

Abstract
cAMP exerts an antiproliferative effect on a number of cell types including lymphocytes. This effect of cAMP is proposed to be mediated by its ability to inhibit G1/S transition. In this report, we provide evidence for a new mechanism whereby cAMP might inhibit cellular proliferation. We show that elevation of intracellular levels of cAMP inhibits DNA replication and arrests the cells in S phase. The cAMP-induced inhibition of DNA synthesis was associated with the increased binding of p21Cip1 to Cdk2-cyclin complexes, inhibition of Cdk2 kinase activity, dephosphorylation of Rb, and dissociation of PCNA from chromatin in S phase cells. The ability of cAMP to inhibit DNA replication and trigger release of PCNA from chromatin required Rb and p21Cip1 proteins, since both processes were only marginally affected by increased levels of cAMP in Rb-/- and p21Cip1-/- 3T3 fibroblasts. Importantly, the implications of cAMP-induced inhibition of DNA synthesis in cancer treatment was demonstrated by the ability of cAMP to reduce apoptosis induced by S phase–specific cytotoxic drugs. Taken together, these results demonstrate a novel role for cAMP in regulation of DNA synthesis and support a model in which activation of cAMP-dependent signaling protects cells from the effect of S phase–specific antitumor agents.
INTRODUCTION
The second messenger cyclical AMP (cAMP) plays an important role in regulation of numerous cellular functions. Elevation of intracellular cAMP levels either stimulates or inhibits proliferation of cells depending on the cell type (Pastan et al., 1975; Dumont et al., 1989). Generally, cAMP has an antiproliferative effect on most cells of mesenchymal origin (Blomhoff et al., 1988; Dugan et al., 1999). We have observed that increase in intracellular cAMP leads to accumulation of lymphoid cells in the G1 phase and correlates with rapid dephosphorylation of the retinoblastoma tumor suppressor protein (Rb; Blomhoff et al., 1987, 1988; Christoffersen et al., 1994; Naderi and Blomhoff, 1999; Gutzkow et al., 2002). Furthermore, this antiproliferative effect of cAMP in lymphocytes has been shown to be mediated through protein kinase A type I (Skalhegg et al., 1992; Tasken et al., 1994).
Rb, is an important inhibitor of cell cycle progression (Wang et al., 1994; Harbour and Dean, 2000). In its hypophosphorylated active form, Rb inhibits transition of cells from G1 into S phase of the cell cycle. In addition, Rb has also been shown to negatively regulate DNA replication and block S phase progression (Chew et al., 1998; Knudsen et al., 1998). The negative effect of Rb on cell cycle progression is countered by its phosphorylation by cyclin-dependent kinases (Cdks). When complexed to their regulatory cyclin subunits, Cdks phosphorylate and thereby inactivate the antiproliferative function of Rb (Mittnacht, 1998). Specifically, inactivation of Rb in G1 is achieved through its sequential phosphorylation by Cdk4/6-cyclin D, Cdk2-cyclin E, and Cdk2-cyclin A complexes (Zarkowska and Mittnacht, 1997; Lundberg and Weinberg, 1998). Later in S phase, phosphorylation of Rb is maintained by the combined actions of Cdk2-cyclin E and Cdk2-cyclin A (Mittnacht, 1998). The activity of Cdk-cyclin complexes is in turn regulated by a number of different mechanisms. Specifically, Cdk-cyclin kinase activity is blocked by its association with small inhibitory proteins referred to as cyclin-dependent kinase inhibitors (CKIs; Morgan, 1995; Sherr and Roberts, 1995). CKIs fall into two families; INK4 proteins that inhibit Cdk4 and Cdk6 (Serrano et al., 1993; Hannon and Beach, 1994; Hirai et al., 1995), and Cip1/Kip1 proteins such as p21Cip1 and p27Kip1 that inhibit a broad range of Cdks including Cdk2-cyclin E and Cdk2-cyclin A complexes (el Deiry et al., 1993; Harper et al., 1993; Xiong et al., 1993; Polyak et al., 1994; Toyoshima and Hunter, 1994; Resnitzky et al., 1995).
Accurate and timely duplication of the genome once each cell cycle ensures transmission of genetic information from one cell generation to the next (Kelly and Brown, 2000). In eukaryotes, this process begins in G1 when a preinitiation complex, consisting of ORC, MCM, and Cdc6 proteins is formed at the origins of replication (Dimitrova and Gilbert, 2000; Kelly and Brown, 2000). On activation of Cdk2-cyclin and Cdc7-Dbf4 kinases at the G1/S transition, DNA replication starts from discrete origins of replication (Diffley, 1996; Dutta and Bell, 1997). Initiation of replication is accompanied by unwinding of the replication origins and displacement of MCM and Cdc6 from the origin (Todorov et al., 1995; Dimitrova et al., 1999; Jiang et al., 1999; Petersen et al., 1999; Dimitrova and Gilbert, 2000). Elongation of replication then begins when proliferating cell nuclear antigen (PCNA) recruits DNA polymerase δ (pol δ) onto chromatin (Waga and Stillman, 1998). PCNA serves as DNA sliding clamp for pol δ, whose function is essential for chromosomal replication (Kelman, 1997). Interruption of either initiation or elongation of replication during S phase leads to block of DNA replication.
In this article, we present evidence for the involvement of cAMP in regulation of S phase progression. We show that elevation of intracellular cAMP levels in S phase leads to inhibition of DNA synthesis and arrest of cells in S phase. The inhibitory effect of cAMP on DNA synthesis is dependent on Rb and p21Cip1 proteins, because both Rb- and p21Cip1-deficient cells were unable to halt DNA synthesis in response to treatment by the cAMP-elevating agent, forskolin. Furthermore, we show that cAMP diminishes the cytotoxic effect of S phase–specific antitumor drugs, suggesting that intracellular cAMP levels could affect the ability of this class of drugs to treat cancer.
MATERIALS AND METHODS
Reagents and Antibodies
Forskolin, aphidicolin (Aph), thymidine, roscovitine, cholera toxin (CTX), prostaglandin E2 (PGE2), camptothecin (CPT), 5-fluorouracil (5-FU), hydroxyurea (HU), paclitaxel (PAC), and vinblastine (VB) were purchased from Calbiochem (La Jolla, CA). 3-isobutyl-1-methylxanthine (IBMX), 5-bromo-2-deoxyuridine (BrdU), propidium iodide (PI), RNase A, Hoechst 33342, and micrococcal nuclease (Mnase) were obtained from Sigma (St. Louis, MO). 8-CPT-cAMP was from Biolog (Hayward, CA). Antibodies against cyclin A (C-19), cyclin E (HE12), Cdk2 (M2-G), p21Cip1 (C-19), p27Kip1 (C-19), and PCNA (PC10) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-MCM2 antibody (610700) was from BD Transduction Laboratories (Lexington, KY). Anti-Rb (G3–245) and FITC-conjugated anti-BrdU (556028) antibodies were obtained from PharMingen (San Diego, CA).
Cloning, Transfection, and Retroviral Infection
Human p21Cip1 cDNA was cloned by PCR into the BamHI and EcoRI sites of the pcDNA3.1(+) vector (pcDNA-p21Cip1; Invitrogen, Carlsbad, CA). The CalPhos Mammalian Transfection Kit (Clontech, Palo Alto, CA) was used to transfect p21Cip1-/- 3T3 cells plated at 50% confluence 12 h before transfection. Twelve hours after transfection, the cells were washed three times with phosphate-buffered saline (PBS) and fed with fresh medium before returning to the incubator. At 36 h posttransfection, the cells were treated and analyzed. Control transfection of cells with pcDNA-EGFP revealed a transfection efficiency of ∼65%. Wt Rb cDNA was subcloned from pCMV-Rb(WT) plasmid (Chen and Wang, 2000) into the unique BamHI site of the pBABE-puro (Morgenstern and Land, 1990) retroviral vector to produce pBABE-wt Rb vector. The day before transfection, the Phoenix packaging cells were seeded at 2 × 106 cells per 6-cm plate with 5 ml of medium per plate. Cells were grown to 80% confluency and transfected with 25 μg of pBABE-puro or pBABE-wt Rb using the CalPhos Mammalian Transfection Kit. The viral supernatants were collected at 48 h after transfection and centrifuged at 250 × g to remove cells and debris. For retroviral infection of cells, 3T3 cells were plated at ∼105 cells per well on a six-well plate. After 24 h, medium was removed and 2 ml of the culture supernatant containing retroviral vectors was added to the wells together with Polybrene (Sigma) at a final concentration of 16 μg/ml. Infection was allowed to proceed for 24 h. The viral supernatant was then removed and replaced with fresh medium containing puromycin at 2 mg/ml. Cells were selected in puromycin for 48 h at which time 100% of mock-infected 3T3 cells were killed.
Cell Culture, Synchronization, and Drug Treatment
The B-lymphoid precursor cell line Reh was originally derived from a patient with acute lymphoblastic leukemia (Rosenfeld et al., 1977). Cells were cultured at a density between 0.4 × 106 and 1.0 × 106 cells/ml in RPMI 1640, supplemented with 10% heat-inactivated fetal bovine serum (FBS; Life Technologies, Rockville, MD), 2 mM glutamine, 125 U/ml penicillin, and 125 μg/ml streptomycin. Wt, Rb-/- and p21Cip1-/- 3T3 fibroblasts were generated from wt, Rb-/- and p21Cip1-/- MEFs, respectively, as previously described (Chau et al., 2002). 3T3 cells were cultured in DMEM supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 125 U/ml penicillin, 62.5 μg/ml streptomycin, and 0.001% β-mercaptoethanol. To synchronize Reh cells in S phase with Aph, cells were cultured in the presence of Aph (1 μg/ml) for 24 h. Cells were released from Aph arrest by two washes with drug-free medium and then were resuspended in drug-free medium. For synchronization of Reh cells by the thymidine double-block method, cells were exposed to 2 mM thymidine for 24 h, released into thymidine-free medium for 9 h, and then reexposed to 2 mM thymidine for 16 h. Cells were released from thymidine block by two washes with drug-free medium and then resuspended in drug-free medium. For synchronization of 3T3 cells in early S phase, cells were first made quiescent by culture in medium containing 0.1% FBS for 72 h and then stimulated with 10% FBS in the presence of 2 μg/ml Aph for 24 h. Cells were released from S phase synchrony by washing once with PBS, followed by two washes with drug-free medium for 5 min.
Flow Cytometry
Cells were incubated with BrdU (10 μM). Cells were harvested by centrifugation, washed once in cold PBS, resuspended in 100 μl PBS, fixed in 4 ml of ice-cold 70% ethanol, and incubated overnight at -20°C. Cells were pelleted at 250 × g for 5 min and incubated in 1 ml of a solution containing 2 M HCl and 0.3 mg/ml pepsin for 30 min at RT. The acid was then neutralized with 3 ml 0.1 M sodium tetraborate (pH 8.5). Cells were pelleted, washed once in IFA buffer (10 mM HEPES [pH 7.4], 25 mM NaCl, 4% fetal calf serum) and then incubated in 1 ml IFA containing 0.5% Tween for 10 min at RT. Cells were then washed once in IFA, pelleted, and incubated in 100 μl of IFA containing 10 μl FITC-conjugated anti-BrdU antibody for 30 min at RT. After antibody incubation, cells were washed once in IFA and incubated for 10 min on ice in 0.5 ml of PBS containing 20 μg/ml PI and 40 μg/ml RNase. Cells were then analyzed on a fluorescene-activated cell sorting (FACS) instrument to determine PI (red) and FITC (green) staining for nuclear DNA and BrdU content, respectively.
