expression
TNF-α and IL-1β, as key proinflammatory cytokines, play a dominant role in inflammatory reactions during the secondary injury phase. To assess the anti-inflammatory effects of DFO, we analyzed protein expression of TNF-α and IL-1β 2 days and 2 weeks post-SCI (Figure 2). We found that TNF-α and IL-1β protein levels were significantly increased 2 days and 2 weeks post-SCI (P < 0.001). By contrast, injured rats treated with DFO showed TNF-α and IL-1β protein levels similar to those in sham-operated rats and significantly lower than those in saline-treated injured rats. These results suggested a prolonged inhibitory effect of DFO on the expression of TNF-α and IL-1β during both the acute and chronic phases post-SCI (P < 0.001) and provided evidence for the effective anti-inflammatory activity of DFO after SCI.
Figure 2.

DFO effects on TNF-α, IL-1β, and caspase-3 protein expression levels in the injured spinal cord of rats.
Expression profiles for the pro-inflammatory cytokines IL-1 β (A, B) and TNF-α (C, D). Expression was analyzed by western blot assays 2 days and 2 weeks after spinal cord injury. Compared with the injury group, DFO-treated animals have significantly lower protein levels of IL-1β and TNF-α (***P < 0.001) but similar levels to those in the sham-operated rats 2 days and 2 weeks post injury. Data are expressed as the mean ± SEM (n = 3 per group, one-way analysis of variance followed by Tukey's post hoc test). Grayscale values of the immunopositive bands were analyzed using ImageJ and normalized to β-actin. DFO: Deferoxamine; TNF-α: tumor necrosis factor-α; IL-1β: interleukin 1β.
DFO ameliorated SCI-induced pathological morphology
To determine the effects of DFO on the histological changes post-SCI, hematoxylin and eosin staining was conducted 2 days and 8 weeks post injury on tissue obtained from each group (Figure 3). Compared with spinal cord tissue from rats in the sham group, tissue from both DFO- and saline-treated rats with SCI showed obvious morphological changes, leaked erythrocytes, and severe spinal cord structural disorder 2 days post injury. However, 8 weeks post-SCI, we observed a substantial and significant increase in cell number (P < 0.001 compared with sham-operated rats) as well as increased neovascularization in the injury group, whereas neovascularization in injured rats treated with DFO was decreased.
Figure 3.

DFO effects on pathological morphology in injured spinal cord of rats.
Images of hematoxylin and eosin stained transverse spinal cord sections at the center of the injury were obtained using an inverted fluorescence microscope. Scale bars: 50 μm. (A–E) Injured spinal cord tissue sections 2 days post injury in the sham (A), injury (B) and injury + DFO groups (C), and at 8 weeks post injury in the injury (D) and injury + DFO groups (E). (A) Sham group shows normal spinal cord and structure. The injury (B) and injury + DFO groups (C) reveal damaged structure and hemorrhagic spinal cord 2 days post injury. (F) At 8 weeks post injury, the injury group presents considerable cell proliferation and new blood vessel formation, whereas cell proliferation is low in the injury + DFO group (***P < 0.001). Data are expressed as the mean ± SEM (n = 3 per group, one-way analysis of variance followed by Tukey's post hoc test). DFO: Deferoxamine.
DFO treatment reduced glial scar formation post-SCI
To analyze the effects of DFO on scar formation post-SCI, we examined the expression of GFAP, a major component of the scar matrix (Figure 4). Compared with that in the injury group, the percentage of the GFAP-positive area in the injured rats treated with DFO was significantly lower, and it was only slightly higher than that observed in the sham group 2 days post-SCI (P < 0.001). At 8 weeks post-SCI, glial scar formation was observed surrounding the crushed area in the injured saline-treated rats. The percentage of the area with glial scars was significantly lower in the DFO-treated rats than in the saline-treated rats (P < 0.001). These results indicated that DFO treatment decreased scar formation after SCI.
Figure 4.

DFO effects on glial scars in the injured spinal cord of rats.
