

Abstract
Ovarian cancer is particularly aggressive once it has metastasized to the abdominal cavity (stage III). Intraperitoneal (IP) as compared to intravenous (IV) administration of chemotherapy improves survival for stage III ovarian cancer, demonstrating that concentrating chemotherapy at tumor sites has therapeutic benefit; unfortunately, IP therapy also increases toxic side effects, thus preventing its completion in many patients. The ability to target chemotherapy selectively to ovarian tumors while sparing normal tissue would improve efficacy and decrease toxicities. We have previously shown that tumor-tropic neural stem cells (NSCs) dramatically improve the intratumoral distribution of nanoparticles (NPs) when given intracerebrally near an orthotopic brain tumor or into a flank xenograft tumor. Here we show that NPs either conjugated to the surface of NSCs or loaded within the cells are selectively delivered to and distributed within ovarian tumors in the abdominal cavity following IP injection, with no evidence of localization to normal tissue. IP administration is significantly more effective than IV administration and NPs carried by NSCs show substantially deeper penetration into tumors than free NPs. The NSC/NPs target and localize to ovarian tumors within 1 hr of administration. Pt-loaded silica NPs (SiNP[Pt]) were developed that can be transported in NSCs and it was found that NSC delivery of SiNP[Pt] (NSC/SiNP[Pt]) results in higher levels of Pt in tumors as compared to free drug or SiNP[Pt]. To the best of our knowledge, this work represents the first demonstration that cells given IP can target the delivery of drug-loaded NPs.
Keywords: Neural Stem Cells, Ovarian Cancer, Targeted Drug Delivery, Silica Nanoparticles, Cisplatin, Chemotherapy
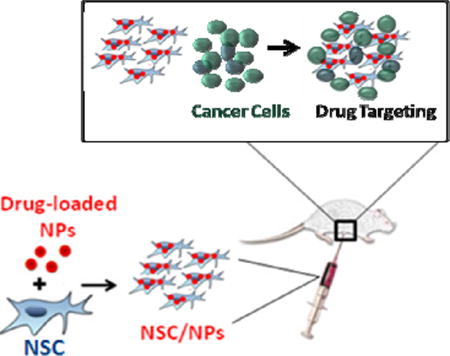
Introduction
Ovarian cancer is a deadly disease that afflicts approximately 22,000 women per year in the US. Sixty-nine percent of all patients with ovarian carcinoma will succumb to their disease. The high mortality of this cancer is largely explained by the fact that the majority (75%) of patients present at an advanced stage, with widespread metastatic disease within the peritoneal cavity.1 Patients with tumors limited to the peritoneal cavity are classified as stage III, and have a 5-year survival rate of only 34%. Frontline therapy consists of surgery combined with chemotherapy. A decade ago, a landmark clinical trial reported a significant survival benefit for ovarian cancer patients who had undergone a combination of intravenous (IV) and intraperitoneal (IP) cisplatin and paclitaxel as compared to IV paclitaxel and cisplatin alone.2 Since, combination IV and IP chemotherapy has been considered the standard of care in stage III ovarian cancer patients who have undergone successful surgery for complete or near complete removal of their tumor burden. However, the IP chemotherapy regimen also has significant toxicities which reduce short-term quality of life and can prevent completion of this regimen in many ovarian cancer patients. A targeted delivery system to concentrate chemotherapy specifically at ovarian tumor sites could substantially enhance both efficacy and reduce side effects for patients undergoing IP chemotherapy.
We have previously shown that the clonal human HB1.F3 neural stem cell (NSC) line is tumor tropic and selectively migrates to a number of malignant solid tumors, including glioma, neuroblastoma, and metastatic breast carcinoma.3–5 These NSCs have been shown to penetrate hypoxic tumor regions, overcoming high interstitial pressures and stiff extracellular matrices.6 NSCs have also been modified to distribute various therapeutic payloads for tumor killing.7–9 One therapeutic approach involves an enzyme-prodrug strategy in which the NSCs are genetically engineered to produce the prodrug activating enzyme cytosine deaminase (CD) which locally converts the prodrug 5-flurocytosine (5-FC) to the active chemotherapeutic 5-fluorouracil (5-FU).3 We recently completed a first-in-human clinical trial with this NSC-mediated treatment for recurrent glioma patients.10 Intracerebral administration of CD-NSCs followed by 5-FC treatment demonstrated safety and proof of concept for brain localized conversion of 5-FC to 5-FU.