Immunoblotting, Immunoprecipitation, and Kinase Assay
For immunoblot analysis, cells were lysed in RIPA buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5 mM EDTA, 10 mM NaF, 5 mM β-glycerophosphate, 0.1 mM Na3VO4, 0.2 mM phenylmethylsulfonyl fluoride [PMSF], 10 μg/ml leupeptin, and 0.5% aprotinin) and equal amount of proteins were separated on a 7.5% (for Rb) or 10% (for other proteins) SDS-PAGE. After transfer to nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ), proteins were detected by use of standard immunoblotting procedures. For detection of Rb in 3T3 cells, cell lysates containing 500 μg protein were immunoprecipitated with 2 μg anti-Rb antibodies. The immunocomplexes were then absorbed to protein G-Sepharose (Amersham Biosciences) and washed four times in RIPA buffer. Proteins were then resuspended in SDS sample buffer, boiled, and subjected to Western blot analysis with anti-Rb antibodies. For immunoprecipitation of Cdk2 from Reh cell extracts, cells were resuspended in Triton X-100 lysis buffer (20 mM Tris [pH 7.5], 250 mM NaCl, 0.1% Triton X-100, 10 mM NaF, 5 mM β-glycerophosphate, 0.1 mM Na3VO4, 0.2 mM PMSF, 10 μg/ml leupeptin, and 0.5% aprotinin). The samples were placed on ice and vortexed at 5-min intervals for 20 min. To immunoprecipitate Cdk2 from 3T3 cell extracts, cells were resuspended in Triton X-100 lysis buffer and incubated on ice for 1 h with intermittent vortexing, followed by sonication. After removal of insoluble materials by centrifugation, 300 μg cell extracts were immunoprecipitated with 2 μg anti-Cdk2 for 2 h at 4°C. The immunocomplexes were then absorbed to 30 μl of a 1:1 slurry of protein G-Sepharose for 1 h at 4°C, collected by centrifugation, and washed twice with Triton X-100 lysis buffer. The beads were then resuspended in SDS sample buffer, boiled, and subjected to Western blot analysis. For Cdk2 kinase assay, immunocomplexes recovered from 300 μg lysates were washed twice with Triton X-100 lysis buffer, and once with kinase buffer (50 mM Tris [pH 7.5], 10 mM MgCl2, 1 mM dithiothreitol [DTT], 2 mM EGTA, 1 mM NaF, 0.1 mM Na3VO4, 5 mM β-glycerophosphate). The beads were resuspended in 20 μl of kinase buffer containing 30 μM ATP, 5 μg histone H1 (Upstate Biotechnology, Lake Placid, NY), 10 μCi of [γ-32P]ATP per reaction mixture and incubated for 30 min at RT. Reactions were stopped with addition of 10 μl 3× SDS sample buffer. The samples were boiled and subjected to SDS-PAGE. After electrophoresis, gels were stained with Coomassie blue, dried, and subjected to autoradiography.
Northern Blot Analysis
Total cellular RNA was isolated using RNeasy (Qiagen, Chatsworth, CA) as outlined by the manufacturer, and 15 μg RNA per lane was fractionated on a 1.2% formaldehyde/agarose gel. After staining the gel to verify equal loading in each lane, RNA was transferred onto Hybond-N filter (Amersham Biosciences) in 20× SSC, and cross-linked by UV illumination. The filter was then hybridized with p21Cip1 cDNA probe following the Amersham Rapid Hybridization Protocol at 65°C in a buffer containing 10% dextran sulfate, 2% SDS, 5× SSPE, and 1× Denhardt's solution; washed to a final stringency of 1× SSC, 0.1% SDS; and autoradiographed. Human p21Cip1 cDNA probe was labeled with [α-32P]dCTP (Amersham Megaprime Labeling System; Amersham Biosciences) according to the manufacturer's protocol.
Immunofluorescence Microscopy
Reh cells were cytospun onto poly-L-lysine–coated coverslips. 3T3 cells were seeded on poly-L-lysine–coated coverslips in six-well dishes. The coverslips were then washed once in cold PBS and extracted by incubation with buffer B (10 mM Tris [pH 7.5], 10 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, 0.1% Triton X-100, 250 mM sucrose, 0.1 mM PMSF, 1 mM DTT) for 15 min at RT before fixing in 3% paraformaldehyde. Anti-PCNA and FITC-conjugated secondary antibodies (Sigma) were used at 1:50 and 1:100 dilutions, respectively. DNA was stained with 0.1 μg/ml Hoechst 33342. Observations were made on an Olympus AX70 epifluorescence microscope (Lake Success, NY) using a 100× objective, and photographs were taken with a Photonic Science CCD camera (Millham, UK) and OpenLab software (Improvision Inc., Lexington, MA).
Nuclear Fractionation and Chromatin Isolation
To isolate chromatin-bound proteins, cells were resuspended in buffer A (10 mM Tris [pH 7.5], 10 mM NaCl, 3 mM MgCl2, 0.1 mM PMSF, 1 mM DTT). NP-40 (0.05%) was added, and the cells were incubated for 20 min on ice. Nuclei were collected by low speed centrifugation (200 × g) for 5 min at 4°C. To elute the weakly bound proteins, nuclei were resuspended in buffer B (10 mM Tris [pH 7.5], 10 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, 0.1 mM PMSF, 1 mM DTT) plus 0.1% Triton X-100 for 15 min at RT. Nuclei were collected by low-speed centrifugation and washed once with buffer B. To distinguish between proteins that are attached to chromatin and those that are bound to some other insoluble structure in the nuclei, the nuclei pellet was resuspended in buffer B plus 5 U/ml MNase. After incubation at 37°C for 5 min, the nuclease reaction was stopped by addition of 5 mM EDTA. Nuclei were pelleted by centrifugation at 10,000 × g for 10 min at 4°C, and the MNase-solubilized fraction containing chromatin-bound proteins was analyzed by Western blotting with antibodies against PCNA and MCM2 proteins.
Measurement of Apoptosis
Typically cells exhibit a decrease in forward light scatter (FSC; caused by cytoplasmic shrinkage) and an increase in side light scatter (SSC; caused by increased granularity) when they undergo apoptosis. For measurement of apoptosis in Reh cells, we used an FSC and SSC gating strategy to gate apoptotic and viable cell populations (Darzynkiewicz et al., 1992; Ferlini et al., 1996). To validate the FSC/SSC quantitation method, duplicate samples were assessed using the PI exclusion method (Ormerod et al., 1992; Darzynkiewicz et al., 1994). Apoptosis in 3T3 cells was assessed by detection of subdiploid population. In brief, adherent and floating cells were harvested by centrifugation, washed once in cold PBS, resuspended in 100 μl PBS, and fixed in 4 ml of ice-cold 70% ethanol. After incubation overnight at -20°C, cells were washed once in cold PBS, twice in phosphate-citrate buffer (192 mM Na2HPO4, 4 mM citric acid [pH 7.8]), and then resuspended in PBS containing 50 μg/ml PI and 100 μg/ml RNase. After incubation for 30 min at RT, cells were analyzed on a FACS instrument for determination of cells with sub-G1 DNA content.
RESULTS
Forskolin Retards S phase Progression
We and others have previously reported that activation of the cAMP signaling pathway inhibits proliferation of lymphoid cells (Blomhoff et al., 1988; Skalhegg et al., 1992; Gutzkow et al., 2002). To investigate in more detail cell cycle perturbations induced by cAMP, we treated the B-precursor cell line Reh with 100 μM forskolin, an activator of adenylyl cyclase, and pulse-labeled with BrdU for 30 min before collection of samples at regular intervals for a total of 36 h. Cells were then fixed, stained with PI and FITC-coupled anti-BrdU antibody, and analyzed by bivariant flow cytometry. After 1-h treatment with forskolin, the overall cell cycle distribution was not affected, whereas the rate of BrdU incorporation was slightly reduced compared with that of untreated cells (Figure 1A). By 2 h, forskolin led to a significant reduction in the rate of BrdU incorporation without having a pronounced effect on the cell cycle distribution of the cells. Within 4 h after forskolin treatment, cells had resumed to incorporate BrdU, albeit at a reduced rate relative to untreated cells. By 24 h, forskolin had led to a strong reduction in the number of S phase cells accompanied by accumulation of cells in G1. The cells remained arrested in G1 by the endpoint of the experiment at 36 h after treatment. Taken together, these results showed that treatment of cells with 100 μM forskolin led to a transient inhibition of BrdU incorporation into DNA. Similar results were obtained with human T and B lymphocytes, mouse embryo fibroblasts (MEFs), and the Jurkat cell line (unpublished data), suggesting that the inhibition of BrdU incorporation observed here represents a general action of forskolin.
Figure 1.

Inhibition of DNA replication by cAMP. (A) Asynchronously proliferating Reh cells were cultured in the absence or presence of 100 μM forskolin. Cells were pulse-labeled with BrdU during the last 30 min of treatment before harvesting at the indicated times. Cells were fixed, stained with FITC-coupled anti-BrdU antibody and PI, and analyzed by flow cytometry to determine BrdU incorporation and cell cycle distribution. Top: representative scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-A). Cell cycle distribution of cells was calculated using the gates shown in the leftmost plot. 1, G1 phase; 2, S phase; 3, G2/M phase. Bottom: the relative rate of BrdU incorporation was visualized by plotting FITC anti-BrdU staining in S phase cells. Gray lines, no treatment; black lines, treatment with forskolin. (B) Reh cells were treated with increasing concentrations of forskolin for 2 h and pulsed with BrdU for the final 30 min of treatment. Cells were processed as in A for detection of BrdU incorporation and cell cycle phase determination. Top: representative scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-A). Bottom: the relative rate of BrdU incorporation was visualized by plotting FITC anti-BrdU staining in S phase cells. Gray lines, treatment with vehicle; black lines, treatment with forskolin. (C) Reh cells were synchronized in early S phase with Aph as described in Materials and Methods. Cells were then washed to remove Aph, incubated in drug-free medium for 2 h, and then pulse-labeled with BrdU for 20 min. Subsequently, BrdU was removed by extensive washing, and cells were incubated in the absence or presence of 100 μM forskolin for 2 h. Cells were then harvested and processed for visualization of BrdU-positive cells to monitor progression through S phase. (D) Reh cells were treated with forskolin (20 μM), or IBMX (100 μM) alone or the combination of two, CTX (0.5 or 2 μg/ml), or 8-CPT-cAMP (200 or 400 μM) for 2 h, and pulsed with BrdU for the final 30 min of treatment. Cells were then processed as in A for detection of DNA synthesis and cell cycle phase determination. Top: representative scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-A). Bottom: the relative rate of DNA synthesis was visualized by plotting FITC anti-BrdU staining in S phase cells. Gray lines, no treatment; black lines, treatment with indicated agents.
We next wanted to determine the dose-response relationship for this inhibitory effect of forskolin on BrdU incorporation. To this end, Reh cells were treated with various concentrations of forskolin, pulse-labeled with BrdU for 30 min before collection of samples after 2 h, and evaluated for their ability to incorporate BrdU by flow cytometry. As shown in Figure 1B, inhibition of BrdU incorporation in Reh cells was dose-responsive to forskolin. Cells exhibited maximal inhibition of BrdU incorporation at 100 μM forskolin, and this concentration was therefore used in the following experiments.
