

SUMMARY
The distribution of sense and antisense strand DNA mutations on transcribed duplex DNA contributes to the development of immune and neural systems along with the progression of cancer. Because developmentally matured B cells undergo biologically programmed strand-specific DNA mutagenesis at focal DNA/RNA hybrid structures, they make a convenient system to investigate strand-specific mutagenesis mechanisms. We demonstrate that the sense and antisense strand DNA mutagenesis at the immunoglobulin heavy chain locus and some other regions of the B cell genome depends upon localized RNA processing protein complex formation in the nucleus. Both the physical proximity and coupled activities of RNA helicase Mtr4 (and Senataxin) with the noncoding RNA processing function of RNA exosome determine the strand specific distribution of DNA mutations. Our study suggests that strand-specific DNA mutagenesis-associated mechanisms will play major roles in other undiscovered aspects of organismic development.
TOC image
Limitation of local RNA-DNA structures restricts DNA mutational asymmetry.
INTRODUCTION
Patterns of DNA mutagenesis in somatic cells are implicated in many biological events, including the development of immunity, neurogenesis, and the onset of cancer (Alexandrov et al., 2013a; Alexandrov et al., 2013b; Basu et al., 2011; Haradhvala et al., 2016; Keim et al., 2013; Lodato et al., 2015; Madabhushi et al., 2015; Wei et al., 2016). Regulated mutagenesis on both strands of DNA directs the formation of programmed DNA double strands breaks (DSBs) during immunoglobulin (Ig) locus recombination events. In many cancers and in several neurodegenerative diseases, an enhanced bias of mutations to the non-template (or sense) strand of DNA has been observed (Haradhvala et al., 2016). Cancer genomes may generate asymmetric strand mutagenesis via transcription-coupled and/or replication-coupled mechanisms. Since both transcription and replication are essential requirements for cellular function, robust preventive measures are required to forestall strand asymmetric mutations (Haradhvala et al., 2016). The formation of secondary DNA structures such as R-loops or G-quadruplexes that attract various DNA mutator proteins and likewise pathophysiological insults that have affinity for single-strand DNA substrates have been considered to skew transcription-associated strand asymmetric DNA mutagenesis toward the sense strand of coding genes (Santos-Pereira and Aguilera, 2015). Stalled RNA polymerase II occurring at sites of strand asymmetric DNA mutagenesis and associated secondary DNA structures, if not rapidly resolved, may cause the replication complex to collide with the transcription complex and create genomic instability and, occasionally, chromosomal translocations (Kim and Jinks-Robertson, 2012).
Activation Induced Deaminase (AID) mediated DNA hypermutation occurs in various regions of the immunoglobulin loci, catalyzing proper antibody diversification via class switch recombination (CSR) and somatic hypermutation (SHM) and occurring with relatively small asymmetry on both the sense and the antisense strands of DNA (Alt et al., 2013; Basu et al., 2011; Keim et al., 2013; Pefanis and Basu, 2015; Pefanis et al., 2014). In the variable regions of the immunoglobulin gene loci, AID mutates both strands of DNA to engineer high affinity antibodies (Pefanis et al., 2014; Wang et al., 2014). Via a related but different mechanism, in the switch region, AID mutates both strands of DNA to generate DNA double strand breaks which permit class switch recombination (Basu et al., 2011). Thus, evolutionary mechanisms that promote mutagenesis to both antisense and sense strands of DNA may be coupled with AID’s function in B cells. These reasons allow us to follow AID’s activity as an ideal model system to understand regulatory events associated with antisense DNA strand mutagenesis.
In this study, we report that the noncoding RNA processing activity of RNA exosome, a 3′–5′ RNA exonuclease, controls sense and antisense strand DNA mutagenesis and prevents an increased asymmetric mutagenesis burden using a mechanism that relies on the unexpected DNA/RNA hybrid unwinding activities of the Mtr4 and Senataxin RNA helicases. We unravel the role of non-coding RNA helicases in regulating transcription-associated DNA template (antisense) strand mutagenesis in mammalian genomes. Our data demonstrate that RNA helicases, RNA exosome, and mutator are exquisitely positioned relative to each other in the nucleus of B cells to control DNA strand specific mutagenesis. Thus, we propose a new mechanism of DNA mutagenesis regulation which, if not functional, places the burden of strand asymmetric mutation mediated genomic instability within a particular locus.