Although NSCs can be modified to produce therapeutic genes, or oncolytic viruses, the number of drugs that are amenable to an enzyme/prodrug strategy is limited. Recently, we demonstrated a complementary payload strategy in which NSCs carry nanoparticles (NPs) to tumors. For an intratumoral (IT) injection into an established orthotopic brain tumor, NPs that were conjugated to the surface of NSCs had dramatically improved tumor distribution and retention relative to free NPs.11 Drug-loaded polymeric micelles that were surface conjugated to NSCs showed increased efficacy relative to free NPs following an IT injection in a xenograft model of breast cancer.12 Furthermore, relative to free nanorods, IT injection of gold nanorods loaded inside of the NSCs had improved distribution throughout the tumor, resulting in significantly increased tumor killing following laser application in a xenograft model of breast cancer.13, 14 This work is part of an emerging body of literature demonstrating that many types of tumor tropic cells can be used to improve NP delivery and retention within tumors, including mesenchymal stem cells,15 macrophages,16 and T-cells.17 Of particular note, mesenchymal stem cells administered IP have been used for the targeted delivery of measles virus for treatment of stage III ovarian cancer and there is an ongoing phase I clinical trial for this work.18 This demonstrates the potential of using stem cells given IP for targeted therapy delivery to stage III ovarian cancer. For our studies, we continue to use the HB1.F3.CD21 NSC cell line, because this cell line has been found to be chromosomally and functionally stable, non-tumorigenic, minimally immunogenic (HLA Class II negative), and has already demonstrated clinical safety in brain tumor patients.19
Here we demonstrate that IP administration of these NSCs can deliver both surface bound and internalized NPs to ovarian tumors with remarkable selectivity making them an attractive vehicle for drug targeting (Figure 1). IP administration is significantly more effective than IV administration and NPs carried by NSCs show substantially deeper penetration into tumors than free NPs. The NSC/NPs arrive at ovarian tumors within 1 hr of administration. For drug transport, the NPs must be designed to prevent the drug cargo from affecting the NSCs during ex-vivo loading and during in vivo migration. Pt-containing silica NPs (SiNP[Pt]) were developed that met these criteria and it was demonstrated that when these novel particles were transported in NSCs, the amount of Pt delivered to ovarian tumors was significantly increased. Future work will both optimize the delivery of Pt-drugs with the current system and explore the delivery of other agents using the NSC/NP platform.
Figure 1.

(A) Schematic of desired targeting. (B) Particles used in this manuscript, note only two surface functional groups are shown for each particle type. In reality there are many surface functional groups.
Results and Discussion
First, a relevant orthotopic mouse model of human stage III ovarian cancer was established. Ovarian cancer cells typically spread from the primary tumor site into the ascites fluid of the peritoneal space, followed by secondary tumor seeding onto the serosal surfaces of abdominal organs.20 To approximate this, human OVCAR8.eGFP.ffluc cells were inoculated into the peritoneal cavity of mice, where they seeded predominantly to the greater omentum and serosal surfaces of the liver, kidney, intestines, diaphragm and pancreas. OVCAR8 cells were selected so that the model is relevant to high-grade serous ovarian cancer, which is the most frequent histotype of the patients diagnosed with stage III peritoneal disseminated disease.21 In order to identify an appropriate time for treatment, progression of engrafted tumors was monitored for 4 weeks after IP injection of 2 × 106 ffluc-expressing OVCAR8 cells using bioluminescence imaging (Figure 2A) and by counting the number of visible macrometastases within the peritoneal cavity upon harvest (Figure 2B). The distribution of metastatic ovarian tumors was confirmed on the surfaces of many organs in the IP cavity (Figure 2C)
Figure 2. OVCAR8 ovarian cancer model.

(A) Mice injected IP with 2e6 OVCAR8.ffluc cells developed widespread metastatic disease in the abdomen. (B) Tumor progression over 4 weeks observed in n=3 mice/time point. Micrometastases are observed by week 1. Macrometastases are first seen at week 2, and increase in number, size and distribution over time. Data represents average number of macroscopic metastases +/− SEM. (C) Organ pluck at 3 weeks showing ovarian metastases (small white nodules; red arrows) on liver, pancreas and intestinal surfaces.
Using this model, IP administration of NSCs was tested to determine if the NSCs would selectively localize to and penetrate ovarian tumors. 21 days after injection of the tumor cells, NSCs labeled with the red-fluorescent, hydrophobic membrane dye CellTracker CM-diI, were administered IP. Four days after IP injection, animals were euthanized and tissue sections were prepared. In all cases, red signal corresponding to NSCs was only found in tumors and never in normal tissues even when those tissues were adjacent to the tumors (Figure 3A–D and Figure S1). Unlabeled NSCs carrying surface-bound red fluorescent polystyrene particles were evaluated next as this construct showed excellent tumor-tropic migration in our previous brain tumor study.22 Again, red signal corresponding to the NPs was only found in tumors and never in normal tissue (Figure 3E–H and Figure S1). Finally, NSCs carrying internalized red-fluorescent SiNPs were tested. Once again, red signal corresponding to NPs was only found in tumors (Figure 3I–L and Figure S1).
Figure 3. Neural stem cell tropism to peritoneal ovarian cancer metastasis.

(A,E,I) Representative confocal z-stack images of native neural stem cells (A), and two different NSC/NP constructs in which the NPs are either localized to the cell surface (E) or internalized (I). Cell nuclei and cytoskeleton are respectively visualized using DAPI (blue) and phalloidin staining (pseudo-colored white). Tumor cells expressed GFP (green). Note: The kidney has a very strong background signal when visualizing GFP. (B–D) NSCs labeled with CellTracker CM-DiI demonstrate good distribution in tumor but not in adjacent normal kidney (2 million NSC.DiI in 200 µL PBS injected IP on Day 38; then harvested 4 days post-NSC injection). (F–H) NSCs with surface conjugated NPs demonstrate good distribution in tumor but not adjacent normal liver (4 million NSC.NP in 1 mL PBS injected IP on Day 13: harvested 4 days post-NSC injection). (J–I) NSCs with internalized NPs demonstrate good distribution in tumor but not adjacent normal liver (4 million NSC/NPs in 1 mL PBS injected IP on Day 25: harvested 4 days post-NSC injection). Scale bars: B,F,J: 1000 microns; C,G,K: 500 microns; D,H,L: 200 microns.