BrdU is a thymidine analog that is incorporated in place of thymidine into synthesized DNA strands of actively proliferating cells. Incorporation of BrdU is therefore used as evidence of DNA replication. The ability of forskolin to reduce the incorporation of BrdU into DNA can thus be interpreted as the ability of forskolin to inhibit DNA synthesis. However, it can also be argued that forskolin may inhibit incorporation of BrdU into DNA but not DNA replication per se. To resolve this issue, we examined the effect of forskolin on cells that had already incorporated BrdU, thus circumventing the possible inhibitory effect of forskolin on incorporation of BrdU into DNA. We first synchronized Reh cells in S phase using an aphidicolin (Aph) synchronization protocol (Materials and Methods). The synchronized cells were washed, cultured in drug-free medium for 2 h, and then pulse-labeled with BrdU for 20 min. After incubation with BrdU, cells were washed and BrdU was chased by culturing the cells in the absence or presence of 100 μM forskolin for 2 h. Examination of cells by flow cytometry revealed that forskolin inhibited the progression of BrdU-positive cells through S phase, indicating that forskolin inhibited DNA synthesis (Figure 1C).
Overall, the results presented in Figure 1, A–C, suggest that forskolin leads to a transient cessation of DNA synthesis. Notably, the fraction of cells in G1, S, and G2/M phases of the cell cycle remained essentially unchanged during the transient inhibition of DNA synthesis induced by forskolin. This indicates that, in addition to S phase, forskolin also inhibits progression of cells through other phases of the cell cycle. Taken together, these results show that forskolin induces an immediate but transient multiphase cell cycle arrest in Reh cells.
Inhibition of DNA Synthesis by Forskolin Is Mediated via cAMP
In addition to its ability to increase intracellular levels of cAMP through direct activation of adenylyl cyclase, forskolin has also been shown to affect certain cellular processes through a mechanism that does not involve production of cAMP (Joost and Steinfelder, 1987; Hoshi et al., 1988; Laurenza et al., 1989; Beavo and Reifsnyder, 1990). To determine if the inhibitory effect of forskolin on DNA synthesis is mediated via cAMP, we treated Reh cells with 3-isobutyl-1-methylxanthine (IBMX), cholera toxin (CTX), and 8-CPT-cAMP. IBMX induces accumulation of cAMP by inhibiting phosphodiesterases, CTX ADP-ribosylates the α subunit of Gs protein, a GTP-binding protein that regulates the activity of adenylyl cyclase, thus causing intracellular increase of cAMP, and 8-CPT-cAMP is a membrane-permeable analog of cAMP (Beavo et al., 1970; Seamon et al., 1984; Beavo and Reifsnyder, 1990; Scott, 1991; Spangler, 1992; Negishi et al., 1993; Coleman et al., 1994). Cells were exposed to forskolin, IBMX, CTX, or 8-CPT-cAMP, pulse-labeled with BrdU for 30 min before collection of samples after 2 h, and examined for BrdU incorporation by flow cytometry. Exposure of cells to either 20 μM forskolin or 100 μM IBMX alone had marginal effect on DNA synthesis (Figure 1D). However, treatment of cells with a combination of forskolin and IBMX, which increases cAMP production by stimulating adenylyl cyclase and inhibiting phosphodiesterases, respectively, led to a substantial inhibition of DNA synthesis. Treatment of Reh cells with CTX led to inhibition of DNA synthesis in a dose-dependent manner. When exposed to 0.5 μg/ml CTX, cells exhibited a slight reduction of DNA synthesis, whereas treatment of cells with 2 μg/ml CTX led to a significant inhibition of DNA synthesis. Similarly, exposure of cells to 8-CPT-cAMP led to a dose-dependent inhibition of DNA synthesis. These observations showed that 1) cAMP led to attenuation of DNA synthesis and 2) the forskolin-mediated inhibition of DNA synthesis was mediated via cAMP.
cAMP Induces Rb Dephosphorylation in S phase Cells
In addition to its role in regulation of G1/S transition, Rb has also been documented to inhibit S phase progression (Chew et al., 1998; Knudsen et al., 1998). To assess the involvement of Rb in cAMP-mediated inhibition of DNA synthesis, we first synchronized Reh cells in S phase using the Aph synchronization protocol. The synchronized cells were washed and cultured in the absence or presence of 100 μM forskolin for 2 or 4 h. Two hours after release of cells from Aph arrest, 85% of cells contained S phase DNA content and ∼83% of cells incorporated BrdU (Figure 2A). In the presence of forskolin for 2 h, cells retained S phase DNA content, whereas only 12% of the cells incorporated BrdU. By 4 h after release of cells from Aph arrest, 65% of cells were in S phase and ∼62% of cells incorporated BrdU. In the presence of forskolin for 4 h, 80% of cells were in S phase whereas only 25% of cells incorporated BrdU. We then examined the status of Rb phosphorylation in S phase–synchronized cells after 2 or 4 h treatment with forskolin. The Aph-arrested cells contained exclusively the hyperphosphorylated forms of Rb (Figure 2B). The phosphorylation of Rb remained unchanged in cultures that were released from Aph arrest for 2 or 4 h. Treatment of Aph-released cells with forskolin for 2 h led to accumulation of the hypophosphorylated form of Rb. The Aph-released cells that were treated with forskolin for 4 h maintained further the expression of hypophosphorylated Rb. These results demonstrate a correlation between the dephosphorylation of Rb and inhibition of DNA synthesis in cells that are treated with forskolin.
Figure 2.


cAMP induces dephosphorylation of Rb and inhibits Cdk2 kinase activity in S phase cells. (A) Reh cells were synchronized in S phase by treatment with Aph. After synchronization, cells were treated with vehicle or forskolin for 2 or 4 h, and pulsed with BrdU for the final 30 min of treatment. Cells were then processed as in Figure 1A for detection of DNA synthesis and cell cycle phase determination. Shown are scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-A). The diagonal bar in each scatter plot separates BrdU-positive from BrdU-negative cells. (B) S phase–synchronized Reh cells were treated with vehicle or forskolin for the indicated times. Cells were harvested, lysates prepared, resolved by SDS-PAGE, and detected by immunoblotting with anti-Rb antibodies. pRb, hypophosphorylated Rb; ppRb, hyperphosphorylated Rb. Rb detected in the lysate of resting human B lymphocytes was used as control for pRB. (C) S phase–synchronized Reh cells were treated with vehicle or forskolin and harvested at the indicated times. Whole-cell extracts were prepared, immunoprecipitated with anti-Cdk2 antibodies, and assayed for H1 kinase activity. (D) S phase–synchronized Reh cells were treated with vehicle or forskolin and harvested at the indicated times. Each sample was split in two. Whole-cell extracts were prepared from one-half of each sample, immunoprecipitated with anti-Cdk2 antibodies, and assayed for H1 kinase activity (top panel). Lysates prepared from the other half of each sample were analyzed by immunoblotting with anti-Rb antibodies (bottom panel). pRb, hypophosphorylated Rb; ppRb, hyperphosphorylated Rb. (E) S phase–synchronized Reh cells with treated with vehicle or 50 μM roscovitine for 2 h. Cell lysates were then prepared and processed for examination of Rb phosphorylation by Western blotting. pRb, hypophosphorylated Rb; ppRb, hyperphosphorylated Rb. (F) S phase–synchronized Reh cells were treated as in A. After treatment, each sample was split in two. Total cell lysates from one-half of each sample were resolved on SDS-PAGE and then subjected to immunoblotting with the indicated antibodies (left panel). Whole-cell extracts were prepared from the other half of each sample and immunoprecipitated with anti-Cdk2 antibodies. The recovered proteins were resolved on SDS-PAGE and then subjected to immunoblotting with the indicated antibodies (right panel). W, Western blotting; IP, immunoprecipitation. (G) Reh cells were synchronized in S phase by the thymidine double-block method as described in Materials and Methods. After synchronization, cells were treated with vehicle or forskolin for 2 h and pulsed with BrdU for the final 30 min of treatment. Each sample was split in two. Cells in one-half of each sample were processed as in Figure 1A for detection of DNA synthesis. Left: scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-Al). The relative rate of DNA synthesis was visualized by plotting FITC anti-BrdU staining in S phase cells as depicted in the histogram. Gray line, no treatment; black line, treatment with forskolin. Right: total cell lysates from the other half of each sample were resolved on SDS-PAGE and analyzed by immunoblotting with the indicated antibodies.
Induction of p21Cip1 and Inhibition of Cdk2-cyclin Kinase Activity by cAMP in S phase Cells
Cdk2 in complex with cyclin E or cyclin A is the kinase responsible for Rb phosphorylation in S phase (Mittnacht, 1998). To assess the mechanism responsible for cAMP-mediated Rb dephosphorylation in S phase cells, we measured the Cdk2 kinase activity in Aph-released Reh cells that were treated with forskolin for 2 or 4 h. The Aph-arrested cells contained substantial amount of Cdk2 kinase activity (Figure 2C). Cdk2 activity increased in cells that were released from Aph arrest for 2 h and remained at this level in cells 4 h after release from Aph arrest. Exposure of cells to forskolin for 2 h after Aph release led a reduction of Cdk2 kinase activity. Similarly, Aph-released cells that were treated with forskolin for 4 h expressed less Cdk2 kinase activity than did untreated cells. This result showed that forskolin inhibited Cdk2 kinase activity in S phase cells, which could account for forskolin-mediated Rb dephosphorylation in these cells. Next, we wanted to verify that forskolin-induced inhibition of Cdk2 activity was responsible for dephosphorylation of Rb in S phase. To do so, we first examined the time-course of Cdk2 kinase inhibition and Rb dephosphorylation after treatment of S phase–synchronized Reh cells with forskolin. Densitometric analysis of the result shown in the top panel of Figure 2D revealed that forskolin inhibited the activity of Cdk2 by ∼20% between 60 and 90 min after treatment. Cdk2 kinase activity decreased further with longer exposure of cells to forskolin, so that by 2 h after forskolin treatment, Cdk2 activity was inhibited by ∼75%. As shown in the bottom panel of Figure 2D, by 90 min after forskolin treatment, cells expressed slightly higher levels of hypophosphorylated Rb than did control cells, and by 2 h after forskolin treatment, cells contained almost exclusively the hypophosphorylated form of Rb. This result establishes a close temporal correlation between inhibition of Cdk2 activity and dephosphorylation of Rb by forskolin in S phase Reh cells. To further demonstrate that inhibition of Cdk2 activity is responsible for dephosphorylation of Rb in S phase cells, Aph-synchronized Reh cells were treated with the specific Cdk2 inhibitor, roscovitine (Gray et al., 1999), and examined for the phosphorylation of Rb. As shown in Figure 2E, treatment of S phase Reh cells with roscovitine for 2 h led to dephosphorylation of Rb. Thus, in agreement with previous reports, Cdk2 is the major enzyme responsible for phosphorylation of Rb in S phase cells (Mittnacht, 1998). Taken together, these results strongly suggest that in S phase cells, forskolin leads to dephosphorylation of Rb through inhibition of Cdk2 kinase activity.