RESULTS
A model system to investigate nuclear RNA exosome complex and associated factors, in vivo
In vitro studies demonstrate that the RNA exosome by itself has potent RNA degradation activity but lacks the ability to access DNA-associated nascent RNA (Januszyk and Lima, 2014). Since it is proposed that RNA exosome’s role involves rapid degradation/processing of noncoding RNAs generated from various transcription complexes, it is likely that there are associated protein factors that provide rapid access to ncRNA substrates that are otherwise present inside DNA heteroduplexes. The RNA exosome complex consists of 11 subunits, the core complex formed from 9 subunits (Exosc1–9) and two additional subunits (Exosc10, Dis3) providing 3′–5′ RNA exonuclease activity (Januszyk and Lima, 2014). Without the Exosc3 subunit, the RNA exosome fails to function in cells. To purify the RNA exosome complex from B cells, we generated a mouse model in which we introduced a Tandem Affinity Purification tag (TAP–tag) consisting of a FLAG-epitope and a biotinylation site. Details of the Exosc3TAP-tag/TAP-tag mouse model may be found in STAR methods and Fig. S1A and S1B. Knowing that activity of the Exosc3 gene is important for catalysis of class switch recombination in B cells (Pefanis et al., 2014), CSR efficiency in B cells from Exosc3TAPtag/TAPtag mice was evaluated and found to be comparable with that seen in Exosc3WT/WT B cells, firmly establishing that the epitope tagged Exosc3 forms a complex with a functional RNA exosome moiety in B cells (Fig. S1C). B cells isolated from the Exosc3TAPtag/TAPtag mouse model were activated for CSR and used for immunoprecipitation experiments with high stringency conditions using two steps, first with anti-Flag antibody and second with a streptavidin column used for eluting biotinylated protein complexes (approach outlined in Fig. S1D). Purification of the complex was first verified by direct western blotting of the immunoprecipitate with anti-Exosc3 antibody, streptavidin (to detect biotinylated Exosc3) and anti-Flag antibody (Fig. S1E). The double tagged purified RNA exosome complex (and a parallel purification preparation obtained from Exosc3WT/WT B cells expressing BirA enzyme, an engineered lysine residue biotinylation enzyme) were analyzed by mass spectrometry for composition (details in supplementary materials and methods). The purified Exosc3 TAP-tagged RNA exosome complex consisted of all 11 subunits of the RNA exosome complex (Fig. 1A–C; details of protein peptides in Suppl. Table 1a and 1b).
Figure 1. TAP-tagged RNA exosome complex in antigen activated B cells is localized in the nucleus and associates with RNA/DNA helicase activity of Mtr4.