Immunohistofluorescent images of GFAP-positive cells in transverse sections captured using an inverted fluorescence microscope at the center of injured spinal cord. Scale bars: 50 μm. (A–C) Results 2 days post injury. GFAP-positive (+) cells are red, and cell nuclei are blue (DAPI) in sham (A), injury (B), and injury + DFO (C) groups. The GFAP(+) area in the injury group is substantially larger than that in the sham group, which is similar to that in the injury + DFO group. (B, C) At 8 weeks post injury, the percentage of the GFAP (+) area in each group is similar to that at 2 days post injury, with the percentages of the GFAP(+) areas in both the sham (image not showed) and injury + DFO (E) groups lower than that in the injury group (D). (F) Quantitation of the percentage of the area that is glial scar as assessed by the percentage of GFAP (+) cells. ***P < 0.001 for the indicated comparisons. Data are expressed as the mean ± SEM (n = 3 per group, one-way analysis of variance followed by Tukey's post hoc test). GFAP: Glial fibrillary acidic protein; DFO: deferoxamine.
DFO treatment protected neurons and increased expression of CNPase
Neurons are critical for the normal functioning of the central nervous system, and neuronal death results in permanent functional loss. The myelin sheath increases the speed of impulse propagation along myelinated neuronal fibers. In the sham group, a large number of NeuN-positive (+) cells were detected and appeared to have normal morphology (Figure 5). Although NeuN(+) cells were also present in the injury group 2 days post-SCI, they were smaller in size and appeared markedly degenerated. In comparison, the number of surviving neurons was higher in the DFO-treated rats 2 days post-SCI (P < 0.05). At 8 weeks post-SCI, few NeuN(+) cells were found in the injured saline-treated rats, and a large glial scar was observed. By contrast, numerous NeuN(+) cells were present at 8 weeks in rats treated with DFO (P < 0.01).
Figure 5.

DFO effects on the number of NeuN(+) neurons in injured spinal cord of rats.
(A–E) Images of immunohistochemistry sections obtained using an inverted fluorescence microscope. Scale bars: 100 μm. (A) Normal neurons are observed 2 days post injury in the sham group. (B) By contrast, the neurons appeared to have shrunken, the tissue structure is damaged, and there is inflammatory cell infiltration 2 days post injury in the injury group. (C) Residual neurons are observed in the injury + DFO group 2 days post injury. (D) However, there are fewer neurons in the injury group compared with injury + DFO group 8 weeks post injury. (E) In the injury + DFO group 8 weeks post injury, there is an evident restoration of neuronal number and morphology compared with injury group. (F) The number of NeuN-positive cells was counted in the three groups and found to be significantly different between the injured saline- and DFO-treated rats at both 2 days and 8 weeks post injury (*P < 0.05, **P < 0.01, ***P < 0.01 for the comparisons indicated). Data are expressed as the mean ± SEM (n = 3 per group, one-way analysis of variance followed by Tukey's post hoc test). DFO: Deferoxamine.
Compared with that from the sham group, spinal cord tissue derived from both DFO- and saline-treated injured rats displayed similar reductions in the percentage of area that was CNPase(+) 2 days post-SCI (P < 0.001). However, DFO treatment was associated with a significant increase in the CNPase(+) area at 8 weeks compared with that for rats in the injury group (P < 0.01; Figure 6). These results indicated that DFO had a protective effect on neuronal survival and maintenance of the myelin sheath post-SCI.
Figure 6.

DFO effects on the number of oligodendrocytes in injured spinal cord of rats (inverted fluorescence microscope).