Next, delivery and tumor penetration by NSC/SiNPs was compared to SiNPs alone. In these experiments, red fluorescent SiNPs were used and the NSCs were stained with CellMask Deep Red plasma membrane stain (magenta color) to allow for simultaneous tracking of SiNPs and NSCs. Lacking a method to quantitatively measure the number of fluorescent SiNPs taken up by the NSCs, dosing was conservatively set at an equal amount of SiNPs for injection in the SiNP only group as was used for cell loading in the NSC/SiNP group. Since NSCs only take up a small fraction of SiNPs they are exposed to, the NSC/SiNP group will clearly contain fewer SiNPs than the SiNP only group. Thus, this experiment is biased against the NSC/SiNP group. When IP administration of SiNPs alone was compared to NSC/SiNPs, it was found that while the SiNPs did show selective binding to ovarian tumors, the NSC/SiNPs demonstrated substantially deeper penetration into the tumors (Figure 4). This was expected since NSCs are known to migrate towards hypoxic regions6 while NPs are known to have difficulty penetrating dense tumor matrices23.
Figure 4. NSC-delivery improves silica nanoparticle penetration.

(A–B) Representative fluorescent images of tumor sections collected 4 days after IP administration of 2 million NSC/SiNPs or free SiNPs on Day 21. (A) Isolated Far Red, and red channels demonstrate NSC/SiNPs exhibit excellent penetration of peritoneal tumors. Tumor signal (eGFP) is visible in the merged image. (B) Isolated Far Red, and red channels demonstrate SiNPs only surface-localized distribution at peritoneal tumors. Scale bars: 200 microns.
The route of administration was tested to see how IV vs IP administration affected NSC distribution. It was found that when NSC were administered IP, NSCs selectively localized to and penetrated tumors. However, when they were administered IV, little to no signal was observed in either normal tissue or tumors in the IP cavity (Figure 5A,B for OVCAR8 cells and Figure S2 for SKOV3 cells).
Figure 5. Neural stem cell administration route to peritoneal ovarian cancer metastasis.

NSCs labeled with CellMask Deep Red plasma membrane stain (magenta) demonstrate good distribution when (A) administered IP but not when (B) administered IV in a mouse model of peritoneal ovarian metastasis established using OVCAR8.eGFP.ffluc cells. (2 million NSC.CellMask in 200 µL PBS injected IP on Day 21; then harvested 4 days post-NSC injection). Scale bars = 500 microns.
Encouraged by the selective distribution shown by NSC/SiNPs, our next focus was developing a NP formulation of chemotherapy that was amenable for transport by the NSCs. The design criteria for NPs that will be transported by NSCs are different than for NPs that will be used systemically. NPs are loaded into or on NSCs several hours before use, so the NPs need to not release toxic drug that will compromise the NSCs during that preparation period or following IP administration. Pt-based drugs were chosen as an initial payload since these are DNA-damaging agents and less likely to compromise migration than drugs that affect cell components involved in migration (ie. paclitaxel is a microtubule stabilizer and microtubules are involved in cell migration24). It was found that Pt could be encapsulated throughout non-porous SiNPs by including cisplatin during the particle-forming reaction (Figure 6A). By TEM, the particles had similar spherical morphology to control SiNPs with no cargo. By high-resolution TEM, small islands of Pt were visible throughout the loaded SiNPs and not in the control particles (Figure 6B). The SiNP[Pt]s had a mean diameter of 52 nm as measured by Nanosight, which was in good agreement with TEM imaging, and a zeta potential of −17 mV. The mean Pt loading was 14% by weight percent. It was found that the Pt-loading was highly stable when the particles were stored in water but significant leakage was observed in PBS and media (Figure 6C). This is in agreement with previous studies demonstrating etching of SiNPs in PBS and media25 and, indeed, an etched morphology was observed after incubating in these solutions (Figure S3). We denoted the Pt remaining in the SiNPs after this burst release as stably encapsulated. Interestingly, when the activity of Pt released by the NPs after 24 hrs of incubation in PBS was tested, it was found that it had no effect on NSCs even at concentration where free drug is toxic (Figure 6D). We speculate that when the Pt is encapsulated in the SiNPs, the Cl ligands are replaced by siloxane ligands and thus Pt that leaks into solution is in an inactive form. This is supported by the lack of a Cl signal in XPS analysis of the Pt-loaded SiNPs (Figure S4). Collectively, this led us to hypothesize that the NSCs could be loaded with these particles as some Pt would be released during the loading conditions, but this Pt would be inactive and the stably encapsulated Pt would be loaded into the NSCs.
Figure 6.

Preparation of Pt-loaded SiNPs. (A) Synthetic scheme (B) TEM and high resolution TEM imaging of control SiNPs (i, iii) and Pt-loaded SiNPs (ii, iv), (Scale bar = 50 nm in (i, ii), and 10 nm in (iii. iv). (C) After 24 hrs preincubation in PBS, 48 % of the Pt remains stably bound in the SiNPs (pellet) (D) Pt released from the SiNPs after 24 hrs (supernatant in C) showed no toxicity on the NSCs up to 72 hrs after treatment.
