

Abstract
Since low JAK2V617F allele burden (AB) has been detected also in healthy subjects, its clinical interpretation may be challenging in patients with chronic myeloproliferative neoplasms (MPNs). We tested 1087 subjects for JAK2V617F mutation on suspicion of hematological malignancy. Only 497 (45.7%) patients were positive. Here we present clinical and laboratory parameters of a cohort of 35/497 patients with an AB ≤ 3%.
Overall, 22/35 (62.9%) received a WHO-defined diagnosis of MPN and in 14/35 cases (40%) diagnosis was supported by bone marrow (BM) histology (‘’Histology-based’’ diagnosis). In patients that were unable or refused to perform BM evaluation, diagnosis relied on prospective clinical observation (12 cases, 34.3%) and molecular monitoring (6 cases, 17.1%) (‘’Clinical-based’’ or ‘’Molecular-based’’ diagnosis, respectively). In 11/35 (31.4%) patients, a low JAK2V617F AB was not conclusive of MPN. The probability to have a final hematological diagnosis (ET/PV/MF) was higher in patients with thrombocytosis than in patients with polyglobulia (73.7% vs 57.1%, respectively). The detection of AB ≥ 0.8% always corresponded to an overt MPN phenotype. The repetition of JAK2V617F evaluation over time timely detected the spontaneous expansion (11 cases) or reduction (4 cases) of JAK2V617F-positive clones and significantly oriented the diagnostic process.
Our study confirms that histology is relevant to discriminate small foci of clonal hematopoiesis with uncertain clinical significance from a full blown disease. Remarkably, our data suggest that a cut-off of AB ≥ 0.8% is very indicative for the presence of a MPN. Monitoring of the AB over time emerged as a convenient and non-invasive method to assess clonal hematopoiesis expansion.
Keywords: JAK2, V617F mutation, allele burden, myeloproliferative neoplasms, MPN
INTRODUCTION
In 2008, the World Health Organization (WHO) classification indicated the positivity of the JAK2V617F mutation as a major criterion for the diagnosis of chronic myeloproliferative neoplasms (MPNs), specifically Essential Thrombocytemia (ET), Polycythemia Vera (PV) and Myelofibrosis (MF) [1–7]. The JAK2V617F mutation is detected in around 50–60% of ET and MF patients and in most (95%) patients with PV [8, 9]. Also, the JAK2V617F mutation may be found in other hematological malignancies. Infrequent occurrence of this unique JAK2 mutation has been reported in chronic myelomonocytic leukemia (CMML), atypical or unclassified myeloproliferative disorder (MPD), myelodysplastic syndrome (MDS), systemic mastocytosis (SM), and chronic neutrophilic leukemia (CNL) [10–16]. A JAK2V617F allele burden (AB) above 50% identifies patients with a higher thrombotic risk, both in ET and in PV [17–22]. Conversely, a low AB seems to correlate with significantly shortened survival and leukemia-free survival in MF [23–25]. As a result, the determination of the mutation load is becoming a standard diagnostic procedure in most molecular laboratories, though WHO criteria do not specify a cut-off value for the diagnosis of a MPN. In the last years, the extensive and generalized use of molecular techniques with high sensitivity, specifically allele-specific Real-Time Quantitative Polymerase Chain Reaction (RQ-PCR), has significantly increased our ability to detect small mutated clones, with AB below 1% [26–32]. Many recent studies have shown that a small clonal hematopoiesis may be present also in otherwise healthy subjects at low level (0.03–1%) [33–38]. Accordingly, a study of the Myeloproliferative Neoplasms and Related Disorders European Network (MPN&MPNr-EuroNet) on 36 subjects carrying low JAK2V617F AB has further suggested that the detection of a small JAK2V617F-mutated clone cannot represent a sufficient evidence to establish malignant myeloproliferation [39]. As a result, the clinical interpretation of a low AB of the JAK2V617F mutation may be challenging. Here, we analyzed the results of JAK2V617F molecular tests performed at our Hematology Department in subjects with a suspected hematological disease over a 2-year period. Specifically, we analyzed clinical and laboratory data of patients with a suspected MPN that presented a low (≤ 3%) JAK2V617F mutation burden, with the aim to define the frequency and the significance of a low AB in the everyday clinical management.
RESULTS
Study plan
We tested 1087 subjects for JAK2V617F mutation due to clinical suspicion of hematological malignancy. A total of 716 (65.9%) out of 1087 tests were performed due to a suspect of classical MPN, including ET (299 cases, 41.8%), PV (272 cases, 38%), MF (133 cases, 18.6%) and MPN underling atypical splanchnic vein thrombosis (12 cases, 1.6%). The remaining 371 (34.1%) out of 1087 tests were performed on suspicion of Myeloproliferative Disease (MPDs; 23 cases) or other causes/not specified (348 cases) (data not shown).
Figure 1 depicts the study population and the study plan. Overall, 497 (45.7%) of the 1087 subjects that were tested for the JAK2V617F mutation resulted positive for the mutation.
Figure 1. Schematic representation of the study population and the study plan.
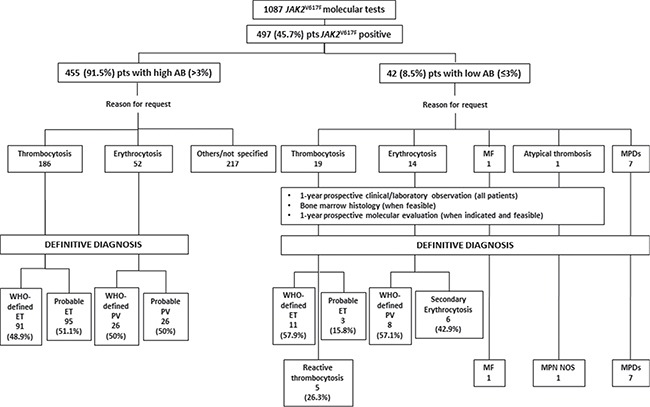
ET: Essential Thrombocythemia; PV: Polycythemia Vera; MF: primary Myelofibrosis; MPN NOS: Myeloproliferative Neoplasm Not Otherwise Specified; MPDs: Myeloproliferative Diseases.
A total of 455/497 (91.5%) patients had an AB > 3% (IC 95%: 88.75–93.71, p = 0.05): 52 (11.4%) were tested because of erythrocytosis, 186 (40.9%) because of thrombocytosis and 217 (47.7%) for other causes (specifically 94 suspicion of MF, 3 MPN underling atypical thrombosis and 120 others/not specified). Overall, the final diagnosis of these patients was: WHO-defined ET (91/186, 48.9%), probable ET (95/186, 51.1%), PV (26/52, 50%), probable PV (50%).