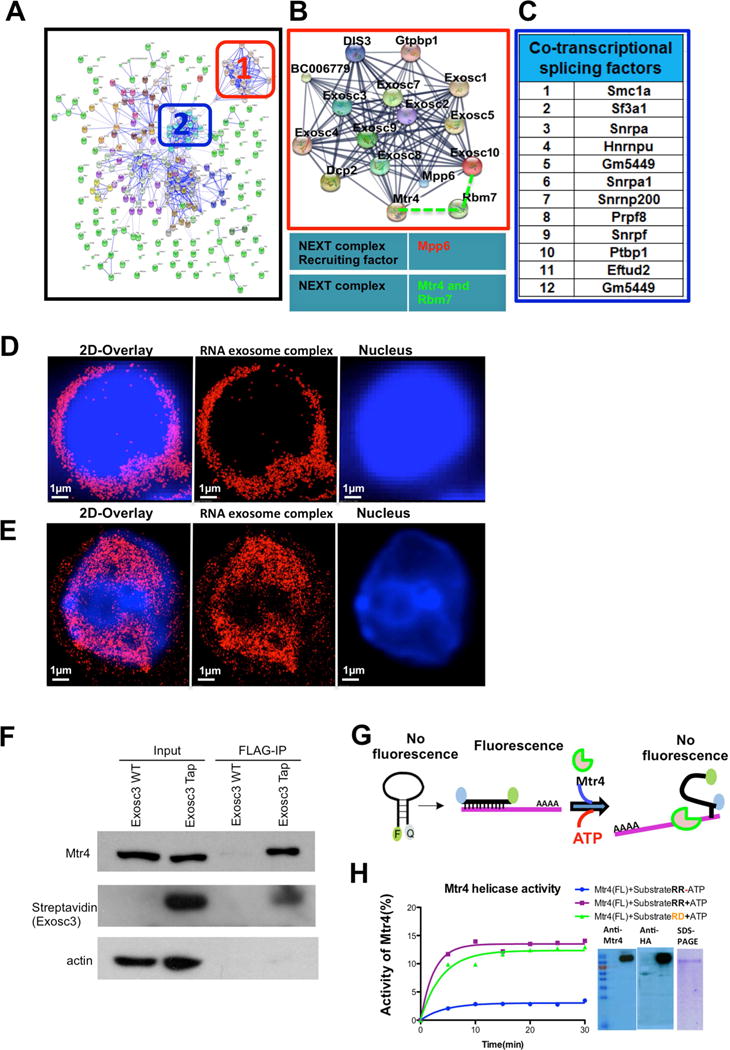
(A) Interaction network of RNA exosome complex associated proteins. In-silico network analysis of proteins identified by mass-spectrometry was performed using the STRING database combined with experimental data. (B) Higher magnification view of the boxed region (1) shows RNA exosome complex associating with NEXT complex components Mtr4 and RBM7 along with Mpp6 (recruiter of the NEXT complex), respectively. (C) Identification of the most stringent interacting partners shown in panel A cluster 2; these proteins are related with cotranscriptional splicing pathways. (D) Reconstructed single color 3D-STORM image from a data set of 50,000 frames of fixed splenic B cells before CSR stimulation, in which Exosc3 was labeled with AlexaFluor647 and the nucleus with DAPI. (E) Single color 3D STORM image of B cells after CSR stimulation, in which Exosc3 was labeled with AlexaFluor647 and the nucleus with DAPI. (F) Flag immunoprecipitation reactions (IPs) were performed on Exosc3TAP B cells to demonstrate Exosc3 and Mtr4 interaction. (G) Schematic representation of molecular beacon (6-FAM and Iowa Black) based helicase assay for Mtr4 protein. (H) Two substrates (RNA-RNA and RNA-DNA) were designed with 5′–6FAM and 3′Iowa black linked to 17mer RNA molecule, which annealed with 22mer RNA and DNA complimentary strand to evaluate the helicase activity of Mtr4 for both substrates. hMtr4 full length protein was expressed in HEKA-293T cells and purified by affinity tag. Expression of the protein was confirmed by western blot using anti Mtr4 antibodies. Percentage activity of hMtr4(FL) was calculated by pseudo-first order rate constant describing fluorescence decay upon ATP addition [[k (min-1)]. All reactions were performed in triplicate, and error bars represent standard deviation among independent reactions. All of the 3D STORM imaging was performed in three different B cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is x100. Scale bars: 1μm(D) &(E). Also, see supplementary video 1 and 2.
To identify cofactors that are of immediate interest, pathway analysis (Ingenuity Pathway Analysis (IPA), parameters described in STAR Methods) was performed, grouping cofactors based on their known functions. We identified two major clusters of proteins from the RNA exosome tandem affinity purification (Fig. 1A). The two clusters were; (1) The core RNA exosome complex and associated proteins with details shown in Fig. 1B and (2) Non-core splicing pathway associated proteins that have function in transcription coupled mRNA splicing pathways, with details in Fig. 1C. It is evident that the core RNA exosome physically interacts with components of the nuclear exosome targeting complex (NEXT), Mtr4 and RBM7 (Lubas et al., 2011), and with the NEXT complex recruitment factor, Mpp6 (Fig. 1B).