The activity of Cdks is subject to regulation by a variety of mechanisms including the regulation of cyclin protein levels or complex formation with CKIs (Morgan, 1995; Sherr and Roberts, 1995). We therefore examined the protein levels of Cdk2, cyclin E, cyclin A, p27Kip1 and p21Cip1 proteins in S phase Reh cells after treatment with forskolin. Exposure of cells to forskolin for 2 or 4 h after release from Aph arrest had no effect on the levels of Cdk2, cyclin E, cyclin A or p27Kip1, whereas it led to a moderate increase in the levels of p21Cip1 (Figure 2F, left panel). To ascertain the functional significance of the observed increase in p21Cip1 protein levels in forskolin-mediated inhibition of Cdk2 kinase activity, we immunoprecipitated Cdk2 from cells that were treated with forskolin for 2 or 4 h after Aph release and examined the relative amount of cyclin E, cyclin A, p27Kip1, and p21Cip1 bound to Cdk2 by Western blot analysis. In agreement with the Western blot shown in the left panel of Figure 2F, the relative amount of cyclin E, cyclin A, and p27Kip1 in complex with Cdk2 remained unchanged both in control and forskolin-treated cells (Figure 2F, right panel). In contrast, the level of p21Cip1 coimmunoprecipitated with Cdk2 increased after treatment of Aph-released cells with forskolin. We also analyzed the relative content of phosphotyrosine in Cdk2 by immunoblotting with an antiphosphotyrosine antibody and found that forskolin did not affect the phosphorylation of Cdk2 on tyrosine (unpublished data). Thus, the increase in the level of Cdk2-cyclin-p21Cip1 complexes could account for the forskolin-mediated inhibition of Cdk2 kinase activity in S phase cells.
Finally, to verify that the effect of cAMP was not specific for Aph-synchronized cells, we used the thymidine double-block as an alternative means to synchronize Reh cells in S phase. As shown in Figure 2G, exposure of cells to forskolin for 2 h after release from thymidine double-block led to induction of p21Cip1, dephosphorylation of Rb, and inhibition of DNA synthesis.
cAMP Induces the Level of p21Cip1 mRNA in S phase Cells
To determine the mechanism by which cAMP induces the level of p21Cip1 protein, we examined the mRNA levels and the protein stability of p21Cip1 in S phase cells after treatment with forskolin. As shown in Figure 3, exposure of cells to forskolin for 2 h increased the p21Cip1 mRNA level. By 4 h after treatment of cells with forskolin, the level of p21Cip1 mRNA had declined slightly but remained above that of untreated cells. The changes in the expression of p21Cip1 mRNA correlated with the expression of p21Cip1 protein induced by forskolin (see Figure 2F). Examination of the stabilities of p21Cip1 transcript and protein revealed that forskolin did not affect the turnover of p21Cip1 transcript or protein in Reh cells (unpublished data).
Figure 3.
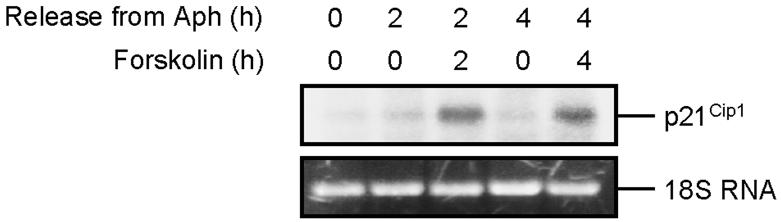
Induction of p21Cip1 mRNA in S phase cells by cAMP. S phase–synchronized Reh cells were treated with vehicle or forskolin and harvested at the indicated times. Total RNA was recovered, and 15 μg of the RNA from each sample was analyzed by Northern blotting using 32P-labeled p21Cip1 probe as described in Materials and Methods. The ethidium bromide-stained 18S RNA was used as a loading control (bottom panel).
Rb and p21Cip1 Are Required for cAMP-mediated Inhibition of DNA Synthesis
Our observation that forskolin-mediated inhibition of S phase progression was associated with the induction of p21Cip1 protein, accumulation of Cdk2-cyclin-p21Cip1 complexes, inhibition of Cdk2 kinase activity, and dephosphorylation of Rb suggested that p21Cip1 and Rb might be involved in inhibition of DNA synthesis by forskolin. To examine this possibility, we utilized 3T3 cells derived from wt, Rb-/-, and p21Cip1-/- MEFs and assessed their response to forskolin-mediated inhibition of DNA synthesis.
Cells were first synchronized in S phase with Aph. These synchronized cells were washed and then treated with 100 μM forskolin, pulse-labeled with BrdU for 30 min before collection of samples after 2 h, and examined for BrdU incorporation by flow cytometry. As can be seen in Figure 4A, wt 3T3 cells exhibited reduced DNA synthesis, whereas p21Cip1-/- cells showed no inhibition of DNA synthesis after forskolin treatment. Rb-/- cells showed a partial inhibition of DNA synthesis in response to forskolin treatment compared with wt cells. This result shows that inhibition of DNA synthesis by forskolin is dependent on Rb and p21Cip1 proteins.
Figure 4.

Rb and p21Cip1 are required for cAMP-induced inhibition of DNA synthesis. (A) Wt, Rb-/- or p21Cip1-/- 3T3 cells were synchronized in early S phase with Aph as described in Materials and Methods. Cells were released from Aph, cultured in drug-free medium in the absence or presence of 100 μM forskolin, and pulsed with BrdU for the final 30 min before harvesting at 2 h. Cells were then processed as in Figure 1A for detection of DNA synthesis and cell cycle phase determination. Shown are scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-A). The relative rate of DNA synthesis was visualized by plotting FITC anti-BrdU staining in S phase cells as depicted in the rightmost panel. Gray lines, treatment with vehicle; black lines, treatment with forskolin. (B) Wt, Rb-/-, or p21Cip1-/- 3T3 cells were treated as in A. Whole-cell extracts were prepared and used for examination of the status of Rb phosphorylation, the level of p21Cip1 protein, and Cdk2-cyclin kinase activity as described in Materials and Methods. pRb, hypophosphorylated Rb; ppRb, hyperphosphorylated Rb. (C) Rb-/- cells harboring pBABE-puro or pBABE-wt Rb vectors were generated by retroviral infection of Rb-/- 3T3 cells. Cells were treated with vehicle or 100 μM forskolin and pulsed with BrdU for the final 30 min before harvesting at 2 h. Each sample was split in two. Left: whole-cell extracts from one-half of each sample were processed for examination of Rb phosphorylation by Western blotting. pRb, hypophosphorylated Rb; ppRb, hyperphosphorylated Rb. Right: cells in the other half of each sample were processed as in Figure 1A for detection of DNA synthesis and cell cycle phase determination. Shown are scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-A). The relative rate of DNA synthesis was visualized by plotting FITC anti-BrdU staining in S phase cells as depicted by the histograms. Gray lines, treatment with vehicle; black lines, treatment with forskolin. (D) p21Cip1-/- cells harboring pcDNA or pcDNA-p21Cip1 vectors were generated by transfection of p21Cip1-/- 3T3 cells. Cells were treated with vehicle or 100 μM forskolin and pulsed with BrdU for the final 30 min before harvesting at 2 h. Each sample was split in two. Left: whole-cell extracts from one-half of each sample were processed for examination of p21Cip1 and Rb phosphorylation by Western blotting. pRb, hypophosphorylated Rb; ppRb, hyperphosphorylated Rb. Right: cells in the other half of each sample were processed as in Figure 1A for detection of DNA synthesis and cell cycle phase determination. Shown are scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-A). The relative rate of DNA synthesis was visualized by plotting FITC anti-BrdU staining in S phase cells as depicted by the histograms. Gray lines, treatment with vehicle; black lines, treatment with forskolin.
Next, we examined the status of Rb phosphorylation, the level of p21Cip1 protein, and the Cdk2 kinase activity in S phase–synchronized wt, Rb-/-, and p21Cip1-/- 3T3 cells after treatment with forskolin. Wt and p21Cip1-/- cells that were released from Aph arrest for 2 h contained both hypophosphorylated and hyperphosphorylated Rb (Figure 4B). Treatment of Aph-released cells with forskolin for 2 h led to accumulation of the hypophosphorylated form of Rb in wt 3T3 cells but not in p21Cip1-/- cells. Exposure of cells to forskolin for 2 h after release from Aph arrest led to an increase in the levels of p21Cip1 in wt and Rb-/- cells. Furthermore, forskolin led to a reduction in Cdk2-cyclin kinase activity only in wt and Rb-/- cells. These results are consistent with a role of p21Cip1 protein in forskolin-mediated inhibition of Cdk2-cyclin kinase activity and Rb dephosphorylation in S phase 3T3 cells.
To further verify the requirement of Rb for forskolin-mediated inhibition of DNA synthesis, we examined whether expression of wt Rb in Rb-/- 3T3 cells would restore forskolin-mediated inhibition of S phase progression. Rb-/- 3T3 cells were infected with retrovirus expressing either wt Rb (pBABE-wt Rb) or empty vector (pBABE-puro) as control, and pools of cells with stably integrated virus were generated. Western blot analysis of Rb-/- 3T3 cells infected with pBABE-wt Rb revealed that the level of Rb expressed in these cells was ∼50% of the level of endogenous Rb expressed in wt 3T3 cells (Figure 4C, left panel). When exposed to forskolin, wt 3T3 cells inhibited DNA synthesis, whereas Rb-/- 3T3 cells infected with pBABE-puro were impaired in their S phase response as expected (Figure 4C, right panel). Restoration of Rb expression in Rb-/- 3T3 cells by pB-ABE-wt Rb partially complemented this defect and caused the cells to partially inhibit DNA synthesis after exposure to forskolin. The lower level of wt Rb expressed in Rb-/- 3T3 infected with pBABE-wt Rb, compared with the level of endogenous Rb expressed in wt 3T3 cells, could account for the partial restoration of S phase response to forskolin in these cells. This result verifies the above observation that Rb is required for inhibition of DNA synthesis in response to cAMP.