The remaining 42/497 (8.5%) patients had a low (0.1–3%, median 0.59%) AB (IC 95%; 0.62–1.15, p = 0.05). In most cases (30 patients, 71.4%), AB was below 1%, while only 8 (28.6%) patients had an AB above 2%. Overall, the final diagnosis of these patients was: WHO-defined MPDs (7 cases), WHO-defined ET/PMF-0 (11/19, 57.9%), probable ET (3/19, 15.8%), reactive thrombocytosis (5/19, 26.3%), PV (8/14, 57.1%), secondary erythrocytosis (6/14, 42.9%).
Here we present clinical and laboratory parameters of this cohort of 42 patients with an AB ≤ 3%.
Low AB patients suspected of MPD
By bone marrow (BM) histology, 7/42 (16.7%) patients had a confirmed diagnosis of a hematological disease that not included classical MPNs, specifically: refractory cytopenia with unilineage dysplasia (RCUD, 2 cases); refractory cytopenia with multilineage dysplasia (RCMD, 1 case); refractory anemia with excess blasts-1 (RAEB-1, 1 case); MDS with isolated del(5q) (1 case); chronic myelomonocytic leukemia (CMML, 2 cases). Median AB of these patients was 0.46% (range, 0.12–2.79%). No additional mutations in JAK2 exon 14, CALR and MPL genes were detected in these 7 patients.
Low AB patients suspected of MPN
Diagnostic workflow of the 35/42 patients with low AB and referred to our Institutions with a suspicion of classical MPNs is shown in Figure 2. Specifically, 19 patients were evaluated for suspected ET, 14 for probable PV, 1 for probable MF and 1 for MPN underling atypical thrombosis. Consequently, they were prospectively followed over time with clinical/laboratory data. In 2/35 cases, BM biopsy confirmed the suspect of a primary MF or MPN not otherwise specified (NOS) in one subject tested after a splanchnic vein thrombosis.
Figure 2. Diagnostic workflow of patients with suspected MPN and low JAK2V617F allele burden.
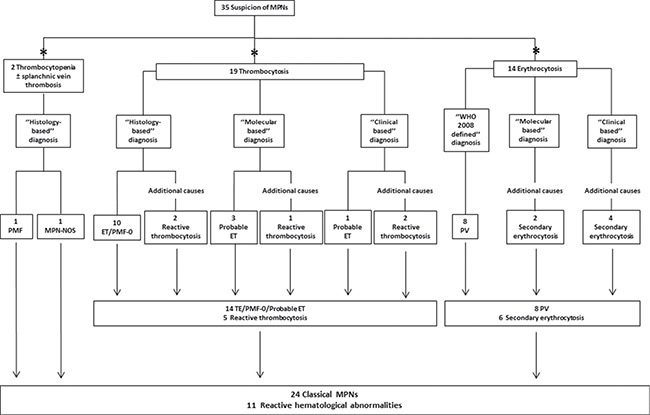
*Main hematological abnormality motivating the JAK2V617F evaluation. ‘’Histology-based’’ diagnosis was made when BM histology was available. In patients that were unable or refused to perform BM evaluation, prospective clinical observation and prospective molecular monitoring were crucial to direct diagnostic uncertainty, allowing to define a ‘’Clinical based’’ or ‘’Molecular based’’ diagnosis, respectively.
Prospective molecular monitoring was performed in 15/35 low AB patients at a 12-month follow-up A significant increase of JAK2V617F AB over time was observed in 11 cases (p < 0.05). Indeed, the median value of JAK2V617F AB at diagnosis and during follow-up was 0.49% (range, 0.12–2.98) and 1.3% (range, 0.28–9.2), respectively. In the remaining 4 cases, a slight decrease of JAK2V617F AB over time was registered (Figure 3).
Figure 3. JAK2V617F allele burden over time in patients with suspected essential thrombocytemia and polycythemia vera.
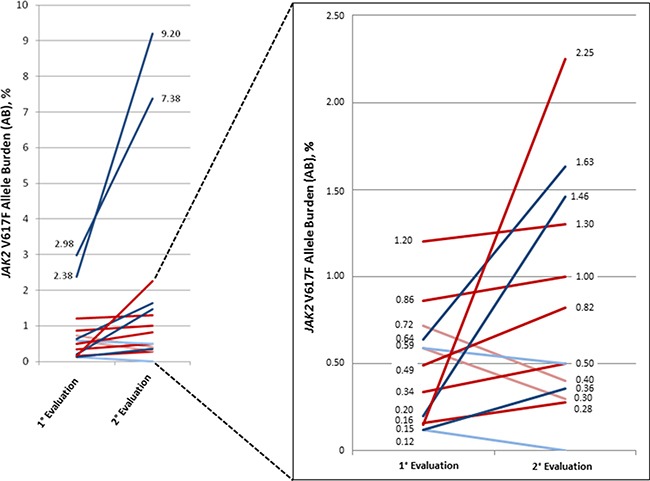
Fifteen patients received the second evaluation of JAK2V617F allele burden after a period of 12 months from the first mutational test. Dark red line: final diagnosis of PV. Light red line: final diagnosis of secondary polyglobulia. Dark blue line: final diagnosis of ET. Light blue line: final diagnosis of secondary thrombocytosis.
Among the 19/35 (54.3%) subjects with low AB that received JAK2V617F evaluation for thrombocytosis, 14 (73.7%) patients finally had a clinical diagnosis of MPN (Table 1). In 10 cases, the diagnosis was confirmed by BM histology (patients T1–T8, T10 and T12). In one case, an additional Type 1 CALR mutation was detected with an AB of 42% (patient T9). Conversely, three additional patients refused or were unable to perform BM biopsy, therefore were classified as ‘’Probable ET’’ because presented a persistent thrombocytosis in absence of other causes (patients T11,T13–T14). In two of these patients (T11 and T14), JAK2V617F mutation load was reassessed after a 12-month follow-up and an increased AB was observed, corroborating the diagnosis of MPN (Table 1 and Figure 3); in the remaining case (T13), prospective molecular evaluation was not performed because cytoreductive therapy was already ongoing. Indeed, despite the absence of BM biopsy, cytoreductive therapy was administered according to standard criteria for treatment start (e.g.: age > 60 years and/or previous thrombosis and/or massive thrombocytosis). In the remaining 5 patients (patients T15–T19), thrombocytosis was transitory and was finally considered as secondary to an inflammatory disease (erysipelas, 1 case; rheumatoid arthritis, 2 cases) or to iron deficiency (2 cases). In two of these cases, BM biopsy excluded a hematological disease (patients T18 and T19). Accordingly, a decrease of AB was observed at second evaluation in patients T17 and T18.