Prior to conducting loading studies, the toxicity on the NSCs of the SiNP[Pt]s and relevant controls was tested, with 5% DMSO treatment serving as a positive control for cell killing (Figure 7A). For IC50 calculations, the level of killing with DMSO was treated as complete killing. Free cisplatin effectively killed NSCs with an IC50 of 30.3 µM one day after treatment, 8.75 µM two days after treatment and 1.90 µM three days after treatment. When NSCs were treated with SiNP[Pt]s, no killing was observed one or two days after treatment for the doses tested. Three days after treatment, killing was finally observed with an IC50 of 21.3 µM. This suggested that NSCs could be loaded with SiNP[Pt]s and maintain their function for at least 24 hrs. No toxicity was observed when NSCs were treated with SiNPs diluted in an identical fashion to the SiNP[Pt]s. When NSCs were loaded by incubating them with SiNP[Pt] for 1 hr, loading of 1.7–3.5 µg Pt/million cells was achieved (Figure 7B). At this loading level, NSC migration was unimpaired when evaluated by an in vitro migration assay (Figure 7C). In this assay, migration was evaluated over 4 hours since that is representative of the time it would take to prepare NSC/NPs, administer them and wait for them to reach the ovarian tumors. NSCs or NSC/SiNP[Pt]s were seeded on a membrane placed above either 5% BSA (negative control) or tumor conditioned media (from U87 glioma cells – a known positive control for NSCs). After 4 hours, cells that had migrated to the bottom of the membrane were quantified. NSCs and NSC/SiNP[Pt]s migrated towards the tumor conditioned media to a similar extent and neither migrated towards the BSA.
Figure 7.

NSC can be loaded with NP[Pt] without compromising viability or migration A) SiNPs show no toxicity and SiNP[Pt] show no toxicity for 2 days B) NSCs can be reliably loaded with SiNP[Pt] C) loaded NSCs have unchanged migration in vitro.
Having demonstrated unimpaired behavior in vitro for NSC carrying SiNP[Pt]s, in vivo distribution was measured for our final experiment. Mice were again implanted with OVCAR8 cells to establish the orthotopic model of stage III ovarian cancer. Three weeks after cells were injected, four groups of 12 mice were treated with either PBS (control), free cisplatin, SiNP[Pt]s or NSC/SiNP[Pt]s. The Pt dose was normalized between treatment groups by measuring Pt concentration by inductively coupled plasma mass spectrometry (ICP-MS) in the NSC/SiNP[Pt]s group immediately before use and then adjusting the other two groups, free cisplatin and SiNP[Pt]s, to exactly match the dose. Three mice per group were euthanized 1, 2, 6 and 24 hrs after treatment and tumors and five major organs (stomach, lungs, mesentery, kidneys, liver) were harvested. Up to six tumors per animal and the major organs were digested using 70% nitric acid at 80 °C so that Pt content could be measured by ICP-MS. For all time points, NSC/SiNP[Pt] delivery resulted in significantly more Pt accumulation in tumors than free Cisplatin and SiNP[Pt] treatments. The variation from tumor to tumor is high for the NSC/NP group, which might be expected since these tumors grow in a wide variety of locations in the IP cavity with variable exposure to the fluid in the cavity. Very little Pt was found in the other organs for any of the treatments and no major differences were observed between the treatments for accumulation in healthy organs (Figure S5). A future goal is to further enhance the delivery, perhaps by physical manipulation of the IP cavity during NSC/SiNP[Pt] administration as is commonly done during clinical treatment of patients. Nonetheless, the NSC/SiNP[Pt] formulation resulted in the highest amount of Pt delivered to tumors.
CONCLUSIONS
Here we demonstrate that NSCs selectively transport NPs to metastatic ovarian tumors in the IP cavity. Following IP injection, the NSCs carry both surface bound and internalized NPs exclusively to tumors, sparing normal tissue. IP injection is significantly more effective than IV administration, which showed very little delivery of NSC/NPs to abdominal ovarian tumors. When given IP, NSC/NPs are more effective than NPs alone at penetrating tumors. Drug-loaded NPs that will be transported by cells must have minimal drug release for an initial period likely greater than 1 hr, which is quite different than NPs designed for IV use. We developed a formulation of Pt in non-porous SiNPs that, after an initial burst release of inactive Pt, contained stably encapsulated Pt such that these drug-loaded NPs could be loaded into NSCs and transported. A head to head comparison of IP injection of free cisplatin, SiNP[Pt] or NSC/SiNP[Pt] showed that the NSC/SiNP[Pt] s resulted in more Pt at the tumor sites. Delivery was accomplished in less than 1 hour and the Pt levels persisted for at least 24 hrs. To the best of our knowledge, this work represents the first demonstration that cells given IP can target the delivery of drug-loaded NPs to tumors.
MATERIALS AND METHODS
Instrumentation
Dynamic light scattering (DLS) and zeta potential (ZP) measurements were performed on a Brookhaven 90 Plus/BI-MAS Instrument (Brookhaven Instruments, New York). DLS measurements were obtained by performing 5 runs at 30 s per run and the ZP values by measuring 10 runs involving 30 cycles per run. All nanoparticle solutions were filtered through a 0.45 µm cellulose filter prior to performing DLS and ZP measurements.
The concentration of nanoparticles as well as their mean diameter were measured using the nanoparticles tracking analysis technique (Malvern NanoSight NS300 instrument, NTA software). Measurement were obtained by performing 3 runs of 60 seconds each and sample flow rate was controlled and kept constant during the acquisition using the NanoSight syringe pump
Transmission electron microscopy (TEM) images were obtained with an FEI Tecnai T12 transmission electron microscope at an accelerating voltage of 120keV and images were taken with a Gatan Ultrascan 2K CCD camera. NPs dispersed in water at an optimal concentration were drop cast onto glow-discharged, 300-mesh carbon/formvar coated grids and allowed to dry before imaging. High resolution TEM characterization was obtained with a JEOL JEM-2100F at an acceleration voltage of 200 kV equipped with Gatan Orius Camera. Grids used were 400 mesh copper with Ultrathin Carbon Film on Lacey Carbon support. Confocal microscopic images were taken on a Zeiss LSM700 confocal microscope at 40×, 20× and 10×. XPS analysis was performed with a Kratos Axis Ultra DLD X-ray photoelectron spectrometer with an operating pressures range from 10−9 to 10−8 torr. Prior to analysis, nanoparticles pellet was dried at 200°C in a vacuum oven and powder was mounted on the instrument’s sample holder.