(A–E) Spinal cord samples collected 2 days (A, B, C) and 8 weeks (D, E) post injury. Scale bars: 100 μm. The CNPase(+) area shows normal morphology in the sham group (A). The tissue structure is damaged, the percentage of the CNPase(+) area is lower compared with the sham group, and the CNPase(+) area is atrophic in the injury group (B). The CNPase(+) area in the sham group has a normal structure, while in the injury group (D), the tissue structure is damaged, with a lower percentage of the area that is CNPase(+). The percentage of the CNPase(+) area and the tissue structure in the CNPase(+) area of the injury + DFO group (E) is larger than that in injury group. (F) There are no differences in the percentages of CNPase(+) areas between the injured saline- and DFO-treated rats at 2 days, but these groups are significantly different at 8 weeks post injury (**P < 0.01, ***P < 0.001 for the comparisons indicated; mean ± SEM, n = 3 per group, one-way analysis of variance followed by Tukey's post hoc test). DFO: Deferoxamine.
DFO inhibited apoptosis post-SCI
To determine whether apoptosis was involved in the response to SCI, we analyzed caspase-3 protein expression and conducted a TUNEL assay (Figure 7). At 2 days post-SCI, caspase-3 expression was markedly and significantly increased in the injury group compared with that in the sham group. However, DFO-treated rats displayed slightly higher caspase-3 protein expression levels than the sham-operated rats and significantly lower expression than the injured saline-treated rats. Furthermore, in contrast to the continued high expression level observed in the injured saline-treated rats, caspase-3 levels in DFO-treated rats were comparable to those in the sham-operated rats 2 weeks post-SCI (Figure 7A, B). The TUNEL assay was conducted to confirm the differences in apoptosis levels among the treatment groups. Consistent with the caspase-3 results, a significantly higher number of TUNEL-positive cells was observed in the injury group relative to both the sham group, which had zero TUNEL-positive cells, and injury + DFO group, which showed a few scattered TUNEL-positive cells 2 days post-SCI. These results provided evidence that DFO treatment strongly inhibits apoptosis in injured spinal cord tissue.
Figure 7.

DFO effects on cell apoptosis in the injured spinal cord of rats.
(A, B) Caspase-3 levels are markedly increased in the injury group 2 days and 2 weeks post injury. DFO treatment (Injury + DFO) reduces the caspase-3 protein expression level compared with that in saline-treated injured spinal cord. Band intensity analysis reveals a significant difference between the injured saline- and DFO-treated rats (***P < 0.001 for the comparisons indicated). (C–F) The TUNEL assay results reveal neurons with normal morphology (black arrows) that are not positive for TUNEL in the sham group. However, 2 days post spinal cord injury, the number of TUNEL-positive cells (red arrows) increases markedly in the injury group (D) (***P < 0.001), whereas the number of TUNEL-positive cells is lower in the injury + DFO group (E). Scale bars: 100 μm. (F) Statistical analysis reveals that the DFO-treated rats have a significantly lower number of TUNEL-positive cells than the saline-treated injured rats 2 days post injury (**P < 0.01; mean ± SEM, n = 3 per group, one-way analysis of variance followed by Tukey's post hoc test). TUNEL: Terminal deoxynucleotidyl transferase dUTP nick endlabeling; DFO: deferoxamine. Deferoxamine.
DFO treatment promotee locomotor recovery after SCI
The BBB scale was used to evaluate the therapeutic effect of DFO on locomotor recovery 0, 7, 14, 21, 28, 35, 42, 49, and 56 days post-SCI (Figure 8). On day 0, the sham group received the highest BBB score possible (21), whereas both the injured groups scored the lowest possible (0), demonstrating the validity of this rat model of SCI. The scores increased in both injured groups over time. However, by 7 days post-SCI, there was a statistically significant difference in BBB scores between the injured DFO- and saline-treated rats (P < 0.05). At 56 days post-SCI, the mean BBB score was 14 ± 0.577 in the saline-treated rats and 18 ± 0.333 in the DFO-treated rats (P < 0.001). These results indicated that DFO exhibited therapeutic efficacy after SCI in rats.
Figure 8.