Table 1. Main baseline characteristics and clinical outcome of patients investigated for thrombocytosis.
| Patient | Age, gender | Prior Thrombosis (Y/N) |
Plt count ≥ 450 (× 109/l) |
BM histology |
JAK2V617F AB, % 1st evaluation |
JAK2V617F AB, % 2nd evaluation |
CALR |
Fulfilled 2008 WHO diagnostic criteria (Y/N) |
Final diagnosis | Therapy | Status at last follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|
| T1 | 20, F | N | Y | Y | 0.12 | 0.36 | WT | Y | ET | IFN | PLT < 400 |
| T2 | 46, M | Y | Y | Y | 0.79 | - | WT | Y | ET | IFN | PLT < 400 |
| T3 | 33, F | N | Y | Y | 0.20 | 1.46 | WT | Y | ET | - | PLT < 1000 |
| T4 | 25, F | N | Y | Y | 2.37 | - | WT | Y | ET | ASA | PLT <1000 |
| T5 | 62, M | Y | Y | Y | 2.98 | 7.38 | WT | Y | ET | HU, ASA | PLT < 600 |
| T6 | 17, M | N | Y | Y | 3.00 | - | WT | Y | ET | - | PLT <1000 |
| T7 | 50, F | N | Y | Y | 0.43 | - | WT | Y | Early-PMF | IFN | PLT < 400 |
| T8 | 31, M | N | Y | Y | 2.36 | - | WT | Y | Early-PMF | IFN | PLT < 400 |
| T9 | 46, M | N | Y | N.A. | 0.56 | - | Type1 (42%) |
N (absence of BM biopsy) | ET | HU | PLT < 600 |
| T10 | 38, F | N | Y | Y | 0.41 | - | WT | Y | ET | HU | PLT < 1000 |
| T11 | 57, F | N | Y | N.A. | 0.64 | 1.63 | WT | N (absence of BM biopsy) | Probable ET | ASA | PLT < 1000 |
| T12 | 66, M | N | Y | Y | 1.05 | - | WT | Y | ET | HU, ASA | PLT < 600 |
| T13 | 71, F | N | Y | N.A. | 1.56 | - | WT | N (absence of BM biopsy) | Probable ET | ASA | PLT < 400 |
| T14 | 91, F | Y | Y | N.A. | 2.38 | 9.20 | WT | N (absence of BM biopsy) | Probable ET | HU, ASA | PLT <400 |
| T15 | 72, F | Y | Y | N.A. | 0.32 | - | WT | N (absence of BM biopsy, evidence of reactive thrombocytosis) | Reactive (reumatoid arthritis) |
- | PLT < 600 |
| T16 | 81, M | N | Y | N.A. | 0.34 | - | WT | N (absence of BM biopsy, evidence of reactive thrombocytosis) | Reactive (reumatoid arthritis) |
- | PLT < 600 |
| T17 | 43, F | N | Y | N.A. | 0.59 | 0.50 | WT | N (absence of BM biopsy, evidence of reactive thrombocytosis) | Reactive (iron deficiency) |
IRON THERAPY | PLT < 400 |
| T18 | 25, F | N | Y | Y (normal) |
0.12 | WT | WT | N (normal BM histology, evidence of reactive thrombocytosis) | Reactive (erysipelas) |
- | PLT < 400 |
| T19 | 35, F | N | Y | Y (normal) |
0.61 | - | WT | N (normal BM histology, evidence of reactive thrombocytosis) | Reactive (iron deficiency) |
- | PLT < 600 |
Patients with persistent thrombocytosis in absence of other causes that did not perform BM biopsy for histological confirmation were classified as ‘’Probable ET’’. PLT: platelet (× 109/l); BM: bone marrow; N.A.: not available; ET: Essential Thrombocythemia; Early-PMF: early-primary myelofibrosis; WT: wild-type; HU: hydroxyurea; IFN: interferon-alpha; ASA: low-dose aspirin. Patient 19 had received splenectomy for a previous diagnosis of immune thrombocytopenia. Only in 5 cases, lactate dehydrogenase (LDH) was elevated. No patients had splenomegaly.
The 14 patients with a final diagnosis of MPN (including WHO-defined ET and “probable ET”) showed higher hemoglobin level (median (range): 13.6 (10.9–16.2) vs 10.3 (7.2–12.1) g/dl) and lower leukocyte count (median (range): 7.4 (4.2–13.3) vs 11.3 (8.2–21) × 109/l) compared to the 5 patients with reactive thrombocytosis (p < 0.001 and p = 0.05, respectively). Conversely, platelet count was similar in the two groups (median (range): 650 (161–1000) vs 587 (490–701) × 109/l; p = N.S.) (data not shown). The mutation load was significantly higher in MPN patients than in subjects finally diagnosed with reactive thrombocytosis (median (range): 1.18 (0.15–2.98) vs 0.34 (0.12–0.61) %; p = 0.02). Overall, no patient with reactive thrombocytosis showed an AB ≥ 0.64% (Table 1).
Among the 14/35 (40.0%) patients with low AB that performed mutational analysis due to erythrocytosis, 8 (57.1%) had a final diagnosis of MPN (Table 2). PV diagnosis was sustained by absence of other causes of polyglobulia, JAK2V617F positivity and reduced baseline erythropoietin (EPO) levels (patients E1-E8). In patients E1-E3 and E6-E8, the diagnosis was also confirmed by the detection of an increased JAK2V617F AB at a 12-month molecular follow-up. In the remaining 6 cases, the hematological abnormality was finally classified as secondary erythrocytosis (patients E9-E14) due to kidney cancer with increased endogenous EPO levels (patient E9) and chronic obstructive pulmonary disease (COPD, 5 patients: E10-E14). In two of these patients, the diagnosis was supported by a decrease in the JAK2V617F AB over time (patients E13 and E14) (Table 2 and Figure 3). In the majority of these patients with diagnosis of secondary erythrocytosis, phlebotomy and/or aspirin were administered after consideration of the causes of polyglobulia and the overall thrombotic risk of the patients [40–42]. No additional mutations were found in none of the patients investigated for erythrocytosis.