Materials
All organic and inorganic compounds and solvents were purchased from Sigma Aldrich.
Cell Culture
All cells were cultured and maintained at 37°C in a humidified incubator (Thermo Electron Corporation, CA, USA) containing 5% CO2. Neural stem cells were cultured in Dulbecco’s Modifed Eagle’s Medium (DMEM; Invitrogen, CA, USA) supplemented with 10% fetal bovine serum (Gemini Bio, CA, USA), 1% L-glutamine (Invitrogen) and 1% penicillin–streptomycin (Invitrogen). OVCAR8 and Skov3 cells were cultured in RPMI 1640 medium (Gibco, CA, USA) supplemented with 10% fetal bovine serum (Gemini Bio, CA, USA), 1% L-glutamine (Invitrogen) and 1% penicillin–streptomycin (Invitrogen). When the cells reached 80% confluency, they were passaged using a 0.25% trypsin/ethylenediaminetetraacetic acid solution (Invitrogen); media was changed every 2–3 days. U87 human glioma cell lines were obtained from American Type Culture Collection and cultured in the same medium as neural stem cells. U87 cells were used to generate glioma-conditioned media by replacing culture media with serum-free media when cells were 80–100% confluent, followed by a 48 hrs incubation period. NSC cell lines including the human, v-myc immortalized, HB1.F3 NSC line, were obtained from Seung Kim (University of British Columbia, Canada).
External conjugation of NPs to NSC surface
NSCs were biotinylated as described previously26 by incubating in 4°C 1 mM NaIO4 /PBS solution for 20 min in the dark, followed by a 90 min incubation in 0.5 mM biotin hydrazide/DMEM (pH 6.5; Sigma, MO, USA) at RT. Rinsed NSCs were used for coupling to nile-red-loaded, streptavidin-conjugated polystyrene NPs (SPHERO™ Streptavidin Coated Fluorescent Particles SVFP-0556-5,Sphereotech, IL, USA). These particles had an effective particle diameter of 798 nm (0.0137 polydispersity index value) and surface charge of (−21.32 ± 3.20 mV) as assessed by Dynamic light scattering and zeta potential measurements (n = 5). Surface-conjugation was achieved by incubating biotinylated cells for 20 min with a suspension of NPs in DMEM at 4°C. Any uncoupled particles remaining in the supernatant were removed by repetitive washing. The NP-coupled NSCs were then trypsinized and concentrated via centrifugation.
Internalization of NPs by NSC
NP internalization was achieved by incubating NSCs for 1 hr with a suspension of NPs (either SiNP[Pt] or red fluorescent SiNPs [30 nm, sicastar®-redF, Micromod, 40-00-301]) in serum-containing DMEM at 37° C. Any particles not internalized were removed by repetitive PBS washing. The NP-loaded NSCs were then trypsinized and concentrated via centrifugation.
Microscopic imaging of surface-associated and internalized NPs in vitro
Suspensions of NSCs and NSC-NP constructs (1 × 107 cells/ml) were fixed in 4% paraformaldehyde, rinsed in 0.1% Tween/PBS, then stained for 15 min at RT in the dark with a PBS solution containing AlexaFluor 488-conjugated phalloidin (1:200; Life Technologies, CA, USA) and 4-,6-diamidino-2-phenylindole (DAPI; 1µg/ml) to stain cellular filamentous-actin and nuclei, respectively. Cells were pelleted, rinsed and then encapsulated within 1% (weight/volume) low-melting-point agarose (Sigma) to stabilize the cells for imaging. The agarose suspension (200 µl) was placed on a glass slide where a coverslip was used to create a thin gel layer that was polymerized upon exposure to 4°C for 10 min. Images were acquired using a confocal microscope (Zeiss, Oberkochen, Germany) equipped with a 100× oil immersion objective. Each image represents a z-stack compiled from 1-µm optical slices spanning the entire thickness of the cell.
In Vivo Orthotopic Ovarian Cancer Model Development
NOD-SCID mice (Charles River) that were 7 weeks old were anesthetized with isoflurane before inoculation with 2 million OVCAR-8.eGFP.ffluc human ovarian cancer cells via intraperitoneal injection. Each week for 4 weeks, tumors from n=3 mice were harvested and sectioned to visualize and quantify the number of both microscopic and macroscopic IP mets.
In Vivo NSC/NSC-NP Tropism in Orthotopic Ovarian Cancer Model
Tropism Assessment
NOD-SCID mice (Charles River) that were 7 weeks old were anesthetized with isoflurane before inoculation with 2 million SKOV3 cells via intraperitoneal injection. After 13 days, mice (n = 5) were injected IP with either diI labeled NSCs, NSCs surface conjugated to polystyrene nanoparticles, or NSCs containing internalized SiNPs. 4 days post NSC injection, tumors were harvested and sectioned to visualize resulting NSC tropism to IP mets.
Administration Route Assessment
NOD-SCID mice that were 7 weeks old were anesthetized with isoflurane before inoculation with 2 million OVCAR8 or SKOV3 cells via intraperitoneal injection. After 21 days, mice (n = 8 per cell line) were injected with CellMask-labeled NSCs either IV or IP. 2 days post NSC injection, tumors were harvested and sectioned to visualize resulting NSC tropism to IP mets.