The requirement of cAMP-mediated induction of p21Cip1 for dephosphorylation of Rb and attenuation of DNA synthesis by cAMP was further demonstrated by examination of p21Cip1 3T3 cells that were transfected with a vector carrying p21Cip1 cDNA driven by the human cytomegalovirus (CMV) major promoter/enhancer (pcDNA-p21Cip1). An 8-base pair core palindrome of CMV promoter has been shown to act as a cAMP-responsive element (CRE; Fickenscher et al., 1989). Elevation of intracellular concentrations of cAMP results in upregulation of CRE and enhancement of the transcriptional activity of the CMV promoter. As shown in the left panel of Figure 4D, the forskolin-mediated induction of p21Cip1 in cells that were transfected with pcDNA-p21Cip1 was comparable to the level of p21Cip1 in wt 3T3 cells after treatment with forskolin (see Figure 4B). Importantly, p21Cip-/- 3T3 cells transfected with empty vector (pcDNA) failed to exhibit inhibition of DNA synthesis after forskolin treatment (Figure 4D, right panel). In contrast, after exposure to forskolin, p21Cip-/- cells harboring pcDNA-p21Cip1 vector expressed dephosphorylated Rb and inhibited DNA synthesis. Thus, p21Cip1 is required for cAMP to induce the dephosphorylation of Rb and inhibit DNA replication.
cAMP Induces Dissociation of PCNA from Chromatin in S phase Cells
The aforementioned results indicate that cAMP, through Rb, negatively regulates the DNA replication machinery. Rb is implicated in transcriptional repression of E2F regulated genes (Kaelin, 1999). Furthermore, both PCNA and MCM2, two essential components of DNA replication machinery, are regulated by E2F (Helin, 1998; Leone et al., 1998). Therefore, we hypothesized that cAMP might inhibit DNA replication through inhibition of PCNA or MCM2 expression. To examine this possibility, S phase–synchronized Reh cells were treated with forskolin for 2 h and then analyzed for the expression of PCNA and MCM2. Surprisingly, forskolin treatment of Reh cells did not affect the levels of PCNA and MCM2 proteins (Figure 5A). Because PCNA and MCM2 exert their effect only when tethered to DNA (Todorov et al., 1995; Dimitrova and Gilbert, 2000), we proceeded to examine the influence of cAMP on association of PCNA and MCM2 with chromatin by two different approaches. First, nuclei were isolated from S phase–synchronized Reh cells that were treated with forskolin for 2 h and subjected to Triton X-100 extraction to remove soluble and weakly bound nuclear proteins. The nuclei were then treated with micrococcal nuclease (MNase) to solubilize the chromatin. The MNase-solubilized fraction containing chromatin-bound proteins was then analyzed by Western blotting with antibodies against PCNA and MCM2 proteins. The MNase-solubilized fraction from cells that were treated with forskolin contained less PCNA, indicating reduced association of PCNA to chromatin in these samples (Figure 5B). In contrast, forskolin treatment of cells had no effect on MCM2 binding to DNA. To further confirm this result, S phase–synchronized Reh cells that were treated with forskolin for 2 h were subjected to in situ extraction to remove unbound proteins. The cells were then examined by immunocyto-chemistry using antibodies against PCNA. As shown in Figure 5C, the chromatin association of PCNA was significantly reduced in cells that were treated with forskolin as evidenced by greatly diminished staining of PCNA.
Figure 5.

cAMP-mediated inhibition of DNA synthesis correlates with the disruption of PCNA tethering to DNA. (A) S phase–synchronized Reh cells were treated with vehicle or forskolin for 2 h. Cells were harvested, and lysates were prepared, resolved by SDS-PAGE, and detected by immunoblotting with PCNA and MCM2 antibodies. (B) Nuclei isolated from S phase–synchronized Reh cells that were exposed to vehicle or 100 μM forskolin for 2 h were treated with Triton X-100 to separate soluble proteins from the chromatin-bound pellet fraction (Materials and Methods). The extracted nuclei were further treated with MNase to solubilize the chromatin. The MNase-solubilized fraction was resolved on SDS-PAGE and immunoblotted with PCNA and MCM2 antibodies. (C) S phase–synchronized Reh cells were treated with vehicle or 100 μM forskolin and harvested after 2 h. Cells were cytospun onto coverslips, extracted, and then fixed in paraformaldehyde before staining with PCNA antibodies. (D) Wt, Rb-/-, and p21Cip1-/- 3T3 cells were synchronized in S phase with Aph and then treated with vehicle or forskolin for 2 h. Nuclei were then isolated and processed as in B for detection of chromatin-bound PCNA by immunoblotting with anti-PCNA antibodies. (E) Wt, Rb-/-, and p21Cip1-/- 3T3 cells were seeded on coverslips, synchronized in S phase with Aph, and then treated with vehicle or forskolin for 2 h. Coverslips were then processed as in C for immunofluorescence staining with PCNA antibodies.
As shown above, Rb- and p21Cip1-deficient 3T3 cells were unable to inhibit DNA replication in response to cAMP. Because disruption of PCNA tethering to chromatin closely correlated with cAMP-mediated abrogation of DNA synthesis, we wanted to examine the association of PCNA to chromatin in Rb-/- and p21Cip1-/- cells after treatment with forskolin. To do so, S phase–synchronized wt, Rb-/-, and p21Cip1-/- 3T3 fibroblasts were treated with forskolin for 2 h. Examination of the MNase-soluble fraction of these cells showed that forskolin treatment of wt 3T3 cells led to disruption of PCNA tethering to chromatin (Figure 5D). In contrast, forskolin did not disrupt binding of PCNA to chromatin in p21Cip1-/- 3T3 cells. Treatment of Rb-/- 3T3 cells with forskolin led to a marginal inhibition of PCNA association with chromatin. The differential response of wt, Rb-/-, and p21Cip1-/- 3T3 fibroblasts to cAMP-induced dissociation of PCNA from chromatin was also demonstrated by immunostaining of cells for PCNA. In agreement with the data presented in Figure 5D, forskolin inhibited association of PCNA with chromatin in wt 3T3 cells, whereas PCNA tethering was unaffected in Rb-/- and p21Cip1-/- 3T3 cells after treatment with forskolin (Figure 5E). Taken together, these results show that forskolin specifically inhibits association of PCNA to chromatin in an Rb- and p21Cip1-dependent manner and establish a link between forskolin-mediated inhibition of DNA synthesis and the DNA replication machinery.
PGE2-mediated Inhibition of DNA Synthesis
Under physiological conditions, generation of cAMP is controlled by the activity of certain receptors, such as prostaglandin E (PGE) receptors, that via regulation of GTP-binding proteins, activate adenylyl cyclase enzyme and thus lead to generation of cAMP (Negishi et al., 1993; Coleman et al., 1994; Regan, 2003). We therefore used PGE2 to examine whether stimulation of such receptors would lead to attenuation of DNA replication. S phase–synchronized Reh cells were treated with PGE2, pulse-labeled with BrdU for 30 min before collection of samples after 2 h, and examined for BrdU incorporation by flow cytometry. As shown in Figure 6A, treatment of cells with PGE2 led to inhibition of DNA synthesis in a dose-dependent manner. When exposed to 30 μM PGE2, cells exhibited a slight reduction of DNA synthesis, whereas treatment of cells with 60 μM PGE2 led to a significant inhibition of DNA synthesis. This result indicates that generation of cAMP through stimulation of G protein–coupled receptors leads to inhibition of DNA synthesis. Next, we wanted to examine whether PGE2-induced attenuation of DNA synthesis occurred via the same mechanism as that found with forskolin. To do so, we treated S phase–synchronized Reh cells with 60 μM PGE2 for 2 h and examined them for the expression of p21Cip1, phosphorylation of Rb, and association of PCNA with chromatin. We used 60 μM PGE2 because, as shown above, this concentration of PGE2 was found to substantially inhibit DNA synthesis in Reh cells. As shown in Figure 6B, PGE2 treatment of Reh cells led to induction of p21Cip1, dephosphorylation of Rb, and reduction of PCNA association with chromatin.
Figure 6.

Inhibition of DNA synthesis by PGE2. (A) S phase–synchronized Reh cells were treated with the indicated concentrations of PGE2 for 2 h and pulsed with BrdU for the final 30 min of treatment. Cells were processed as in Figure 1A for detection of BrdU incorporation and cell cycle phase determination. Top: representative scatter plots with the log FITC anti-BrdU staining (FL1-H) versus PI staining (FL2-A). Bottom: the relative rate of BrdU incorporation was visualized by plotting FITC anti-BrdU staining in S phase cells. Gray lines, treatment with vehicle; black lines, treatment with forskolin. (B) S phase–synchronized Reh cells were treated with 60 μM PGE2 for 2 h or left untreated. Each sample was then split in two. Total cell lysates from one-half of each sample were resolved on SDS-PAGE and then subjected to immunoblotting with the antibodies against p21Cip1, Rb, and PCNA. Nuclei isolated from the other half of each sample were processed as in Figure 5B and then examined for chromatin-bound PCNA by immunoblotting with anti-PCNA antibodies.
cAMP Protects Cells from Apoptosis Induced by S phase–specific Cytotxic Drugs in an Rb- and p21Cip1-dependent Manner
The negative effect of forskolin on DNA synthesis suggested that activation of cAMP signaling pathway would render cells resistant to antitumor agents whose function depends on DNA replication. To assess this assumption, we examined the effect of forskolin on apoptosis induced by the S phase–specific agents, camptothecin (CPT), 5-fluorouracil (5-FU), hydroxyurea (HU), and the microtubule-damaging agents paclitaxel (PAC) and vinblastine (VB) that exhibit G2/M phase–specific cytotoxicity (Yarbro, 1992; Jordan et al., 1998; Liu et al., 2000; Jordan, 2002; Sampath et al., 2003). We exposed asynchronously proliferating Reh cells to a 2-h pulse treatment with the cytotoxic drugs alone or together with 100 μM forskolin and analyzed sensitivity in terms of apoptotic response at regular intervals after recovery. As shown in Figure 7A, both S phase– and G2/M phase–specific drugs induced apoptosis in Reh cells. Interestingly, cotreatment of cells with forskolin led to a significant reduction in the level of apoptosis in response to S phase–specific agents, whereas it had no effect on apoptosis induced by G2/M phase–specific drugs. These results show that increase in intracellular levels of cAMP can protect cells from the cytotoxic effect of S phase–specific anticancer drugs.
Figure 7.

cAMP prevents cell death induced by S phase–specific cytotoxic agents in an Rb- and p21Cip1-dependant manner. (A) Asynchronously proliferating Reh cells were treated with 100 nM CPT, 1 mM 5-FU, 110 mM HU, 1 μM PAC, or 25 nM VB alone or together with 100 μM forskolin for 2 h. Cells were then washed, cultured in drug-free medium, and harvested at the indicated recovery times for evaluation of apoptosis by flow cytometry. (B) Asynchronously proliferating wt, Rb-/-, or p21Cip1-/- 3T3 cells were treated with 500 μM 5-FU alone or together with 100 μM forskolin for 2 h. Cells were then washed, cultured in drug-free medium, and harvested after 72 h for evaluation of apoptosis by flow cytometry. Data in A and B are from four independent experiments. Error bars, mean ± SE.
Given the observation that cAMP-mediated inhibition of DNA synthesis was dependent on Rb and p21Cip1, we next wanted to determine whether the inhibitory effect of cAMP on apoptosis induced by S phase–specific cytotoxic drugs required Rb and p21Cip1. To do so, wt, Rb-/-, and p21Cip1-/- 3T3 cells were exposed to a 2-h pulse treatment with 5-FU alone or together with 100 μM forskolin. Samples were then collected at 72 h after recovery and analyzed for apoptosis. Cotreatment of wt cells with forskolin substantially inhibited apoptosis induced by 5-FU (Figure 7B). By contrast, forskolin had no effect on 5-FU–induced apoptosis in Rb-/- and p21Cip1-/- 3T3 cells. These results indicate that cAMP-mediated inhibition of apoptosis induced by S phase–specific cytotoxic agents is dependent on Rb and p21Cip1.
DISCUSSION
Elevation of intracellular cAMP leads to growth inhibition in a number of cell types including lymphocytes, and this growth inhibition has been linked to G1 arrest (Blomhoff et al., 1987, 1988; Christoffersen et al., 1994; Dugan et al., 1999). We have previously shown that cAMP leads to a transient accumulation of Reh cells in G2, followed by permanent arrest of cells in G1 phase of the cell cycle (Blomhoff et al., 1988). Here, we provide evidence for an inhibitory effect of cAMP on S phase progression. We have shown that elevation of cAMP inhibits DNA synthesis and arrests the cells transiently in S phase. This cAMP-induced S phase arrest requires Rb and p21Cip1 proteins and closely correlates with dissociation of PCNA from chromatin.