Table 2. Main baseline characteristics and clinical outcome of patients investigated for erythrocytosis.
| Patient | Age, gender | Prior Thrombosis (Y/N) |
Hb ≥ 18.5 g/dl (males) or Hb ≥ 16.5 g/dl (females) (Y/N) |
JAK2V617F AB, % 1st evaluation |
JAK2V617F AB, % 2nd evaluation |
Reduced EPO levels |
Fulfilled 2008 WHO diagnostic criteria (Y/N) | Final diagnosis | Therapy | Status at last follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| E1 | 54, M | N | Y | 0.15 | 2.25 | Y | Y | PV | ASA, PHLEBOTOMY | Hct control < 45% |
| E2 | 63, M | N | Y | 0.16 | 0.28 | Y | Y | PV | ASA, PHLEBOTOMY | Hct control < 45% |
| E3 | 58, M | Y | Y | 0.34 | 0.50 | Y | Y | PV | NONE | Hct control < 45% |
| E4 | 50, M | Y | Y | 0.16 | - | Y | Y | PV | HU, ASA, PHLEBOTOMY | Hct control < 45% |
| E5 | 70, M | N | Y | 2.23 | - | Y | Y | PV | HU, ASA, PHLEBOTOMY | Hct control < 45%, PLT< 600 |
| E6 | 81, M | Y | N | 1.20 | 1.30 | Y | No (Hb ≤ 18.5) |
PV | HU, ASA, PHLEBOTOMY | Hct control < 45% |
| E7 | 65, M | N | N | 0.86 | 1.00 | Y | No (Hb ≤ 18.5 & EPO levels not available) |
PV | ASA, PHLEBOTOMY | Hct control < 45% |
| E8 | 41, M | Y | N | 0.49 | 0.82 | Y | No (Hb ≤ 18.5 & EPO levels not available) |
PV | ASA, PHLEBOTOMY | Hct control < 45% |
| E9 | 59, M | N | Y | 0.12 | - | N | No (increased EPO levels) |
Secondary (kidney carcinoma) | ASA, PHLEBOTOMY | Deceased (kidney carcinoma) |
| E10 | 67, M | Y | N | 0.16 | - | N | No (Hb ≤ 18.5 & normal EPO levels) |
Secondary (COPD) | ASA, PHLEBOTOMY | Hct control < 48% |
| E11 | 54, M | Y | N | 0.39 | - | N.A. | No (Hb ≤ 18.5 & EPO levels not available) |
Secondary (COPD) | PHLEBOTOMY | Hct control < 48% |
| E12 | 82, F | N | N | 0.74 | - | N.A. | No (Hb ≤ 18.5 & EPO levels not available) |
Secondary (COPD) | NONE | Hct control < 48% |
| E13 | 77, M | Y | N | 0.59 | 0.30 | N | No (Hb ≤ 18.5 & normal EPO levels) |
Secondary (COPD) | ASA, PHLEBOTOMY | Hct control < 48% |
| E14 | 72, M | N | N | 0.72 | 0.40 | N | No (Hb ≤ 18.5 & normal EPO levels) |
Secondary (COPD) | ASA, PHLEBOTOMY | Hct control < 48% |
Hb: hemoglobin (g/dl); Hct: hematocrit; N.A.: not available; HU: hydroxyurea; ASA: low-dose aspirin; COPD: chronic obstructive pulmonary disease.
The 8 patients with a diagnosis of PV showed higher hemoglobin level compared to the 6 patients with secondary polyglobulia (median (range): 18.8 (17.4–20.1) vs 16.9 (16.2–19.7) g/dl; p = 0.04), but presented comparable leucocyte (median (range): 7.3 (5.4–0.9) vs 6.2 (4.5–9.4) × 109/l; p = N.S.) and platelet counts (median (range): 250 (186–703) vs 235 (166–283) × 109/l; p = N.S.) (data not shown). Median JAK2V617F AB was also similar in the two groups (median (range): 0.42 (0.16–2.25) vs 0.49 (0.12–0.74)%; p = N.S.). Nonetheless, 75% of PV patients carried a mutation load above the median value of 0.44% (vs 16.7% in patients with secondary erythrocytosis, p = 0.03) (Table 2).
As expected, low AB patients investigated for a suspected PV showed significantly higher median hemoglobin levels (median (range): 18.2 (16.2–20.1) vs 12.9 (7.2–16.2) g/dl; p < 0.001) and lower median platelet count (median (range): 238 (166–703) vs 638 (161–1000) × 109/l; p < 0.001) compared to patients with suspected ET (data not shown). Conversely, median JAK2V617F burden was similar in the two cohorts both considering all evaluated patients (median (range): 0.44 (0.12–.25) vs 0.64 (0.12–2.98) %; p = N.S.) and only patients with final diagnosis of PV and ET/early-PMF/MPN NOS/probable ET (median (range): 1.18 (0.16–2.25) vs 0.42 (0.15–2.98) %; p = N.S.) (Tables 1 and 2).
Overall, in 11 out of 35 (31.4%) patients the detection of a low JAK2V617F AB was considered insufficient to make a diagnosis of MPN (Figure 2). In 6 cases, the exclusion of MPN relied only on clinical monitoring over time, with the observation that the hematological abnormalities were transient and dependent on other contributing factors. In 2 cases BM histology revealed no signs of MPNs (patients T18 and T19), and in 3 additional patients the JAK2V617F mutation load spontaneously decreased over time (patients T17, E13 and E14). The probability to have a final hematological diagnosis was higher in patients tested for thrombocytosis who received a diagnosis of ET/early-PMF in 73.7% of the cases (vs 57.1% of patients with polyglobulia finally diagnosed with PV). In the former cohort, BM biopsy was fundamental for diagnosis in 62.9% of the cases; also, the detection of a concomitant CALR mutation was decisive in confirming ET diagnosis in one patient. Conversely, in patients tested for polyglobulia BM biopsy was never performed, thus limiting the diagnostic accuracy. However, in 15 cases the repetition of the JAK2V617F mutation load over time was of remarkable help in the diagnostic process. Nonetheless, in the low AB cohort, the probability to carry a hematological disease directly correlated with a higher mutation load, since all patients with a JAK2V617F AB > 0.8% were finally diagnosed with a MPN. In order to exclude under-estimation of the JAK2V617F AB caused by hampering correct primer or probe annealing, additional mutations in JAK2 exon 14 were investigated; however, they were not detected in any patient.