Free NP vs Internalized NP comparison
6 NOD-SCID mice and 6 nude mice that were 7 weeks old were anesthetized with isoflurane before inoculation with 2 million OVCAR8 cells via intraperitoneal injection. After 21 days, mice (3 NOD-SCID and 3 nude per treatment) were injected IP with 200uL of either free NPs or 2e6 NSCs with internalized SiNPs. 4 days post NSC injection, tumors were harvested and sectioned to visualize resulting NSC tropism to IP mets.
Synthesis of Cisplatin-Loaded Silica Nanoparticles (SiNP-Cis)
Cisplatin loaded Silica Nanoparticles were synthesized using a standard reverse w/o microemulsion protocol for Silica nanoparticles synthesis that we modified by incorporating cis-diamineplatinum(II) dichloride (cisplatin) in the preparation. 12 mg of Cisplatin powder and 40 mL of tetraethyl orthosilicate (TEOS) were added to a quaternary microemulsion system that contains a mixture of 7.7 mL cyclohexane, 2 mL Triton X-100, 1.6 mL hexanol, and 0.34 mL MilliQ water. This mixture was stirred at 750 rpm (position 3 on an IKA-C-MAG-HS stir plate) for 5 h, at room temperature, followed by the addition of 100 µL ammonium hydroxide. The reaction mixture was allowed to stir at 750 rpm for 16 h, at room temperature followed by addition of ethanol to break the microemulsion and precipitate NPs out of solution. SiNP-Cis was collected by centrifugation (3220 g, 10 min). The supernatant was discarded, the NP pellet was resuspended in 2 mL EtOH and transferred to a 2 mL eppendorf. SiNP-Cis was washed via repeated centrifugation 2 more times with EtOH and 3 more times with MilliQ water (21,000 g, 1.5 min). The NP solution was sonicated in between washes to assist their redispersion back into solution. After the final wash, SiNP-Cis was dispersed in 0.5 mL MilliQ water and stored at 4° C. The amount of Pt loaded in SiNP-Cis was quantified by inductively coupled mass spectrometry (ICP-MS). A small fraction of SiNP-Cis was digested in 70% HNO3 and it was diluted with 2% HNO3 prior to injection to ICP-MS. A standard curve was generated using serial dilutions of a 1 ppm solution of Pt standard solution (Spex CertiPrep). Encapsulation efficiency was 16%. Data were analyzed quantitatively in Excel. To prepare empty SiNPs, the exact same procedure was followed except the addition of cisplatin during the reaction.
Quantification of Pt-Loading in NSCs by Inductively Coupled Mass Spectrometry
NSCs were plated at 2×104 cells/cm2 density for 3 days until they reach ~100% confluency. Old media was removed and fresh DMEM was added, followed by the addition of SiNP-Cis. NSCs were incubated with SiNP-Cis at 37 °C for 1 h. After 1 h, the media containing free NPs were removed and cells were washed with PBS once to remove unbound NPs. NSCs were lifted off the surface by incubating with 0.25% trypsin at 37 °C for 1 min, and harvested by centrifugation (450 g, 3 min). Cells were resuspended via pipetting and gentle vortexing in PBS and total number of cells were measured Guava EasyCyte. A small fraction of the cell solution (10µL) was added to a 15 mL flat bottom tube, digested with 70% HNO3 at RT for 1 h, and diluted with 2% HNO3 prior to injection to ICP-MS. A standard curve was generated using serial dilutions of a 1 ppm solution of Pt standard solution (Spex CertiPrep). Data was analyzed quantitatively in Excel.
In Vitro Cytotoxicity Assay
Freshly trypsinized NSCs were plated at 4000 cells per 100 µL DMEM into each well of a 96-well plate and allowed to adhere to the plate overnight. The next day, old media was removed and 95 µL of pre-warmed fresh DMEM was added to each well together with 5 µL of treatment solutions. Free drug treatment stocks contain 14 concentrations of cisplatin solutions ranging from 0 to 5.12 mM of cisplatin; NP-Cis treatments stocks contain the same 14 concentrations of Cisplatin as the free Cisplatin stocks. Those concentrations were matched between both conditions based on the amount of Pt measured by ICPMS in both initial stocks of free cisplatin and NPs Cis. Final concentrations of cisplatin in each well range from 0 to 256 uM. Each treatment concentration was tested in triplicates. Water and DMSO were used as negative and positive controls, repectively. Water was used because the NPs are stored dispersed in water. After each time point (Day1, Day2, and Day3), old media was removed and 100 µL of fresh media containing 0.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to each well. Cells were then incubated at 37 °C for 1 h or until purple formazan crystals formed inside the cells. Media was removed and 100 µL of DMSO was then added to each well followed by measurement of absorbance at 570 nm using a plate reader. Cell viability was calculated based on the amount of formazan produced by live cells and data was normalized to the untreated control condition.
In Vitro Transwell Boyden Migration Assay
Boyden chamber chemotaxis assays were performed to assess the tumor tropism of NSCs after being labeled with SiNP-Cis. As previously described, in a 24-well plate, 500 µL of 5% BSA/DMEM solution (as negative control) or FBS-free U87 condition media was added to the lower chamber in each well. Polycarbonate inserts with pore size of 8.0 µm were placed carefully into the wells and cell suspensions of 1×105 cells in 250 µL 5% BSA/DMEM were added above the Transwell chamber, and they were incubated at 37 °C for 4 h. After the incubation period, SiNP-labeled NSCs that had migrated across the Transwell membrane toward the condition media were quantified and compared to untreated NSCs. Quantification was accomplished by transferring the inserts to a new 24-well plate, with 200 µL of accutase already added into each well. After incubating the chambers in accutase at 37 °C for 10min, the 24-well plate was shaken at r.t. for another 10 min to further release the cells from the membrane. Detached NSCs were collected and washed once with PBS in a 96-well V-bottomed plate. Cells were resuspended in 1:1 of PBS to ViaCount solution, and their cell numbers were counted by Guava ViaCount assay using Guava EasyCyte System.