Our observation that cAMP-induced inhibition of DNA synthesis was associated with dephosphorylation of Rb in S phase cells, suggested the requirement of Rb for inhibition of DNA synthesis by cAMP. Indeed Rb-/- cells were partially defective in halting DNA synthesis after elevation of cAMP levels, and ectopic expression of wt Rb in these cells restored the ability of cAMP to inhibit DNA synthesis. In S phase, phosphorylation and inactivation of Rb is achieved through the action of Cdk2 in complex with cyclin E or cyclin A (Mittnacht, 1998). Thus, the observed cAMP-mediated inhibition of Cdk2-cyclin activity could be considered as the mechanism responsible for dephosphorylation of Rb in S phase cells. The cAMP-induced reduction in Cdk2-cyclin activity correlated with induction of p21Cip1 expression and an increase in Cdk2-cyclin-p21Cip1 complexes, supporting a model wherein elevation of intracellular cAMP leads to induction of p21Cip1 protein expression. p21Cip1 then binds to Cdk2-cyclin complexes and attenuates the Cdk2-associated kinase activity. Reduction in Cdk2 kinase activity in turn leads to expression of dephosphorylated Rb and inhibition of DNA replication. The observation that p21Cip1-/-cells failed to 1) inhibit Cdk2-cyclin kinase activity, 2) dephosphorylate Rb, and 3) abrogate DNA synthesis after exposure to forskolin, together with the finding that cAMP-inducible expression of exogenous p21Cip1 in p21Cip1-/- 3T3 cells restored their ability to dephosphorylate Rb and inhibit DNA replication strongly support this model.
We have previously observed that treatment of Reh cells with forskolin induces the expression of p21Cip1 (within 2 h) and p27Kip1 (within 4–8 h) proteins and leads to dephosphorylation of Rb (Christoffersen et al., 1994; Naderi and Blomhoff, 1999; Gutzkow et al., 2002). In this study, we found that exposure of Reh cells to forskolin led to increase in expression of p21Cip1 and dephosphorylated Rb but had no effect on the levels of p27Kip1 protein. It is important to note that here we have used Aph-synchronized cultures of cells to specifically analyze the effect of forskolin on cells in the S phase of the cell cycle. In contrast, in our previous studies (Naderi and Blomhoff, 1999; Gutzkow et al., 2002), we examined the effect of forskolin on asynchronous cultures of Reh cells, in which cells are present in all phases of the cell cycle. Taken together, our results indicate that cAMP-mediated induction of p27Kip1 is associated with G1 arrest and not with inhibition of DNA replication.
Both Rb (Chew et al., 1998; Knudsen et al., 1998; Saudan et al., 2000) and p21Cip1 (Ogryzko et al., 1997; Niculescu et al., 1998) have previously been implicated in regulation of DNA replication. The mechanism by which Rb inhibits DNA synthesis is currently unknown. Activation of Rb has recently been shown to inhibit DNA replication by two kinetically and functionally different mechanisms; an acute pathway that targets PCNA loading onto chromatin and a more stable arrest program that involves a sustained attenuation of the protein levels of several E2F target genes involved in DNA replication (Angus et al., 2004). Disruption of PCNA activity elicited by Rb is shown to depend on E2F-mediated transcriptional repression and the ability of Rb to inhibit the expression of cyclin A has been suggested to be such an event (Sever-Chroneos et al., 2001; Angus et al., 2004). As demonstrated here, the cAMP-mediated inhibition of DNA synthesis is dependent on Rb function and correlates with dissociation of PCNA from chromatin. It can therefore be suggested that cAMP utilizes Rb in its capacity to acutely disrupt the PCNA function and leads to cessation of DNA replication. This assumption predicts that Rb-mediated transcriptional repression contributes to cAMP-induced dissociation of PCNA from chromatin and inhibition of DNA replication. However, we cannot formally exclude the possibility that cAMP inhibits DNA replication independent of the transcriptional repression function of Rb. This notion is based on two lines of evidence: First, Rb dephosphorylation and inhibition of DNA synthesis occur almost simultaneously in forskolin-treated cells. Second, cAMP does not inhibit the expressions of cyclin E and A in S phase cells. Because cyclin E and A are known targets of Rb, this observation suggests that the ability of Rb to regulate gene transcription is distinct from the cAMP-mediated inhibition of DNA replication. It is conceivable that Rb might directly interfere with PCNA function to inhibit DNA replication at the elongation phase. Our data that cAMP-mediated dephosphorylation of Rb and inhibition of DNA synthesis coincided with dissociation of PCNA from chromatin is consistent with this notion. Further support for this idea is provided by the finding that cAMP failed to induce dissociation of PCNA from chromatin and inhibit DNA replication in Rb-/- cells. Indeed, the phenomenon of disruption of PCNA function and inhibition of DNA replication at the elongation phase has been reported previously. Activation of the S phase checkpoint by DNA damage has been shown to inhibit replication elongation and decrease the association of PCNA with DNA replication foci, an event that appears to depend on Rb signaling (Watanabe, 1974; Knudsen et al., 2000; Sever-Chroneos et al., 2001; Wang et al., 2004). The exact mechanism by which Rb interferes with PCNA function is unclear. However, a potential insight into how Rb exerts its effect on PCNA might be provided by the report showing interaction of Rb with replication factor C (RFC; Pennaneach et al., 2001). This study indicated that Rb interacts with RFC to induce cell cycle arrest and promote cell survival after DNA damage. Considering the function of RFC as the loader of PCNA onto DNA, it is tempting to speculate that Rb might inhibit association of PCNA with chromatin through its interaction with RFC and thus lead to inhibition of DNA replication. Further studies will be required to test this model.
Results from a number of studies suggest a role of Rb in inhibition of replication at the level of initiation. For instance, Rb has been reported to bind MCM7, a component of the prereplication complex, and inhibit in vitro DNA replication in an MCM7-dependent manner (Sterner et al., 1998). In Drosophila, dDP–dE2F1–Rbf in complex with DmORC at the chorion origin of replication has been shown to limit replication initiation at the chorion locus, independent of transcription (Bosco et al., 2001). Furthermore, after irradiation of S phase–synchronized cells, hypophosphorylated Rb has been reported to be directed to certain chromosomal replication initiation sites, at or after the time they had begun to fire, as part of the process whereby Rb suppresses abnormal rereplication activity (Avni et al., 2003). Although the interaction of Rb with proteins at the replication origins or its presence at the replication foci has been disputed (Dimitrova and Berezney, 2002; Angus et al., 2004), we cannot exclude the possibility that after dephosphorylation, Rb might be recruited to replication initiation sites at or after the time they have begun to fire. This might, in turn, control any series of steps in initiation such as origin recognition, DNA unwinding, or it can block the switching from pol α–dependent primer formation to processive replication using pol δ and PCNA, and thus lead to inhibition of DNA replication.
When exposed to forskolin, Rb-/- cells exhibited an incomplete reduction in S phase arrest compared with wt cells. In contrast, p21Cip1-/- cells were irresponsive to the S phase inhibitory effect of forskolin. These observations suggest that inhibition of DNA replication by cAMP is partially dependent on Rb and indicate an Rb-independent role of p21Cip1 in cAMP-mediated inhibition of DNA replication. In addition to binding to Cdk-cyclin complexes, p21Cip1 also associates with PCNA (Flores-Rozas et al., 1994). In vitro, interaction of p21Cip1 with PCNA has been shown inhibit the RFC-mediated loading of PCNA onto DNA and also to prevent binding of pol δ to the PCNA clamp assembled on DNA (Waga et al., 1994; Podust et al., 1995). Similarly, a recent study has shown that, in vivo, direct binding of p21Cip1 to chromatin-bound PCNA results in the loss of interaction with the catalytic subunit of pol δ (Cazzalini et al., 2003). Based on these results, it can be postulated that p21Cip1 contributes to the cAMP-mediated inhibition of DNA replication through its ability to directly interact with PCNA and thus abolish the replication complex formation involving PCNA, pol δ, and RFC. The possible role of p21Cip1, as an inhibitor of Cdk-cyclin complexes, in attenuation of S phase progression should also be addressed. Several reports have suggested that the negative effect of p21Cip1 on DNA synthesis relies primarily on its ability to inhibit Cdk-cyclin–dependent pathways (Luo et al., 1995; Nakanishi et al., 1995a, 1995b; Ogryzko et al., 1997). Moreover, a number of studies have shown that, in addition to phosphorylation of Rb, Cdk2-cyclin complexes also regulate DNA replication in a manner that does not appear to require Rb function (Mimura and Takisawa, 1998; Zou and Stillman, 1998; Furstenthal et al., 2001). One such report has shown that inhibition of Cdk2-cyclin A kinase has the potential to negatively regulate DNA replication through displacement of PCNA from chromatin (Sever-Chroneos et al., 2001). On the basis of these observations and our finding that p21Cip1-deficient cells were unable to disrupt PCNA association with chromatin in response to cAMP, we propose that the Rb-independent function of p21Cip1 in cAMP-mediated inhibition of DNA synthesis might stem from its ability to attenuate Cdk2-cyclin activity and thus lead to disruption of PCNA tethering to chromatin.
In summary, we propose that cAMP inhibits DNA replication through its ability to induce the expression of p21Cip1 protein and inhibit the activity of Cdk2-cyclin kinases (Figure 8). Abrogation of Cdk2-cyclin kinase activity in turn leads to expression of hypophosphorylated, active form of Rb, which leads to disruption of PCNA association with chromatin and thus inhibition of DNA replication. Furthermore, the inability of Rb-/- cells, as opposed to p21Cip1-/- cells, to completely alleviate the inhibitory effect of cAMP on association of PCNA with chromatin and DNA replication, argues for an additional, Rb-independent pathway through which p21Cip1 can regulate PCNA activity and DNA replication.
Figure 8.

Suggested model for the cAMP-mediated inhibition of DNA replication. In S phase cells, elevation of intracellular levels of cAMP induces the expression of p21Cip1 protein. This leads to inhibition of Cdk2-cyclin kinase activity. Abrogation of Cdk2-cyclin kinase activity in turn leads to expression of hypophosphorylated, active form of Rb (pRb), which leads to disruption of PCNA association with chromatin, and thus inhibition of DNA synthesis. Furthermore, data presented here argue for an additional, Rb-independent pathway through which p21Cip1, possibly via inhibition of Cdk2-cyclin kinase, can regulate PCNA activity and DNA replication. Pre-Rc, prereplication complex.
A number of current cancer therapy regimes rely on the use of drugs whose cytotoxic activity depends on DNA replication. Our observation that elevation of intracellular cAMP inhibits DNA replication and blocks apoptosis induced by such S phase–specific anticancer agents may have potential practical implications for cancer therapy. These observations predict that conditions that lead to increase in intracellular cAMP would reduce the efficacy of such drugs against tumor cells. For instance, it is known that lymphocytes and macrophages are attracted to sites of neoplasia (Witz, 2001). At these sites, tumors stimulate macrophages to secrete a number of regulatory molecules including PGE2 (Alleva et al., 1993; Brum-Fernandes et al., 1996; Elgert et al., 1998). Most effects of PGE2 are mediated by the PGE2 receptors whose activation leads to elevation of intracellular cAMP (Brum-Fernandes et al., 1996; Kiriyama et al., 1997). Thus, it can be postulated that tumor-induced secretion of PGE2 by macrophages might elevate cAMP levels inside tumor cells and render them resistant to S phase–specific antitumor drugs. Accordingly, a combinatorial approach, consisting of an S phase–specific antitumor agent and cAMP antagonists, can be considered as an effective regime to increase the sensitivity of tumor cells to such drugs.