Finally, regarding clinical outcome, the frequency of thrombosis was not significantly different according to JAK2V617F AB, in both ET and PV. In addition, when we analyzed the distribution of low and high AB patients according to risk categories, we did not find any significant correlation between the two parameters (data not shown).
DISCUSSION
The present study investigated the role of a low JAK2V617F AB in a cohort of subjects that received the molecular test in the suspect of MPN. All mutational studies were performed according to international recommendations over a 1-year period and patients were homogeneously followed at a single Institution. BM histology was always decisive to direct diagnosis. When histology was unavailable, molecular monitoring together with clinical observation were utmost importance.
Among the 42 patients with low (0.1–3%) JAK2V617F AB, 7 had a confirmed diagnosis of non-classical MPNs by BM histology, 11 were classified as reactive hematological abnormalities due to the presence of additional causes of thrombocytosis (5 cases) or polyglobulia (6 cases) whereas 24 received a diagnosis of classical MPNs. JAK2V617F mutation represents a non-driver and subclonal event in non-classical MPNs, occurring most frequently with low mutation burden [11]. As a consequence, the percentage of subjects with an AB ≤ 3 that were finally diagnosed as MPDs was higher (7/23, 30.4%) compared to MPNs 24/716 (3.3%).
Also, the present study demonstrates that within low AB patients a higher mutation load is associated with a higher probability to receive a hematological diagnosis, with an AB ≥ 0.8% always corresponding to an overt MPN phenotype. Moreover, while higher hemoglobin levels significantly correlated with a diagnosis of WHO-defined PV, platelet count was similar in patients with or without a final diagnosis of ET/early-PMF, and was therefore not indicative per se of a hematological disorder. Another interesting observation is that histology, when performed, was diagnostic for a full-blown disease regardless of the JAK2V617F AB. This not only further confirms the central role of histology in MPN diagnosis, distinguishing true MPNs by small foci of clonal hematopoiesis that are not of clinical significance, but also indicate that the morphologic pattern is not strictly driven by the molecular aberrancy. As a result, evaluation of BM histology could be useful especially for patients with a suspicion of PV and an AB below 0.8% to confirm MPNs diagnosis. Accordingly, revised WHO classifications for myeloproliferative neoplasm indicated BM morphology as one of three major diagnostic criteria, also in PV [43]. Nonetheless, BM biopsy is not feasible in all patients, because of older age and/or patients’ refusal; also, the material may result inadequate for a correct histology evaluation. In these cases, the repetition of the molecular test over time timely detected the spontaneous expansion (11 cases) or reduction (4 cases) of the JAK2V617F-positive clones and significantly oriented the diagnostic process. Finally, the co-existence of additional CALR/MPL or JAK2 exon 14 and exon 12 mutations was routinely excluded in all cases of thrombocytosis or polyglobulia, respectively, that present with a low JAK2V617F AB [44]. Accordingly, many recent studies have demonstrated that CALR and MPL mutations may co-exist with JAK2 mutations in chronic MPNs/MDS [45,46].
From the biological point of view, our clinical results seem to sustain the hypothesis that a low JAK2V617F AB reflects the presence of a small mutated clone within an overall polyclonal hematopoiesis. Indeed, it was recently demonstrated that patients with early stage hematological malignancy may harbor distinct clones, and that such clones may arise independently [47]. Alternatively, we can also draw the assumption that, depending on whether the oncogenic hit marks the stem or the progenitor's compartments, the mutation load might be more or less enlarged. In any case, this mutational event represents an early molecular onset and is probably not sufficient per se to induce the malignant MPN phenotype. Consequently, the detection of low AB in one single occasion is likely not appropriate to determine the diagnosis. Therefore, the molecular monitoring over time may allow knowing whether this is a temporary clone (a condition which is likely to occur in healthy subjects) or may expand and give origin to the disease.
In conclusion, our results highlight that the detection of a JAK2V617F mutation at low levels is difficult to be interpreted in everyday clinical practice, since not all positive patients received a hematological diagnosis. However, all patients with an AB ≥ 0.8% finally received a diagnosis of MPN; therefore, a mutation load above this cut-off may be considered very indicative for the presence of a myeloproliferative disease. Additionally, the study identified the prospective evaluation of JAK2V617F mutation load as a convenient and non-invasive method to evaluate patients with small mutated clones in order to timely detect the expansion of clonal hematopoiesis and diagnose a full blown disease. The study should require validation in larger cohorts of patients prospectively examined with standardized molecular methods.
MATERIALS AND METHODS
Study population
Between January 2013 and January 2015, 1087 JAK2V617F mutational studies were performed at the Institute of Hematology "L. e A. Seràgnoli", Bologna. The clinical suspicions that motivated the request for JAK2V617F evaluation were: essential thrombocytemia (ET) (299 cases, 27.5%); polycythemia vera (PV) (272 cases, 25.1%); myelodysplasia (MDS) (23 cases, 2.1%); atypical thrombosis (12 cases, 1.1%); myelofibrosis (MF) (133 cases, 12.2%); others/not specified (348 cases, 32.0%) (Figure 1). Clinical and laboratory data of patients with a suspected ET, PV or MF and an allele burden ≤ 3% were prospectively monitored for 1 year. All patients provided an informed written consent in accordance with the Declaration of Helsinki for the use of remnant DNA for investigational purposes. The study was approved by the local Ethics Committee.
Patient samples
Polymorphonuclear cells were isolated from peripheral blood samples by density centrifugation with Polymorphoprep (Axis-Shield, Scotland) [48–50]. Genomic DNA was extracted using the QIAamp DNA Blood Mini kit (QIAGEN-Werfen) on QIAcube (QIAGEN GmbH, Hilden, Germany) and was quantified with the NanoDrop spectrophotometer (Wilmington, DE, USA).