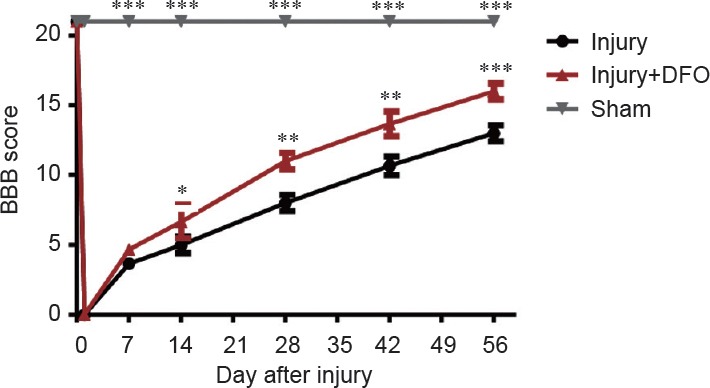
DFO effects on motor function in rats after spinal cord injury.
All groups exhibit increasing BBB scores over time. The first evaluation was conducted immediately after laminectomy in all groups. No statistically significant differences in BBB scores are initially observed between the injured saline- (Injury) and DFO-treated (Injury + DFO) rats (P > 0.05), whereas the BBB scores in the sham-operated rats were significantly different from both the injured saline- and DFO-treated rats (***P < 0.001). However, beginning at the second evaluation 2 weeks-post injury, the BBB scores for the injured saline- and DFO-treated rats were significantly different from each other (*P < 0.05; mean ± SEM, n = 3 per group, one-way analysis of variance followed by Tukey's post hoc test). DFO: Deferoxamine; BBB: Basso, Beattie, Bresnahan locomotor rating scale.
Discussion
Secondary SCI is an important process for early treatment of SCI (Huang et al., 2015; Young et al., 2015; Zhao et al., 2015). SCI-induced inflammatory responses play an important role in the secondary injury phase. Inflammatory cytokines and mediators are the basic effector molecules that participate in and control the inflammatory response. TNF-α, IL-1β, and other inflammatory mediators infiltrate the injured spinal cord tissue, thereby causing a strong inflammatory cascade response (Yin et al., 2012).
Expression of these cytokines and the resulting inflammatory responses are the initiating factors that lead to neuronal apoptosis (Paulson et al., 2013), which is the key form of secondary injury after the initial insult. Therefore, control of the initial inflammatory response is very important for treating SCI and preventing further injury (David et al., 2012). Thus, identification of therapeutic means to inhibit inflammatory responses after SCI could help to reduce secondary SCI and protect the injured spinal cord.
As an important regulator of neuroinflammation, TNF-α is produced by many immune and non-immune cells, including macrophages, T cells, mast cells, granulocytes, natural killer cells, fibroblasts, neurons, and has been shown to play important roles in signal transduction and posttraumatic inflammatory responses (Esposito and Cuzzocrea, 2011; Olmos and Llado, 2014).
Harrington et al. (2005) demonstrated increased expression of TNF-α and its receptors in neurons 6 hours post-SCI. Previous studies have shown that rapid and sustained high expression of TNF-α increases the inflammatory response and promotes secondary injury (Kwon et al., 2012). Thus, preventing the upregulation of TNF-α during the early stage of SCI could reduce inflammatory responses at the injured site. Macrophages are triggered by central nervous system injury and contribute to secondary injury and functional damage (David and Kroner, 2011).
Post-SCI, macrophage phagocytosis of myelin and erythrocytes, released following hemorrhage and tissue damage, results in high iron levels in cells, which could induce overexpression of TNF-α and transformation of macrophages from the M2 to the M1 phenotype and then aggravate secondary injury (Kroner et al., 2014). Importantly, M2 macrophages reduce local inflammation and protect the spinal cord from further injury (Kigerl et al., 2009), whereas M1 macrophages aggravate the inflammatory response and promote secondary injury, suggesting a potential underlying mechanism explaining the positive effects of DFO treatment. As an iron chelator, DFO may reduce the iron ion level, and our results showed that DFO treatment significantly reduced the iron concentration at the site of SCI, especially 2 weeks post injury, consistent with previous studies (de Castro et al., 1999).