Acknowledgments
We thank Camilla Solberg for excellent technical assistance. This work was funded by the Norwegian Cancer Society, the Norwegian Research Council, the Jahre Foundation, Blix Foundation, Rachel and Otto Kr. Bruum's foundation, Nansen foundation (S.N., K.B.G., and H.K.B.) and the National Cancer Institute grant CA58320 (J.Y.J.W.).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-06-0501) on January 12, 2005.
Abbreviations used: cAMP, cyclic AMP; Rb, retinoblastoma protein; Cdk, cyclin-dependent kinase; CKI, cyclin-dependent kinase inhibitor; PCNA, proliferating cell nuclear antigen; RFC, replication factor C; pol δ, DNA polymerase δ; CPT, camptothecin; 5-FU, 5-fluorouracil; HU, hydroxyurea; PAC, paclitaxel; VB, vinblastine; PI, propidium iodide; FITC, fluorescein isothiocyanate; BrdU, bromodeoxyuridine; MEFs, mouse embryo fibroblasts; Aph, aphidicolin; IBMX, 3-isobutyl-1-methylxantine; PGE2, prostaglandin E2; CTX, cholera toxin; MNase, micrococcal nuclease.
References
- Alleva, D. G., Burger, C. J., and Elgert, K. D. (1993). Tumor growth increases Iamacrophage synthesis of tumor necrosis factor-alpha and prostaglandin E2, changes in macrophage suppressor activity. J. Leukoc. Biol. 53, 550-558. [DOI] [PubMed] [Google Scholar]
- Angus, S. P., Mayhew, C. N., Solomon, D. A., Braden, W. A., Markey, M. P., Okuno, Y., Cardoso, M. C., Gilbert, D. M., and Knudsen, E. S. (2004). RB reversibly inhibits DNA replication via two temporally distinct mechanisms. Mol. Cell Biol. 24, 5404-5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avni, D., Yang, H., Martelli, F., Hofmann, F., ElShamy, W. M., Ganesan, S., Scully, R., and Livingston, D. M. (2003). Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell 12, 735-746. [DOI] [PubMed] [Google Scholar]
- Beavo, J. A., and Reifsnyder, D. H. (1990). Primary sequence of cyclic nucleotide phosphodiesterase isozymes and the design of selective inhibitors. Trends Pharmacol. Sci. 11, 150-155. [DOI] [PubMed] [Google Scholar]
- Beavo, J. A., Rogers, N. L., Crofford, O. B., Hardman, J. G., Sutherland, E. W., and Newman, E. V. (1970). Effects of xanthine derivatives on lipolysis and on adenosine 3′,5′-monophosphate phosphodiesterase activity. Mol. Pharmacol. 6, 597-603. [PubMed] [Google Scholar]
- Blomhoff, H. K., Blomhoff, R., Stokke, T., deLange, D. C., Brevik, K., Smeland, E. B., Funderud, S., and Godal, T. (1988). cAMP-mediated growth inhibition of a B-lymphoid precursor cell line Reh is associated with an early transient delay in G2/M, followed by an accumulation of cells in G1. J. Cell Physiol. 137, 583-587. [DOI] [PubMed] [Google Scholar]
- Blomhoff, H. K., Smeland, E. B., Beiske, K., Blomhoff, R., Ruud, E., Bjoro, T., Pfeifer-Ohlsson, S., Watt, R., Funderud, S., and Godal, T. (1987). Cyclic AMP-mediated suppression of normal and neoplastic B cell proliferation is associated with regulation of myc and Ha-ras protooncogenes. J. Cell Physiol. 131, 426-433. [DOI] [PubMed] [Google Scholar]
- Bosco, G., Du, W., and Orr-Weaver, T. L. (2001). DNA replication control through interaction of E2F-RB and the origin recognition complex. Nat. Cell Biol. 3, 289-295. [DOI] [PubMed] [Google Scholar]
- Brum-Fernandes, A. J., Morisset, S., Bkaily, G., and Patry, C. (1996). Characterization of the PGE2 receptor subtype in bovine chondrocytes in culture. Br. J. Pharmacol. 118, 1597-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzalini, O., Perucca, P., Riva, F., Stivala, L. A., Bianchi, L., Vannini, V., Ducommun, B., and Prosperi, E. (2003). p21CDKN1A does not interfere with loading of PCNA at DNA replication sites, but inhibits subsequent binding of DNA polymerase delta at the G1/S phase transition. Cell Cycle 2, 596-603. [PubMed] [Google Scholar]
- Chau, B. N., Borges, H. L., Chen, T. T., Masselli, A., Hunton, I. C., and Wang, J. Y. (2002). Signal-dependent protection from apoptosis in mice expressing caspase-resistant Rb. Nat. Cell Biol. 4, 757-765. [DOI] [PubMed] [Google Scholar]
- Chen, T. T., and Wang, J. Y. (2000). Establishment of irreversible growth arrest in myogenic differentiation requires the RB LXCXE-binding function. Mol. Cell Biol. 20, 5571-5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew, Y. P., Ellis, M., Wilkie, S., and Mittnacht, S. (1998). pRB phosphorylation mutants reveal role of pRB in regulating S phase completion by a mechanism independent of E2F. Oncogene 17, 2177-2186. [DOI] [PubMed] [Google Scholar]
- Christoffersen, J., Smeland, E. B., Stokke, T., Tasken, K., Andersson, K. B., and Blomhoff, H. K. (1994). Retinoblastoma protein is rapidly dephosphorylated by elevated cyclic adenosine monophosphate levels in human B-lymphoid cells. Cancer Res. 54, 2245-2250. [PubMed] [Google Scholar]
- Coleman, R. A., Smith, W. L., and Narumiya, S. (1994). International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev. 46, 205-229. [PubMed] [Google Scholar]
- Darzynkiewicz, Z., Bruno, S., Del Bino, G., Gorczyca, W., Hotz, M. A., Lassota, P., and Traganos, F. (1992). Features of apoptotic cells measured by flow cytometry. Cytometry 13, 795-808. [DOI] [PubMed] [Google Scholar]
- Darzynkiewicz, Z., Li, X., and Gong, J. (1994). Assays of cell viability: discrimination of cells dying by apoptosis. Methods Cell Biol. 41, 15-38. [DOI] [PubMed] [Google Scholar]
- Diffley, J. F. (1996). Once and only once upon a time: specifying and regulating origins of DNA replication in eukaryotic cells. Genes Dev. 10, 2819-2830. [DOI] [PubMed] [Google Scholar]
- Dimitrova, D. S., and Berezney, R. (2002). The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell Sci. 115, 4037-4051. [DOI] [PubMed] [Google Scholar]
- Dimitrova, D. S., and Gilbert, D. M. (2000). Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat. Cell Biol. 2, 686-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova, D. S., Todorov, I. T., Melendy, T., and Gilbert, D. M. (1999). Mcm2, but not RPA, is a component of the mammalian early G1-phase prereplication complex. J. Cell Biol. 146, 709-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan, L. L., Kim, J. S., Zhang, Y., Bart, R. D., Sun, Y., Holtzman, D. M., and Gutmann, D. H. (1999). Differential effects of cAMP in neurons and astrocytes. Role of B-raf. J. Biol. Chem. 274, 25842-25848. [DOI] [PubMed] [Google Scholar]
- Dumont, J. E., Jauniaux, J. C., and Roger, P. P. (1989). The cyclic AMP-mediated stimulation of cell proliferation. Trends Biochem. Sci. 14, 67-71. [DOI] [PubMed] [Google Scholar]
- Dutta, A., and Bell, S. P. (1997). Initiation of DNA replication in eukaryotic cells. Annu. Rev. Cell Dev. Biol. 13, 293-332. [DOI] [PubMed] [Google Scholar]
- el Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E., Kinzler, K. W., and Vogelstein, B. (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817-825. [DOI] [PubMed] [Google Scholar]
- Elgert, K. D., Alleva, D. G., and Mullins, D. W. (1998). Tumor-induced immune dysfunction: the macrophage connection. J. Leukoc. Biol. 64, 275-290. [DOI] [PubMed] [Google Scholar]
- Ferlini, C., Di Cesare, S., Rainaldi, G., Malorni, W., Samoggia, P., Biselli, R., and Fattorossi, A. (1996). Flow cytometric analysis of the early phases of apoptosis by cellular and nuclear techniques. Cytometry 24, 106-115. [DOI] [PubMed] [Google Scholar]
- Fickenscher, H., Stamminger, T., Ruger, R., and Fleckenstein, B. (1989). The role of a repetitive palindromic sequence element in the human cytomegalovirus major immediate early enhancer. J. Gen. Virol. 70(Pt 1), 107-123. [DOI] [PubMed] [Google Scholar]
- Flores-Rozas, H., Kelman, Z., Dean, F. B., Pan, Z. Q., Harper, J. W., Elledge, S. J., O'Donnell, M., and Hurwitz, J. (1994). Cdk-interacting protein 1 directly binds with proliferating cell nuclear antigen and inhibits DNA replication catalyzed by the DNA polymerase delta holoenzyme. Proc. Natl. Acad. Sci. USA 91, 8655-8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furstenthal, L., Kaiser, B. K., Swanson, C., and Jackson, P. K. (2001). Cyclin E uses Cdc6 as a chromatin-associated receptor required for DNA replication. J. Cell Biol. 152, 1267-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, N., Detivaud, L., Doerig, C., and Meijer, L. (1999). ATP-site directed inhibitors of cyclin-dependent kinases. Curr. Med. Chem. 6, 859-875. [PubMed] [Google Scholar]
- Gutzkow, K. B., Naderi, S., and Blomhoff, H. K. (2002). Forskolin-mediated G1 arrest in acute lymphoblastic leukaemia cells: phosphorylated pRB sequesters E2Fs. J. Cell Sci. 115, 1073-1082. [DOI] [PubMed] [Google Scholar]
- Hannon, G. J., and Beach, D. (1994). p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 371, 257-261. [DOI] [PubMed] [Google Scholar]
- Harbour, J. W., and Dean, D. C. (2000). The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 14, 2393-2409. [DOI] [PubMed] [Google Scholar]
- Harper, J. W., Adami, G. R., Wei, N., Keyomarsi, K., and Elledge, S. J. (1993). The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805-816. [DOI] [PubMed] [Google Scholar]
- Helin, K. (1998). Regulation of cell proliferation by the E2F transcription factors. Curr. Opin. Genet. Dev. 8, 28-35. [DOI] [PubMed] [Google Scholar]
- Hirai, H., Roussel, M. F., Kato, J. Y., Ashmun, R. A., and Sherr, C. J. (1995). Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell Biol. 15, 2672-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi, T., Garber, S. S., and Aldrich, R. W. (1988). Effect of forskolin on voltage-gated K+ channels is independent of adenylate cyclase activation. Science 240, 1652-1655. [DOI] [PubMed] [Google Scholar]
- Jiang, W., Wells, N. J., and Hunter, T. (1999). Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc. Natl. Acad. Sci. USA 96, 6193-6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost, H. G., and Steinfelder, H. J. (1987). Forskolin inhibits insulin-stimulated glucose transport in rat adipose cells by a direct interaction with the glucose transporter. Mol. Pharmacol. 31, 279-283. [PubMed] [Google Scholar]
- Jordan, A., Hadfield, J. A., Lawrence, N. J., and McGown, A. T. (1998). Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med. Res. Rev. 18, 259-296. [DOI] [PubMed] [Google Scholar]
- Jordan, M. A. (2002). Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anti.-Canc. Agents 2, 1-17. [DOI] [PubMed] [Google Scholar]