Molecular evaluations
Molecular analyses were assessed at diagnosis or before treatment start on DNA obtained from granulocytes. When clinical, laboratory and/or histological data were not decisive for the diagnosis of MPN, mutational status was prospectively evaluated at a 12-month interval. A second JAK2V617F molecular test was not performed in patients that had already started cytoreductive therapy due to high thrombotic risk. JAK2V617F mutation was evaluated with ipsogen JAK2 MutaQuant Kit, which is based on allele specific real time quantitative polymerase chain reaction (qPCR) technology on 7900 HT Fast Real Time PCR System (Applied Biosystem) [21]. The percentage of mutant JAK2V617F allele was expressed as the ratio of JAK2V617F copies to total copy number (CN) of JAK2 (CN of JAK2V617F + CN of JAK2 wild type). Even if the lower detection limit (LOD) of the assay was 0.01% (See Supplemental File), we identified the 0.1% as cut-off of positivity as also suggested by several studies [23–24, 27–29]. All samples were tested in duplicate with both qPCR and also with digital PCR (ddPCR) to confirm the evaluation [51–52] (See Supplemental File). In addition to JAK2V617F mutation, CALR and MPL mutations were screened for all patients to obtain a comprehensive molecular characterization and to exclude the co-existence of additional mutations [44–46, 53–54]. CALR exon 9 sequencing was performed by Next Generation Sequencing (NGS) approach with GS Junior (Roche 454 platform); analysis was carried out with AVA software (GRCh38 as references). CALR mutations identified by NGS were confirmed by Sanger sequencing. MPL mutations were investigated by ipsogen MPLW515K/L MutaScreen Kit and by Sanger sequencing (for MPLS505N and other secondary exon 10 mutations), as previously described [55]. Additional masking mutations in JAK2 exon 14 were investigated by Sanger sequencing [39]. In case of clinical suspicion of PV, JAK2 exon 12 mutations were also tested by Denaturing High Pressure Liquid Chromatography (DHPLC) and confirmed by Sanger sequencing [55–58]. Diagnoses of all hematological diseases were made according to the WHO2008 criteria [1].
Statistical methods
Numerical variables have been summarized by their median and range, and categorical variables by count and relative frequency (%) of each category. Comparisons of quantitative variables between groups of patients were carried out by the nonparametric Wilcoxon rank-sum test. Association between categorical variables (2-way tables) was tested by the Fisher exact test or χ2, as appropriate. All p values were two-sided and statistical significance was defined as p < 0.05. All statistical analyses were computed with SPSS software (SPSS Inc., Chicago, IL, USA).
SUPPLEMENTARY MATERIALS FIGURES
Footnotes
Authors’ contributions
F.P., M.P., N.P., G.M. designed the study and wrote the paper. L.C., D.F., G.S., C.P., M.C., N.V. contributed to the data collection and interpretation. M.P., E.O.,E.Z., E.F. performed molecular evaluations. E.S. performed bone marrow analysis. All Authors revised the manuscript critically, and gave final approval to submit for publication.
CONFLICTS OF INTEREST
There is no conflicts of interest.
FUNDING
The research leading to these results has received funding from BolognAIL, from the European Union Seventh Framework Programme (FP7/2007-2013) under Grant Agreement n° 306242-NGS-PTL and from Progetto Regione–Università 2012-2012 (L. Bolondi). GM is supported by RFO 2015. FP is supported by RFO 2015. MP is supported by Unibo.
REFERENCES
- 1.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22:14–22. doi: 10.1038/sj.leu.2404955. [DOI] [PubMed] [Google Scholar]
- 2.Wang YL, Vandris K, Jones A, Cross NC, Christos P, Adriano F, Silver RT. JAK2 Mutations are present in all cases of polycythemia vera. Leukemia. 2008;22:1289. doi: 10.1038/sj.leu.2405047. [DOI] [PubMed] [Google Scholar]
- 3.Lippert E, Boissinot M, Kralovics R, Girodon F, Dobo I, Praloran V, Boiret-Dupré N, Skoda RC, Hermouet S. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108:1865–7. doi: 10.1182/blood-2006-01-013540. [DOI] [PubMed] [Google Scholar]
- 4.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, Skoda RC. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–40. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 5.Levine RL. JAK2V617F: you can't have too much. Blood. 2008;111:3913. doi: 10.1182/blood-2008-01-133322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott LM, Campbell PJ, Baxter EJ, Todd T, Stephens P, Edkins S, Wooster R, Stratton MR, Futreal PA, Green AR. The V617F JAK2 mutation is uncommon in cancers and in myeloid malignancies other than the classic myeloproliferative disorders. Blood. 2005;106:2920–1. doi: 10.1182/blood-2005-05-2087. [DOI] [PubMed] [Google Scholar]
- 7.Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, Gilliland DG, Tefferi A. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood. 2005;106:1207–9. doi: 10.1182/blood-2005-03-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–90. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 10.Jelinek J, Oki Y, Gharibyan V, Bueso-Ramos C, Prchal JT, Verstovsek S, Beran M, Estey E, Kantarjian HM, Issa JP. JAK2 mutation 1849G > T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood. 2005;106:3370–3. doi: 10.1182/blood-2005-05-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, Gilliland DG, Tefferi A. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood. 2005;106:1207–9. doi: 10.1182/blood-2005-03-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, Score J, Seear R, Chase AJ, Grand FH, White H, Zoi C, Loukopoulos D, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–8. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 13.Boissinot M, Garand R, Hamidou M, Hermouet S. The JAK2-V617F mutation and essential thrombocythemia features in a subset of patients with refractory anemia with ring sideroblasts (RARS) Blood. 2006;108:1781–2. doi: 10.1182/blood-2006-03-008227. [DOI] [PubMed] [Google Scholar]