In Vivo Pt Biodistribution Study in Orthotopic Ovarian Cancer Model
NOD-SCID mice (Charles River) that were 7 weeks old were anesthetized with isoflurane before inoculation with 2 million OVCAR8 cells via intraperitoneal injection. After 21 days, mice (n = 12) were injected with free cisplatin, Pt-SiNP, or 10 million NSC/SiNP[Pt]. All groups except PBS control had matching amount of Pt. Control animal received PBS injection. After 1, 2, 6, and 24 h, 3 mice per group were euthanized. Up to six tumors per animal and 5 organs (liver, kidney, stomach, intestines, and mesentery) were collected from the peritoneal cavity of each animal and each sample was digested with 70% HNO3 at 80° C for 16 h. Samples were diluted with 2% HNO3 prior to injection to ICP-MS. All animal protocols were approved by the City of Hope Institutional Animal Care and Use Committee. Animals were housed in an AAALAC-accredited facility and were given food and water ad libitum.
Statistical Analysis
Data are presented as mean ± SEM unless otherwise stated. Statistical significance was determined using a two-tailed students t-test (* p <0.05) unless otherwise stated.
Supplementary Material
Figure 8.

NSC/SiNP[Pt] given IP enhance the retention of Pt at tumor sites. At 1, 2, 6 and 24 hours, tumors from NSC/SiNP[Pt]-treated animals had higher levels of Pt than those treated with free cisplatin or drug-loaded NPs. Statistical significance was determined using a two-tailed students t-test (* p <0.05, ** p<0.01, *** p<0.001).
Acknowledgments
We gratefully acknowledge: ICP-MS instrumentation under the supervision of Nathan Dalleska at the Environmental Analysis Center at the California Institute of Technology, High resolution TEM images and XPS data were taken at the Center for Electron Microscopy and Microanalysis at the University of Southern California. TEM imaging was conducted at the Electron Microscopy core at City of Hope with Marcia Millier, Zhuo Li and Ricardo Zerda, and fluorescent imaging was conducted at the Digital and Light Microscopy core at City of Hope with Brian Armstrong and Loren Quintanar, flow cytometry data was collected at the Analytical Cytometry Core at City of Hope with Lucy Brown. Studies were supported by generous funding from STOP Cancer, The Rosalinde and Arthur Gilbert Foundation, the Alvarez Family Foundation, The Anthony F. & Susan M. Markel Foundation, Jeanne and Bruce Nordstrom, the NIH R01CA197359, RM was supported by a fellowship from the California Institute for Regenerative Medicine (CIRM) Grant Number TG2-01150) and the Ladies Auxiliary of the Veterans of Foreign Wars, MG and UN were supported by the CIRM Bridges Training Program (Grant Number TB1-01177). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or any other agency of the State of California. Materials Transfer Information is available from the City of Hope Office of Technology Licensing (http://www.cityofhope.org/research/support/center-for-applied-technology-development/office-of-technology-licensing/Pages/default.aspx). Research reported in this publication included work performed in Biostatistics core with Leanne Goldstein supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Additional images showing NSC-mediated NP delivery to peritoneal metastasis are provided (Figure S1). We further demonstrate superior NSC tropism to ovarian mets upon IP vs. IV administration in another ovarian model made using a SKOV-3 cancer line (Figure S2). TEM images of SiNPs after incubation in serum containing media (Figure S3) are provided, as well as Pt accumulation levels in organs after IP injection of cisplatin, SiNP[Pt] or NSC/SiNP[Pt] (Figure S4). These materials are available free of charge via the Internet at http://pubs.acs.org.
DISCLOSURE
KSA, AJA are officers and board members of TheraBiologics, Inc., a clinical stage biopharmaceutical company supporting development of NSC-mediated cancer treatments.
The remaining authors declare no conflict of interest.