- Kaelin, W. G., Jr. (1999). Functions of the retinoblastoma protein. Bioessays 21, 950-958. [DOI] [PubMed] [Google Scholar]
- Kelly, T. J., and Brown, G. W. (2000). Regulation of chromosome replication. Annu. Rev. Biochem. 69, 829-880. [DOI] [PubMed] [Google Scholar]
- Kelman, Z. (1997). PCNA: structure, functions and interactions. Oncogene 14, 629-640. [DOI] [PubMed] [Google Scholar]
- Kiriyama, M., Ushikubi, F., Kobayashi, T., Hirata, M., Sugimoto, Y., and Narumiya, S. (1997). Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 122, 217-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, E. S., Buckmaster, C., Chen, T. T., Feramisco, J. R., and Wang, J. Y. (1998). Inhibition of DNA synthesis by RB: effects on G1/S transition and S phase progression. Genes Dev. 12, 2278-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, K. E., Booth, D., Naderi, S., Sever-Chroneos, Z., Fribourg, A. F., Hunton, I. C., Feramisco, J. R., Wang, J. Y., and Knudsen, E. S. (2000). RB-dependent S phase response to DNA damage. Mol. Cell Biol. 20, 7751-7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenza, A., Sutkowski, E. M., and Seamon, K. B. (1989). Forskolin: a specific stimulator of adenylyl cyclase or a diterpene with multiple sites of action? Trends Pharmacol. Sci. 10, 442-447. [DOI] [PubMed] [Google Scholar]
- Leone, G., DeGregori, J., Yan, Z., Jakoi, L., Ishida, S., Williams, R. S., and Nevins, J. R. (1998). E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev. 12, 2120-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. F., Desai, S. D., Li, T. K., Mao, Y., Sun, M., and Sim, S. P. (2000). Mechanism of action of camptothecin. Ann. NY Acad. Sci. 922, 1-10. [DOI] [PubMed] [Google Scholar]
- Lundberg, A. S., and Weinberg, R. A. (1998). Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell Biol. 18, 753-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y., Hurwitz, J., and Massague, J. (1995). Cell-cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature 375, 159-161. [DOI] [PubMed] [Google Scholar]
- Mimura, S., and Takisawa, H. (1998). Xenopus Cdc45-dependent loading of DNA polymerase alpha onto chromatin under the control of S phase Cdk. EMBO J. 17, 5699-5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittnacht, S. (1998). Control of pRB phosphorylation. Curr. Opin. Genet. Dev. 8, 21-27. [DOI] [PubMed] [Google Scholar]
- Morgan, D. O. (1995). Principles of CDK regulation. Nature 374, 131-134. [DOI] [PubMed] [Google Scholar]
- Morgenstern, J. P., and Land, H. (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naderi, S., and Blomhoff, H. K. (1999). Mad1 expression in the absence of differentiation: effect of cAMP on the B-lymphoid cell line Reh. J. Cell Physiol. 178, 76-84. [DOI] [PubMed] [Google Scholar]
- Nakanishi, M., Robetorye, R. S., Adami, G. R., Pereira-Smith, O. M., and Smith, J. R. (1995a). Identification of the active region of the DNA synthesis inhibitory gene p21Sdi1/CIP1/WAF1. EMBO J. 14, 555-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, M., Robetorye, R. S., Pereira-Smith, O. M., and Smith, J. R. (1995b). The C-terminal region of p21SDI1/WAF1/CIP1 is involved in proliferating cell nuclear antigen binding but does not appear to be required for growth inhibition. J. Biol. Chem. 270, 17060-17063. [DOI] [PubMed] [Google Scholar]
- Negishi, M., Sugimoto, Y., and Ichikawa, A. (1993). Prostanoid receptors and their biological actions. Prog. Lipid Res. 32, 417-434. [DOI] [PubMed] [Google Scholar]
- Niculescu, A. B., III, Chen, X., Smeets, M., Hengst, L., Prives, C., and Reed, S. I. (1998). Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol. 18, 629-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko, V. V., Wong, P., and Howard, B. H. (1997). WAF1 retards S phase progression primarily by inhibition of cyclin-dependent kinases. Mol. Cell Biol. 17, 4877-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod, M. G., Collins, M. K., Rodriguez-Tarduchy, G., and Robertson, D. (1992). Apoptosis in interleukin-3-dependent haemopoietic cells. Quantification by two flow cytometric methods. J. Immunol. Methods 153, 57-65. [DOI] [PubMed] [Google Scholar]
- Pastan, I. H., Johnson, G. S., and Anderson, W. B. (1975). Role of cyclic nucleotides in growth control. Annu. Rev. Biochem. 44, 491-522. [DOI] [PubMed] [Google Scholar]
- Pennaneach, V. et al. (2001). The large subunit of replication factor C promotes cell survival after DNA damage in an LxCxE motif- and Rb-dependent manner. Mol. Cell 7, 715-727. [DOI] [PubMed] [Google Scholar]
- Petersen, B. O., Lukas, J., Sorensen, C. S., Bartek, J., and Helin, K. (1999). Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 18, 396-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podust, V. N., Podust, L. M., Goubin, F., Ducommun, B., and Hubscher, U. (1995). Mechanism of inhibition of proliferating cell nuclear antigen-dependent DNA synthesis by the cyclin-dependent kinase inhibitor p21. Biochemistry 34, 8869-8875. [DOI] [PubMed] [Google Scholar]
- Polyak, K., Lee, M. H., Erdjument-Bromage, H., Koff, A., Roberts, J. M., Tempst, P., and Massague, J. (1994). Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78, 59-66. [DOI] [PubMed] [Google Scholar]
- Regan, J. W. (2003). EP2 and EP4 prostanoid receptor signaling. Life Sci. 74, 143-153. [DOI] [PubMed] [Google Scholar]
- Resnitzky, D., Hengst, L., and Reed, S. I. (1995). Cyclin A-associated kinase activity is rate limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol. Cell Biol. 15, 4347-4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld, C., Goutner, A., Choquet, C., Venuat, A. M., Kayibanda, B., Pico, J. L., and Greaves, M. F. (1977). Phenotypic characterisation of a unique non-T, non-B acute lymphoblastic leukaemia cell line. Nature 267, 841-843. [DOI] [PubMed] [Google Scholar]
- Sampath, D., Rao, V. A., and Plunkett, W. (2003). Mechanisms of apoptosis induction by nucleoside analogs. Oncogene 22, 9063-9074. [DOI] [PubMed] [Google Scholar]
- Saudan, P., Vlach, J., and Beard, P. (2000). Inhibition of S phase progression by adeno-associated virus Rep78 protein is mediated by hypophosphorylated pRb. EMBO J. 19, 4351-4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, J. D. (1991). Cyclic nucleotide-dependent protein kinases. Pharmacol. Ther. 50, 123-145. [DOI] [PubMed] [Google Scholar]
- Seamon, K. B., Vaillancourt, R., Edwards, M., and Daly, J. W. (1984). Binding of [3H]forskolin to rat brain membranes. Proc. Natl. Acad. Sci. USA 81, 5081-5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano, M., Hannon, G. J., and Beach, D. (1993). A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704-707. [DOI] [PubMed] [Google Scholar]
- Sever-Chroneos, Z., Angus, S. P., Fribourg, A. F., Wan, H., Todorov, I., Knudsen, K.E., and Knudsen, E. S. (2001). Retinoblastoma tumor suppressor protein signals through inhibition of cyclin-dependent kinase 2 activity to disrupt PCNA function in S phase. Mol. Cell Biol. 21, 4032-4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr, C. J., and Roberts, J. M. (1995). Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 9, 1149-1163. [DOI] [PubMed] [Google Scholar]
- Skalhegg, B. S., Landmark, B. F., Doskeland, S. O., Hansson, V., Lea, T., and Jahnsen, T. (1992). Cyclic AMP-dependent protein kinase type I mediates the inhibitory effects of 3′,5′-cyclic adenosine monophosphate on cell replication in human T lymphocytes. J. Biol. Chem. 267, 15707-15714. [PubMed] [Google Scholar]
- Spangler, B. D. (1992). Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol. Rev. 56, 622-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner, J. M., Dew-Knight, S., Musahl, C., Kornbluth, S., and Horowitz, J. M. (1998). Negative regulation of DNA replication by the retinoblastoma protein is mediated by its association with MCM7. Mol. Cell Biol. 18, 2748-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasken, K., Andersson, K. B., Erikstein, B. K., Hansson, V., Jahnsen, T., and Blomhoff, H. K. (1994). Regulation of growth in a neoplastic B cell line by transfected subunits of 3′,5′-cyclic adenosine monophosphate-dependent protein kinase. Endocrinology 135, 2109-2119. [DOI] [PubMed] [Google Scholar]
- Todorov, I. T., Attaran, A., and Kearsey, S. E. (1995). BM28, a human member of the MCM2–3-5 family, is displaced from chromatin during DNA replication. J. Cell Biol. 129, 1433-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima, H., and Hunter, T. (1994). p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78, 67-74. [DOI] [PubMed] [Google Scholar]
- Waga, S., Hannon, G. J., Beach, D., and Stillman, B. (1994). The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 369, 574-578. [DOI] [PubMed] [Google Scholar]
- Waga, S., and Stillman, B. (1998). The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 67, 721-751. [DOI] [PubMed] [Google Scholar]
- Wang, J. Y., Knudsen, E. S., and Welch, P. J. (1994). The retinoblastoma tumor suppressor protein. Adv. Cancer Res. 64, 25-85. [DOI] [PubMed] [Google Scholar]
- Wang, X., Guan, J., Hu, B., Weiss, R. S., Iliakis, G., and Wang, Y. (2004). Involvement of Hus1 in the chain elongation step of DNA replication after exposure to camptothecin or ionizing radiation. Nucleic Acids Res. 32, 767-775. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Watanabe, I. (1974). Radiation effects on DNA chain growth in mammalian cells. Radiat. Res. 58, 541-556. [PubMed] [Google Scholar]
- Witz, I. P. (2001). Presence and functions of immune components in the tumor microenvironment. Adv. Exp. Med. Biol. 495, 317-324. [DOI] [PubMed] [Google Scholar]
- Xiong, Y., Hannon, G. J., Zhang, H., Casso, D., Kobayashi, R., and Beach, D. (1993). p21 is a universal inhibitor of cyclin kinases. Nature 366, 701-704. [DOI] [PubMed] [Google Scholar]
- Yarbro, J. W. (1992). Mechanism of action of hydroxyurea. Semin. Oncol. 19, 1-10. [PubMed] [Google Scholar]
- Zarkowska, T., and Mittnacht, S. (1997). Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem. 272, 12738-12746. [DOI] [PubMed] [Google Scholar]
- Zou, L., and Stillman, B. (1998). Formation of a preinitiation complex by S phase cyclin CDK-dependent loading of Cdc45p onto chromatin. Science 280, 593-596. [DOI] [PubMed] [Google Scholar]