In activated B cells, our preferred experimental system, the TAP-tagged RNA exosome complex could be evaluated by microscopy, making the system tractable to imaging experiments. Strikingly, following activation with a cocktail of stimuli, the RNA exosome complex was found to translocate into the nucleus of B cells, thereby providing a unique opportunity to investigate the function of the nuclear RNA exosome complex. As shown by three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy (3D-STORM) images, unstimulated B cells have very low levels of TAP-tagged nuclear RNA exosome (Fig. 1D) but a significant amount of the RNA exosome complex translocates to the nucleus following 72 hrs of stimuli cocktail treatment (Fig. 1E). Thus, using the Exosc3TAP/TAP B cells, we developed a unique system wherein the nuclear RNA exosome complex could be biochemically and functionally evaluated.
Mtr4 possesses DNA/RNA hybrid helicase activity
In the core RNA exosome complex (Complex 1; Fig. 1, B) we found interaction with Mtr4. We validated the RNA exosome-Mtr4 interaction with co-immunoprecipitation experiments in B cells (Fig. 1F) and also via co-expression with FLAG-tagged RNA exosome complex in 293T cells (Fig. S1F). Mammalian Mtr4 possess RNA helicase activity to unwind RNA hybrids (Johnson and Jackson, 2013), although its DNA/RNA hybrid unwinding activity has not been reported. However, its ortholog in prokaryotes has been reported to unwind RNA secondary structures and DNA/RNA hybrids (Uson et al., 2015). We expressed HA-tagged Mtr4 in HEK293T cells. As shown in Fig. 1H, the purified HA-Mtr4 can be detected by western blotting and runs as a single purified band in a Coomassie Brilliant Blue stained SDS-PAGE. To interrogate the DNA/RNA hybrid helicase activity, we developed an assay in which an RNA strand flanked with a fluorophore (F=FAM) and a quencher (Q=Iowa black) was duplexed with a DNA or RNA strand to form a DNA/RNA or RNA/RNA hybrid substrate. This DNA/RNA or RNA/RNA heteroduplex was analyzed for HA-Mtr4 mediated unwinding in vitro, as schematized in Fig. 1G. Increased unwinding leads to quenching of FAM due to its interaction with Q (Iowa black), permitting evaluation of Mtr4 helicase activity. As shown in Fig. 1H, we find that in these conditions of assay Mtr4 has substantial RNA/DNA helicase activity (in green) and this activity is comparable to RNA/RNA helicase activity (in purple).
Increased proximity of RNA exosome with Mtr4 in the nucleus of activated B cells
Co-immunoprecipitation of RNA exosome with Mtr4 points toward a role of Mtr4 in RNA exosome function. However, Mtr4-RNA exosome interactions may be restricted in the nucleus or occur via different sub-complexes in the nucleus versus the cytoplasm and such variations in the mode of interaction cannot be evaluated by gross co-immunoprecipitation assays. We envision that fine regulation of Mtr4 and RNA exosome interaction can be unraveled and better understood by studying their proximity in the nucleus of activated B cells. We used 3D-STORM super resolution microscopy to evaluate RNA exosome/Mtr4 proximity in primary B cells (detailed in STAR Methods). In the conditions of our assays, we set closest proximity of two proteins to be 36nm (based on the interaction of Exosc3 and Exosc5 subunits of RNA exosome; Fig. S2A, S2B, S2C, and 2G) and distance between two non-interacting proteins (Exosc3 with RFP-HA; Fig. S2D, S2E and 2G) as 205nm. We collected two color 3D STORM images and performed nearest neighbor analysis of Mtr4 and Exosc3 in the nucleus and cytoplasm of activated B cells. As shown in Figs. 2A–D, in the nuclei of B cells there is close proximity of Mtr4 with the RNA exosome complex. It is possible that the increased frequency in proximity of RNA exosome with Mtr4 is influenced by the translocation of the RNA exosome complex to the nucleus of a B cell after stimulation (Fig. 1D) (details of assay in STAR methods). Mtr4 and RNA exosome are located 45±26 nm from each other in the nucleus (Fig. 2D) versus a distance of 68±33 nm in the cytoplasm (Fig. 2E). As shown in Fig. 2F and Figs. S2A–E, the mean proximity of two interacting proteins is 36nm and of two non-interactors is 205 nm. Thus, RNA exosome and Mtr4 may form a complex both in the nucleus and cytoplasm, but based on proximity analysis the conformation and constitution of the complex is likely to be different and may employ alternative mechanisms for ncRNA processing in both compartments.