Our data also confirmed that DFO inhibited the expression of TNF-α, as well as IL-1β. Thus DFO reduced the concentration of iron ions in the injured segment and inhibited TNF-α expression, which may block the transformation of M2 macrophages to the M1 phenotype, thereby reducing inflammation and protecting against further SCI.
Our results demonstrated that DFO reduced the SCI-induced expression of IL-1β, as well as caspase-3, and decreased the neuronal apoptosis observed after SCI. Interleukin-1β has been shown to inhibit nerve regeneration after SCI (David and Kroner, 2011), and IL-1 receptor antagonists reduce neuronal apoptosis, alleviating damage after injury and serving as protective agents (Nesic et al., 2001).
After injury, pro-inflammatory cytokines induce primary inflammation and promote tissue remodeling. Soluble alarmins and high-mobility group box 1 are expressed, activating toll-like receptor-4 and the receptor for advanced glycation end-products. These receptors activate signaling pathways that increase expression of IL-1β, activate the nuclear factor-κB cell signaling pathway, and lead to secondary inflammation in SCI. This vicious cycle results in persistent inflammation (Didangelos et al., 2016).
Previous studies have demonstrated that DFO can inhibit nuclear factor-κB signaling pathways and reduce infiltration of neutrophils into the spinal cord, thereby diminishing the local inflammatory response (Paterniti et al., 2010). However, the present study did not address the effect of DFO on inflammatory factors, which are the major mediators of the inflammatory response. Therefore, in future studies, it will be important to determine DFO effects on these inflammatory factors to better understand the protective mechanisms underlying the benefits of DFO after SCI. Our results demonstrated that DFO decreased IL-1β expression and reduced the inflammatory response after injury, decreasing neuronal apoptosis and protecting against further SCI. The mechanism by which DFO reduces the inflammatory response may be through inhibition of high mobility group box 1 production or decreased expression of toll-like receptor-4 and the receptor for advanced glycation end-products, interrupting the vicious inflammatory response cycle and protecting against SCI. Further experiments will be required to test these hypotheses.
DFO directly decreased iron levels at the injured site and reduced apoptosis. The underlying relationship of reduced iron levels and apoptosis may be affected by myriad factors. For example, reactive oxygen species, induced by iron, usually leads to cell death upon injury; apoptosis, which is a form of programmed cell death, is reduced by DFO treatment post-SCI (Fankhauser et al., 2000). Additionally, TNF-α and IL-1β have been implicated as signals to induce apoptosis in neurons, astrocytes, and oligodendrocytes after SCI. The reduction of proinflammatory cytokines after DFO treatment abolishes the apoptotic cascade (Ehrlich et al., 1999). DFO promotes neuronal survival, as we showed in the present study using a TUNEL assay. The neuroprotective effect can also be observed as the protection of oligodendrocytes, which create the neuronal myelin sheaths (Takahashi et al., 2003). Therefore, DFO showed a protective effect on nerve function following SCI. Recent studies indicate that other forms of cell death are also found after SCI, such as necroptosis, ferroptosis and autophagy. The relationship between iron levels and other types of cell death in SCI remains to be elucidated.
The glial scar, which serves as a physiological barrier that blocks neuronal axon regeneration and inhibits functional recovery, is associated with astrocyte proliferation. The glial scar also contributes to the inhibition of the posttraumatic cavitation and inflammation in the early stage after SCI. However, only moderate astrocyte proliferation may prevent the spread of harmful factors in the early stage of SCI and provide protection for SCI repair. Excessive activated astrocytes will eventually form a dense glial scar, which obstructs the extension of the regenerated axons. Iron, as an essential factor involved in cell proliferation, is chelated by DFO and thereby inhibits astrocyte proliferation. The anti-proliferation effect of iron chelators has been well studied. DFO causes the arrest of the G1/S phases of the cell cycle and inhibits the gliogenesis associated with proliferation of activated astrocytes (Yu et al., 2007). Thus, DFO might effectively control glial scar formation by inhibiting the proliferation of astrocytes. The present study found that DFO treatment reduced the expression of proinflammatory cytokines, which are also inducers of glial scar formation.