- 14.Ohyashiki K, Aota Y, Akahane D, Gotoh A, Miyazawa K, Kimura Y, Ohyashiki JH. The JAK2 V617F tyrosine kinase mutation in myelodysplastic syndromes (MDS) developing myelofibrosis indicates the myeloproliferative nature in a subset of MDS patients. Leukemia. 2005;19:2359–60. doi: 10.1038/sj.leu.2403989. [DOI] [PubMed] [Google Scholar]
- 15.Boissinot M, Lippert E, Girodon F, Dobo I, Fouassier M, Masliah C, Praloran V, Hermouet S. Latent myeloproliferative disorder revealed by the JAK2-V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood. 2006;108:3223–4. doi: 10.1182/blood-2006-05-021527. [DOI] [PubMed] [Google Scholar]
- 16.Wang SA, Hasserjian RP, Loew JM, Sechman EV, Jones D, Hao S, Liu Q, Zhao W, Mehdi M, Galili N, Woda B, Raza A. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis harbors JAK2 mutation and shows overlapping myeloproliferative and myelodysplastic features. Leukemia. 2006;20:1641–4. doi: 10.1038/sj.leu.2404316. [DOI] [PubMed] [Google Scholar]
- 17.Zhao S, Zhang X, Xu Y, Feng Y, Sheng W, Cen J, Wu D, Han Y. Impact of JAK2V617F Mutation Burden on Disease Phenotype in Chinese Patients with JAK2V617F-positive Polycythemia Vera (PV) and Essential thrombocythemia (ET) Int J Med Sci. 2016;13:85–91. doi: 10.7150/ijms.10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vannucchi AM, Antonioli E, Guglielmelli P, Rambaldi A, Barosi G, Marchioli R, Marfisi RM, Finazzi G, Guerini V, Fabris F, Randi ML, De Stefano V, Caberlon S, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–6. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 19.Antonioli E, Guglielmelli P, Poli G, Bogani C, Pancrazzi A, Longo G, Ponziani V, Tozzi L, Pieri L, Santini V, Bosi A, Vannucchi AM. Myeloproliferative Disorders Research Consortium (MPD-RC). Influence of JAK2V617F allele burden on phenotype in essential thrombocythemia. Haematologica. 2008;93:41–48. doi: 10.3324/haematol.11653. [DOI] [PubMed] [Google Scholar]
- 20.Lussana F, Caberlon S, Pagani C, Kamphuisen PW, Büller HR, Cattaneo M. Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic review. Thrombosis research. 2009;124:409–417. doi: 10.1016/j.thromres.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 21.De Stefano V, Za T, Rossi E, Vannucchi AM, Ruggeri M, Elli E, Micò C, Tieghi A, Cacciola RR, Santoro C, Gerli G, Vianelli N, Guglielmelli P, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica. 2008;93:372–380. doi: 10.3324/haematol.12053. [DOI] [PubMed] [Google Scholar]
- 22.Barbui T, Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E, Ruggeri M, Rodeghiero F, Randi ML, Bertozzi I, Gisslinger H, Buxhofer-Ausch V, De Stefano V, et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis) Blood. 2012;120:5128–5133. doi: 10.1182/blood-2012-07-444067. [DOI] [PubMed] [Google Scholar]
- 23.Tefferi A, Lasho TL, Huang J, Finke C, Mesa RA, Li CY, Wu W, Hanson CA, Pardanani A. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008;22:756–61. doi: 10.1038/sj.leu.2405097. [DOI] [PubMed] [Google Scholar]
- 24.Guglielmelli P, Barosi G, Specchia G, Rambaldi A, Lo-Coco F, Antonioli E, Pieri L, Pancrazzi A, Ponziani V, Delaini F, Longo G, Ammatuna E, Liso V, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009;114:1477–83. doi: 10.1182/blood-2009-04-216044. [DOI] [PubMed] [Google Scholar]
- 25.Campbell PJ, Baxter EJ, Beer PA, Scott LM, Bench AJ, Huntly BJ, Erber WN, Kusec R, Larsen TS, Giraudier S, Le Bousse-Kerdilès MC, Griesshammer M, Reilly JT, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. 2006;108:3548–55. doi: 10.1182/blood-2005-12-013748. [DOI] [PubMed] [Google Scholar]
- 26.Bench AJ, White HE, Foroni L, Godfrey AL, Gerrard G, Akiki S, Awan A, Carter I, Goday-Fernandez A, Langabeer SE, Clench T, Clark J, Evans PA, et al. British Committee for Standards in Haematology. Molecular diagnosis of the myeloproliferative neoplasms: UK guidelines for the detection of JAK2 V617F and other relevant mutations. Br J Haematol. 2013;160:25–34. doi: 10.1111/bjh.12075. [DOI] [PubMed] [Google Scholar]
- 27.Jovanovic JV, Ivey A, Vannucchi AM, Lippert E, Oppliger Leibundgut E, Cassinat B, Pallisgaard N, Maroc N, Hermouet S, Nickless G, Guglielmelli P, van der Reijden BA, Jansen JH, et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: a joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) study. Leukemia. 2013;27:2032–9. doi: 10.1038/leu.2013.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lippert E, Girodon F, Hammond E, Jelinek J, Reading NS, Fehse B, Hanlon K, Hermans M, Richard C, Swierczek S, Ugo V, Carillo S, Harrivel V, et al. Concordance of assays designed for the quantification of JAK2V617F: a multicenter study. Haematologica. 2009;94:38–45. doi: 10.3324/haematol.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mason J, Akiki S, Griffiths MJ. Pitfalls in molecular diagnosis in haemato-oncology. J Clin Pathol. 2011;64:275–8. doi: 10.1136/jcp.2010.081349. [DOI] [PubMed] [Google Scholar]
- 30.McClure R, Mai M, Lasho T. Validation of two clinically useful assays for evaluation of JAK2 V617F mutation in chronic myeloproliferative disorders. Leukemia. 2006;20:168–71. doi: 10.1038/sj.leu.2404007. [DOI] [PubMed] [Google Scholar]
- 31.Campbell PJ, Scott LM, Baxter EJ, Bench AJ, Green AR, Erber WN. Methods for the detection of the JAK2 V617F mutation in human myeloproliferative disorders. Methods Mol Med. 2006;125:253–64. doi: 10.1385/1-59745-017-0:253. [DOI] [PubMed] [Google Scholar]
- 32.Cankovic M, Whiteley L, Hawley RC, Zarbo RJ, Chitale D. Clinical performance of JAK2 V617F mutation detection assays in a molecular diagnostics laboratory: evaluation of screening and quantitation methods. Am J Clin Pathol. 2009;132:713–21. doi: 10.1309/AJCPFHUQZ9AGUEKA. [DOI] [PubMed] [Google Scholar]
- 33.Martinaud C, Brisou P, Mozziconacci MJ. Is the JAK2(V617F) mutation detectable in healthy volunteers? Am J Hematol. 2010;85:287–8. doi: 10.1002/ajh.21627. [DOI] [PubMed] [Google Scholar]
- 34.Sidon P, Housni H El, Dessars B, Heimann P. The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia. 2006;20:1622. doi: 10.1038/sj.leu.2404292. [DOI] [PubMed] [Google Scholar]