Figure 2. Analysis of spatial distribution of RNA exosome complex and RNA helicase Mtr4 in the nucleus of mouse B cells.
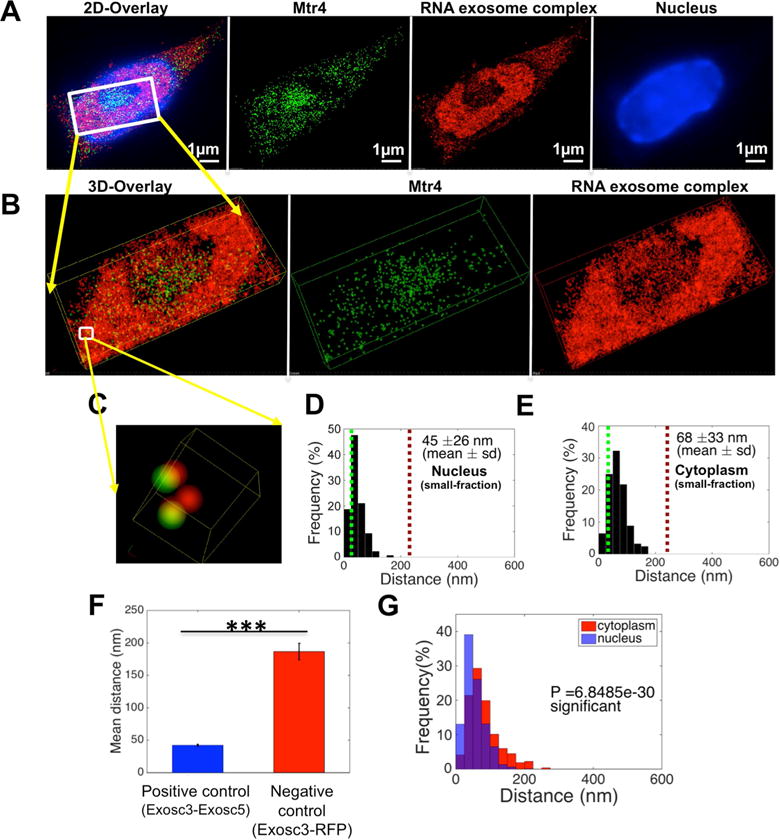
B cells were harvested from Exosc3 TAP-tagged mice and treated with stimulation cocktail for 72h. (A) Reconstructed two color STORM image from: a data set of 50,000 frames with Atto488 labeled Mtr4, AlexaFluor647 labeled RNA exosome complex and DAPI labeled nucleus. (B) Three dimensional views of the boxed region. (C) Higher magnification three dimensional [3D] views of the boxed region in panel B. (D), (E): Histogram plot of distribution of pair interaction of Mtr4 and RNA exosome complex calculated in (D) the nuclear sub-compartment and (E) the cytoplasm by using custom written algorithm “Nearest Neighbors Search” in Matlab (2014b, MathWorks) software. (F) Comparison of the distribution of paired interaction of Exosc3 and 5 in the B cell for positive control and RFP-HA tag & RNA exosome complex in HEK293 cells for negative control. (G) Comparison of the distribution of paired interactions in the nucleus and cytoplasm for RNA exosome complex and Mtr4 was calculated using a Student’s t-test in Matlab (2014b, MathWorks) software and P values are indicated in the graph. All of the 3D STORM imaging was performed in three separate B cells (obtained from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is ×100. Scale bar: 1μm(A). Error bars indicate S.D. (P value: *** <0.001)
AID in close proximity to the RNA exosome complex in the nucleus of activated B cells
We postulated that functional requirements for Mtr4 and RNA exosome proximity could be fruitfully investigated in the context of AID mediated DNA mutagenesis and coupled ncRNA degradation mechanisms. We evaluated the proximity of AID and Exosc3TAP in stimulated B cells. AID localizes predominantly in the cytoplasm and in various regions of the nucleoplasm of activated B cells. When analyzed at single molecule resolution, nuclear AID was in close proximity with RNA exosome complex (Fig. 3A–C). Nearest neighbor analyses show that AID and RNA exosome are separated by 68±34 nm in the nucleus (Fig. 3D) but 143±64 nm in the cytoplasm (Fig. 3E). An independent set of experiments is shown in Fig. S3A–F, where the nuclear proximity of RNA exosome with AID was