References
- 1.Lengyel E. Ovarian cancer development and metastasis. The American journal of pathology. 2010;177:1053–64. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA, Gynecologic Oncology G. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 3.Aboody KS, Najbauer J, Metz MZ, D’Apuzzo M, Gutova M, Annala AJ, Synold TW, Couture LA, Blanchard S, Moats RA, et al. Neural Stem Cell–Mediated Enzyme/Prodrug Therapy for Glioma: Preclinical Studies. Science translational medicine. 2013;5:184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aboody KS, Najbauer J, Danks MK. Stem and progenitor cell-mediated tumor selective gene therapy. Gene therapy. 2008;15:739–52. doi: 10.1038/gt.2008.41. [DOI] [PubMed] [Google Scholar]
- 5.Zhao D, Najbauer J, Annala AJ, Garcia E, Metz MZ, Gutova M, Polewski MD, Gilchrist M, Glackin CA, Kim SU, et al. Human neural stem cell tropism to metastatic breast cancer. Stem cells. 2012;30:314–25. doi: 10.1002/stem.784. [DOI] [PubMed] [Google Scholar]
- 6.Zhao D, Najbauer J, Garcia E, Metz MZ, Gutova M, Glackin CA, Kim SU, Aboody KS. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Molecular cancer research : MCR. 2008;6:1819–29. doi: 10.1158/1541-7786.MCR-08-0146. [DOI] [PubMed] [Google Scholar]
- 7.Auffinger B, Ahmed AU, Lesniak MS. Oncolytic virotherapy for malignant glioma: translating laboratory insights into clinical practice. Frontiers in oncology. 2013;3:32. doi: 10.3389/fonc.2013.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnarr K, Mooney R, Weng Y, Zhao D, Garcia E, Armstrong B, Annala AJ, Kim SU, Aboody KS, Berlin JM. Gold Nanoparticle-Loaded Neural Stem Cells for Photothermal Ablation of Cancer. Advanced Healthcare Materials. 2013;2:976–982. doi: 10.1002/adhm.201300003. [DOI] [PubMed] [Google Scholar]
- 9.Frank RT, Edmiston M, Kendall SE, Najbauer J, Cheung CW, Kassa T, Metz MZ, Kim SU, Glackin CA, Wu AM, et al. Neural stem cells as a novel platform for tumor-specific delivery of therapeutic antibodies. PloS one. 2009;4:e8314. doi: 10.1371/journal.pone.0008314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Portnow J, Synold TW, Badie B, Tirughana R, Lacey SF, D'Apuzzo M, Metz MZ, Najbauer J, Bedell V, Vo T, et al. Neural stem cell-based anti-cancer gene therapy: a first-in-human study in recurrent high grade glioma patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rachael Mooney YW, Tirughana-Sambandan Revethiswari, Garcia Elizabeth, Hernandez Valerie, Aramburo Soraya, Annala Alexander, Berlin Jacob, Aboody Karen. Human Neural Stem Cell-Mediated Targeting of Nanoparticles to Brain Tumors. Future Oncology Accepted. 2014 doi: 10.2217/fon.13.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mooney R, Weng Y, Garcia E, Bhojane S, Smith-Powell L, Kim SU, Annala AJ, Aboody KS, Berlin JM. Conjugation of pH-responsive nanoparticles to neural stem cells improves intratumoral therapy. Journal of controlled release : official journal of the Controlled Release Society. 2014;191:82–9. doi: 10.1016/j.jconrel.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mooney R, Roma L, Zhao D, Van Haute D, Garcia E, Kim SU, Annala AJ, Aboody KS, Berlin JM. Neural stem cell-mediated intratumoral delivery of gold nanorods improves photothermal therapy. ACS nano. 2014;8:12450–60. doi: 10.1021/nn505147w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnarr K, Mooney R, Weng Y, Zhao D, Garcia E, Armstrong B, Annala AJ, Kim SU, Aboody KS, Berlin JM. Gold nanoparticle-loaded neural stem cells for photothermal ablation of cancer. Advanced healthcare materials. 2013;2:976–82. doi: 10.1002/adhm.201300003. [DOI] [PubMed] [Google Scholar]
- 15.Li L, Guan Y, Liu H, Hao N, Liu T, Meng X, Fu C, Li Y, Qu Q, Zhang Y, et al. Silica nanorattle-doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS nano. 2011;5:7462–70. doi: 10.1021/nn202399w. [DOI] [PubMed] [Google Scholar]
- 16.Madsen SJ, Baek SK, Makkouk AR, Krasieva T, Hirschberg H. Macrophages as cell-based delivery systems for nanoshells in photothermal therapy. Annals of biomedical engineering. 2012;40:507–15. doi: 10.1007/s10439-011-0415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephan MT, Stephan SB, Bak P, Chen J, Irvine DJ. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials. 2012;33:5776–87. doi: 10.1016/j.biomaterials.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mader EK, Butler G, Dowdy SC, Mariani A, Knutson KL, Federspiel MJ, Russell SJ, Galanis E, Dietz AB, Peng K-W. Optimizing patient derived mesenchymal stem cells as virus carriers for a Phase I clinical trial in ovarian cancer. Journal of Translational Medicine. 2013;11:1–14. doi: 10.1186/1479-5876-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aboody KS, Najbauer J, Metz MZ, D'Apuzzo M, Gutova M, Annala AJ, Synold TW, Couture LA, Blanchard S, Moats RA, et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Science translational medicine. 2013;5:184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le O. Patterns of peritoneal spread of tumor in the abdomen and pelvis. World journal of radiology. 2013;5:106–12. doi: 10.4329/wjr.v5.i3.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nature communications. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mooney R, Weng Y, Tirughana-Sambandan R, Valenzuela V, Aramburo S, Garcia E, Li Z, Gutova M, Annala AJ, Berlin JM, et al. Neural stem cells improve intracranial nanoparticle retention and tumor-selective distribution. Future oncology. 2014;10:401–15. doi: 10.2217/fon.13.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliver Rev. 2001;46:149–168. doi: 10.1016/s0169-409x(00)00131-9. [DOI] [PubMed] [Google Scholar]
- 24.Etienne-Manneville S. Microtubules in Cell Migration. Annual Review of Cell and Developmental Biology. 2013;29:471–499. doi: 10.1146/annurev-cellbio-101011-155711. [DOI] [PubMed] [Google Scholar]
- 25.Park S-J, Kim Y-J, Park S-J. Size-Dependent Shape Evolution of Silica Nanoparticles into Hollow Structures. Langmuir. 2008;24:12134–12137. doi: 10.1021/la8028885. [DOI] [PubMed] [Google Scholar]
- 26.Krishnamachari Y, Pearce ME, Salem AK. Self-Assembly of Cell–Microparticle Hybrids. Advanced Materials. 2008;20:989–993. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.