In summary, DFO exhibited treatment efficacy in an animal model of SCI by ameliorating pathophysiological morphology and improving locomotor function. The underlying mechanism may involve reduction of local iron concentrations resulting from hemorrhage in the injured spinal cord and subsequent decreases in TNF-α and IL-1β levels released by macrophages and microglia as a result of the reduced iron levels. These changes in cytokine expression may facilitate macrophage shifts from the M1 to the M2 phenotype, thereby inhibiting inflammatory reactions during secondary SCI. Consequent reductions in levels of apoptosis post-SCI and rescue of remaining neurons and oligodendrocytes likely underlie both the ameliorated pathophysiology and recovery of locomotor function. This study provided a potential new pharmacologic strategy for treatment of SCI, began to elucidate the detailed underlying mechanism for this therapy, and provided evidence indicating that further preclinical and clinical investigation for the therapeutic use of DFO in SCI is warranted.
Footnotes
Funding: This study was supported by the National Natural Science Foundation of China, No. 81672171, 81330042; the International Cooperation Program of National Natural Science Foundation of China, No. 81620108018; a grant from the Ministry of Science and Technology of China, No. 2014DFR31210; a grant from the Tianjin Science and Technology Committee of China, No. 13RCGFSY19000, 14ZCZDSY00044; the Youth Foundation of Tianjin Medical University General Hospital of China, No. ZYYFY2015008.
Conflicts of interest: None declared.
Research ethics: The study protocol was approved by the Ethics Committee of Tianjin Medical University (Approval No. TMUAMEC 20170004). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animal (NIH Publication No. 85-23, revised 1986), and “Consensus Author Guidelines on Animal Ethics and Welfare” produced by the International Association for Veterinary Editors (IAVE). The article was prepared in accordance with the “Animal Research: Reporting of In Vivo Experiments Guidelines” (ARRIVE Guidelines).
Contributor agreement: A statement of “Publishing Agreement” has been signed by an authorized author on behalf of all authors prior to publication.
Plagiarism check: This paper has been checked twice with duplication-checking software iThenticate.
Peer review: A double-blind and stringent peer review process has been performed to ensure the integrity, quality and significance of this paper.
Copyedited by Smith T, Pack M, Wang J, Li CH, Qiu Y, Song LP, Zhao M
References
- Ahn YH, Lee G, Kang SK. Molecular insights of the injured lesions of rat spinal cords: Inflammation, apoptosis, and cell survival. Biochem Biophys Res Commun. 2006;348:560–570. doi: 10.1016/j.bbrc.2006.07.105. [DOI] [PubMed] [Google Scholar]
- Barros Filho TE, Molina AE. Analysis of the sensitivity and reproducibility of the Basso, Beattie, Bresnahan (BBB) scale in Wistar rats. Clinics (Sao Paulo) 2008;63:103–108. doi: 10.1590/s1807-59322008000100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnacki A, Weiss S. Production of neurons, astrocytes and oligodendrocytes from mammalian CNS stem cells. Nat Protoc. 2008;3:935–940. doi: 10.1038/nprot.2008.55. [DOI] [PubMed] [Google Scholar]
- David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- David S, Zarruk JG, Ghasemlou N. Inflammatory pathways in spinal cord injury. Int Rev Neurobiol. 2012;106:127–152. doi: 10.1016/B978-0-12-407178-0.00006-5. [DOI] [PubMed] [Google Scholar]