- 35.Xu X, Zhang Q, Luo J, Xing S, Li Q, Krantz SB, Fu X, Zhao ZJ. Prevalence in a large Chinese hospital population. Blood. 2007;109:339–42. doi: 10.1182/blood-2006-03-009472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nielsen C, Birgens HS, Nordestgaard BG, Bojesen SE. Diagnostic value of JAK2 V617F somatic mutation for myeloproliferative cancer in 49 488 individuals from the general population. Br J Haematol. 2013(160):70–9. doi: 10.1111/bjh.12099. [DOI] [PubMed] [Google Scholar]
- 37.Rapado I, Albizua E, Ayala R, Hernández JA, Garcia-Alonso L, Grande S, Gallardo M, Gilsanz F, Martinez-Lopez J. Validity test study of JAK2 V617F and allele burden quantification in the diagnosis of myeloproliferative diseases. Ann Hematol. 2008;87:741–9. doi: 10.1007/s00277-008-0512-x. [DOI] [PubMed] [Google Scholar]
- 38.Passamonti F, Rumi E, Pietra D, Lazzarino M, Cazzola M. JAK2 (V617F) mutation in healthy individuals. Br J Haematol. 2007;136:678–9. doi: 10.1111/j.1365-2141.2006.06483.x. [DOI] [PubMed] [Google Scholar]
- 39.Lippert E, Mansier O, Migeon M, Denys B, Nilsson A, Rosmond C, Lodé L, Ugo V, Lascaux A, Bellosillo B, Martinez-Lopez J, Naguib D, Gachard N, et al. Clinical and biological characterization of patients with low (0.1-2%) JAK2V617F allele burden at diagnosis. Haematologica. 2014;99:e98–101. doi: 10.3324/haematol.2014.107656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMullin MF. Investigation and Management of Erythrocytosis. Curr Hematol Malig Rep. 2016;11:342–7. doi: 10.1007/s11899-016-0334-1. [DOI] [PubMed] [Google Scholar]
- 41.Assi TB, Baz E. Current applications of therapeutic phlebotomy. Blood Transfus. 2014;12:s75–83. doi: 10.2450/2013.0299-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim KH, Oh KY. Clinical applications of therapeutic phlebotomy. J Blood Med. 2016;7:139–44. doi: 10.2147/JBM.S108479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 44.Malcovati L, Rumi E, Cazzola M. Somatic mutations of calreticulin in myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms. Haematologica. 2014;99:1650–2. doi: 10.3324/haematol.2014.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGaffin G, Harper K, Stirling D, McLintock L. JAK2 V617F and CALR mutations are not mutually exclusive; findings from retrospective analysis of a small patient cohort. Br J Haematol. 2014;167:276–8. doi: 10.1111/bjh.12969. [DOI] [PubMed] [Google Scholar]
- 46.Zamora L, Xicoy B, Cabezón M, Fernandez C, Marcé S, Velez P, Xandri M, Gallardo D, Millá F, Feliu E, Boqué C. Co-existence of JAK2 V617F and CALR mutations in primary myelofibrosis. Leuk Lymphoma. 2015;56:2973–4. doi: 10.3109/10428194.2015.1015124. [DOI] [PubMed] [Google Scholar]
- 47.Beer PA, Jones AV, Bench AJ, Goday-Fernandez A, Boyd EM, Vaghela KJ, Erber WN, Odeh B, Wright C, McMullin MF, Cullis J, Huntly BJ, Harrison CN, et al. Clonal diversity in the myeloproliferative neoplasms: independent origins of genetically distinct clones. Br J Haematol. 2009;144:904–8. doi: 10.1111/j.1365-2141.2008.07560.x. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi K, Patel KP, Kantarjian H, Luthra R, Pierce S, Cortes J, Verstovsek S. JAK2 p.V617F detection and allele burden measurement in peripheral blood and bone marrow aspirates in patients with myeloproliferative neoplasms. Blood. 2013;122:3784–6. doi: 10.1182/blood-2013-07-515676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lange T, Edelmann A, Siebolts U, Krahl R, Nehring C, Jäkel N, Cross M, Maier J, Niederwieser D, Wickenhauser C. JAK2 p.V617F allele burden in myeloproliferative neoplasms one month after allogeneic stem cell transplantation significantly predicts outcome and risk of relapse. Haematologica. 2013;98:722–8. doi: 10.3324/haematol.2012.076901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larsen TS, Pallisgaard N, Møller MB, Hasselbalch HC. Quantitative assessment of the JAK2 V617F allele burden: equivalent levels in peripheral blood and bone marrow. Leukemia. 2008(22):194–5. doi: 10.1038/sj.leu.2404861. [DOI] [PubMed] [Google Scholar]
- 51.Waterhouse M, Follo M, Pfeifer D, von Bubnoff N, Duyster J, Bertz H, Finke J. Sensitive and accurate quantification of JAK2 V617F mutation in chronic myeloproliferative neoplasms by droplet digital PCR. Ann Hematol. 2016;95:739–44. doi: 10.1007/s00277-016-2623-0. [DOI] [PubMed] [Google Scholar]
- 52.Fontanelli G, Baratè C, Ciabatti E, Guerrini F, Grassi S, Del Re M, Morganti R, Petrini I, Arici R, Barsotti S, Metelli MR, Danesi R, Galimberti S. Real-Time PCR and Droplet Digital PCR: two techniques for detection of the JAK2(V617F)mutation in Philadelphia-negative chronic myeloproliferative neoplasms. Int J Lab Hematol. 2015;37:766–73. doi: 10.1111/ijlh.12404. [DOI] [PubMed] [Google Scholar]
- 53.Kim SY, Im K, Park SN, Kwon J, Kim JA, Lee DS. CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable. Am J Clin Pathol. 2015;143:635–44. doi: 10.1309/AJCPUAAC16LIWZMM. [DOI] [PubMed] [Google Scholar]
- 54.Broséus J, Park JH, Carillo S, Hermouet S, Girodon F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood. 2014;124:3964–6. doi: 10.1182/blood-2014-06-583161. [DOI] [PubMed] [Google Scholar]
- 55.Palandri F, Latagliata R, Polverelli N, Tieghi A, Crugnola M, Martino B, Perricone M, Breccia M, Ottaviani E, Testoni N, Merli F, Aversa F, Alimena G, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia. 2015;29:1344–9. doi: 10.1038/leu.2015.87. [DOI] [PubMed] [Google Scholar]
- 56.Scott LM. The JAK2 exon 12 mutations: a comprehensive review. Am J Hematol. 2011;86:668–76. doi: 10.1002/ajh.22063. [DOI] [PubMed] [Google Scholar]
- 57.Percy MJ, Jones FGC, Green AR, Reilly JT, McMullin MF. The incidence of the JAK2 V617F mutation in patients with idiopathic erythrocytosis. Haematologica. 2006;91:413–4. [PubMed] [Google Scholar]
- 58.Percy MJ, Scott LM, Erber WN, Harrison CN, Reilly JT, Jones FG, Green AR, McMullin MF. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica. 2007;92:1607–14. doi: 10.3324/haematol.11643.t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.