- de Castro R, Jr, Alcock NW, McAdoo DJ. Sampling of low molecular weight iron by microdialysis following spinal cord injury. J Neurosci Res. 1999;57:735–739. [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28:3404–3414. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, Puglia M, Iberl M, Sanchez-Bellot C, Roschitzki B, Bradbury EJ. High-throughput proteomics reveal alarmins as amplifiers of tissue pathology and inflammation after spinal cord injury. Sci Rep. 2016;6:21607. doi: 10.1038/srep21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich LC, Peterson PK, Hu S. Interleukin (IL)-1beta-mediated apoptosis of human astrocytes. Neuroreport. 1999;10:1849–1852. doi: 10.1097/00001756-199906230-00009. [DOI] [PubMed] [Google Scholar]
- Emerit J, Beaumont C, Trivin F. Iron metabolism, free radicals, and oxidative injury. Biomed Pharmacother. 2001;55:333–339. doi: 10.1016/s0753-3322(01)00068-3. [DOI] [PubMed] [Google Scholar]
- Esposito E, Cuzzocrea S. Anti-TNF therapy in the injured spinal cord. Trends Pharmacol Sci. 2011;32:107–115. doi: 10.1016/j.tips.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Fankhauser C, Friedlander RM, Gagliardini V. Prevention of nuclear localization of activated caspases correlates with inhibition of apoptosis. Apoptosis. 2000;5:117–132. doi: 10.1023/a:1009672411058. [DOI] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Crisafulli C, Esposito E, Di Paola R, Muia C, Di Bella P, Meli R, Bramanti P, Cuzzocrea S. Combination of dexamethasone and etanercept reduces secondary damage in experimental spinal cord trauma. Neuroscience. 2007;150:168–181. doi: 10.1016/j.neuroscience.2007.06.059. [DOI] [PubMed] [Google Scholar]
- Harrington JF, Messier AA, Levine A, Szmydynger-Chodobska J, Chodobski A. Shedding of tumor necrosis factor type 1 receptor after experimental spinal cord injury. J Neurotrauma. 2005;22:919–928. doi: 10.1089/neu.2005.22.919. [DOI] [PubMed] [Google Scholar]
- Huang H, Mao G, Chen L, AL. Progress and challenges with clinical cell therapy in neurorestoratology. J Neurorestoratol. 2015;3:91–95. [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koozekanani SH, Vise WM, Hashemi RM, McGhee RB. Possible mechanisms for observed pathophysiological variability in experimental spinal cord injury by the method of Allen. J Neurosurg. 1976;44:429–434. doi: 10.3171/jns.1976.44.4.0429. [DOI] [PubMed] [Google Scholar]
- Koszyca B, Manavis J, Cornish RJ, Blumbergs PC. Patterns of immunocytochemical staining for ferritin and transferrin in the human spinal cord following traumatic injury. J Clin Neurosci. 2002;9:298–301. doi: 10.1054/jocn.2001.0969. [DOI] [PubMed] [Google Scholar]
- Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 2014;83:1098–1116. doi: 10.1016/j.neuron.2014.07.027. [DOI] [PubMed] [Google Scholar]
- Kwon BK, Ghag A, Dvorak MF, Tetzlaff W, Illes J. Expectations of benefit and tolerance to risk of individuals with spinal cord injury regarding potential participation in clinical trials. J Neurotrauma. 2012;29:2727–2737. doi: 10.1089/neu.2012.2550. [DOI] [PubMed] [Google Scholar]
- Leal-Filho MB. Spinal cord injury: From inflammation to glial scar. Surg Neurol Int. 2011;2:112. doi: 10.4103/2152-7806.83732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XG, Lin XJ, Du JH, Xu SZ, Lou XF, Chen Z. Combination of methylprednisolone and rosiglitazone promotes recovery of neurological function after spinal cord injury. Neural Regen Res. 2016;11:1678–1684. doi: 10.4103/1673-5374.193250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Liu J, Sun D, Alcock NW, Wen J. Spinal cord injury increases iron levels: catalytic production of hydroxyl radicals. Free Radic Biol Med. 2003;34:64–71. doi: 10.1016/s0891-5849(02)01184-x. [DOI] [PubMed] [Google Scholar]
- Liu J, Tang T, Yang H. Protective effect of deferoxamine on experimental spinal cord injury in rat. Injury. 2011;42:742–745. doi: 10.1016/j.injury.2010.08.028. [DOI] [PubMed] [Google Scholar]
- Nesic O, Xu GY, McAdoo D, High KW, Hulsebosch C, Perez-Pol R. IL-1 receptor antagonist prevents apoptosis and caspase-3 activation after spinal cord injury. J Neurotrauma. 2001;18:947–956. doi: 10.1089/089771501750451857. [DOI] [PubMed] [Google Scholar]
- Olmos G, Llado J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm 2014. 2014:861231. doi: 10.1155/2014/861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyinbo CA. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp (Wars) 2011;71:281–299. doi: 10.55782/ane-2011-1848. [DOI] [PubMed] [Google Scholar]
- Paterniti I, Mazzon E, Emanuela E, Paola RD, Galuppo M, Bramanti P, Cuzzocrea S. Modulation of inflammatory response after spinal cord trauma with deferoxamine, an iron chelator. Free Radic Res. 2010;44:694–709. doi: 10.3109/10715761003742993. [DOI] [PubMed] [Google Scholar]
- Paulson TA, Goosey-Tolfrey VL, Lenton JP, Leicht CA, Bishop NC. Spinal cord injury level and the circulating cytokine response to strenuous exercise. Med Sci Sports Exerc. 2013;45:1649–1655. doi: 10.1249/MSS.0b013e31828f9bbb. [DOI] [PubMed] [Google Scholar]
- Routhe LJ, Moos T. Handling iron in restorative neuroscience. Neural Regen Res. 2015;10:1558–1559. doi: 10.4103/1673-5374.165316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Q, Nakamura Y, Kataoka K. A serum factor enhances production of nitric oxide and tumor necrosis factor-alpha from cultured microglia. Exp Neurol. 2000;162:89–97. doi: 10.1006/exnr.2000.7334. [DOI] [PubMed] [Google Scholar]
- Takahashi JL, Giuliani F, Power C, Imai Y, Yong VW. Interleukin-1beta promotes oligodendrocyte death through glutamate excitotoxicity. Ann Neurol. 2003;53:588–595. doi: 10.1002/ana.10519. [DOI] [PubMed] [Google Scholar]
- Wang CY, Chen JK, Wu YT, Tsai MJ, Shyue SK, Yang CS, Tzeng SF. Reduction in antioxidant enzyme expression and sustained inflammation enhance tissue damage in the subacute phase of spinal cord contusive injury. J Biomed Sci. 2011;18:13. doi: 10.1186/1423-0127-18-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Kong KM, Qi WL, Ye WL, Song PS. Interleukin-1 beta induction of neuron apoptosis depends on p38 mitogen-activated protein kinase activity after spinal cord injury. Acta Pharmacol Sin. 2005;26:934–942. doi: 10.1111/j.1745-7254.2005.00152.x. [DOI] [PubMed] [Google Scholar]
- Yin X, Yin Y, Cao FL, Chen YF, Peng Y, Hou WG, Sun SK, Luo ZJ. Tanshinone IIA attenuates the inflammatory response and apoptosis after traumatic injury of the spinal cord in adult rats. PLoS One. 2012;7:e38381. doi: 10.1371/journal.pone.0038381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young W, AlZoubi ZM, Saberi H, Sharma A, Muresanu D, Feng S, Chen L, HH. Beijing declaration of international association of neurorestoratology. J Neurorestoratol. 2015;3:121–122. [Google Scholar]
- Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle. 2007;6:1982–1994. doi: 10.4161/cc.6.16.4603. [DOI] [PubMed] [Google Scholar]
- Zhao WT, Yuan HB, Li PP, Zhang HF, Liu MQ. Methylprednisolone for acute spinal cord injury: a meta-analysis of therapeutic efficacy and adverse reactions. Zhongguo Zuzhi Gongcheng Yanjiu. 2015;19:6868–6874. [Google Scholar]
- Zoppi S, Perez Nievas BG, Madrigal JL, Manzanares J, Leza JC, Garcia-Bueno B. Regulatory role of cannabinoid receptor 1 in stress-induced excitotoxicity and neuroinflammation. Neuropsychopharmacology. 2011;36:805–818. doi: 10.1038/npp.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]