found to be 64±36 nm (Fig. S3D) versus a cytoplasmic proximity of 147±66 nm (Fig. S3E). An overlap analysis of the nuclear and cytoplasmic proximities between AID and RNA exosome demonstrates that the two factors have the tightest proximity in the nucleus of an activated B cell (Fig. 3F and Fig. S3F; controls staining shown in Fig. S3G). We do note that more than one AID molecule has the potential to be present in a nuclear complex, bridged by other factors protein (Keim et al., 2013), DNA (Goodman et al., 2007) or ncRNAs (Pefanis et al., 2014), given that the distance of two AID molecules in the nucleus is close (95 nm) compared to that seen in the cytoplasm (135 nm) (Fig. S3H). It should be noted that the distribution of nuclear AID in its physiological environment (B cells) (Fig. S3I) differs from that seen in conditions of transient overexpression in the non-physiological environment of HEK293T cells (Fig. S3J). The separation of AID/RNA exosome in the nucleus of 293T cells is 95nm and in the cytoplasm is 114 nm (Figs. S3K–M). Taken together, AID and RNA exosome exist in closest proximity in the nucleus of B cells, and this B cell specific AID/RNA exosome proximity is not observed in a non-B cell environment.
Figure 3. Analysis of spatial distribution of AID and RNA exosome in the nucleus of mouse B cells.
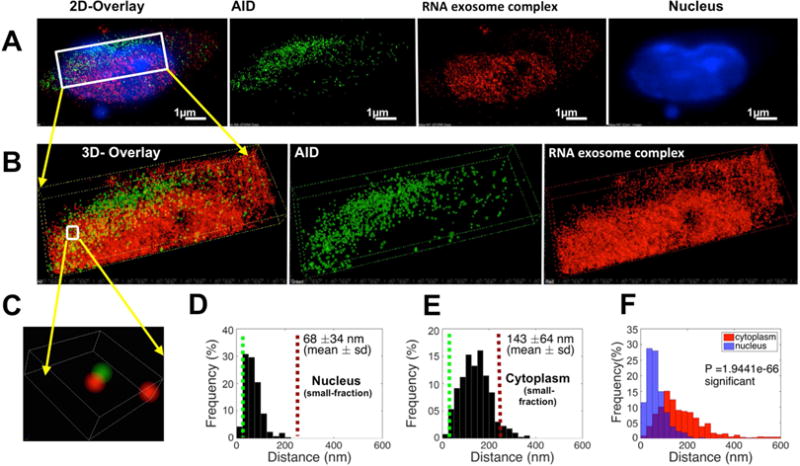
B cells were harvested from Exosc3 TAP-tagged mice following 72 hr of stimulation cocktail treatment (A) Reconstructed two color 3D STORM image from a data set of 50,000 frames with Atto488 labeled AID, AlexaFluor647 labeled Exosc3 and DAPI labeled nucleus. (B) Three dimensional views of the boxed region. (C) Higher magnification three dimensional [3D] views of the boxed region. (D) Histogram of the distribution of interactions of AID and the RNA exosome complex was calculated in the B cell nucleus and (E) in the cytoplasm, by Matlab (2014b, MathWorks) software. (F) Comparison of the distribution of paired interaction of AID and RNA exosome in the nucleus and cytoplasm were calculated by using a Student’s T-test in Matlab (2014b, MathWorks) software and P values are noted in the graph. All of the 3D STORM imaging was performed in three different B cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is ×100 Scale bar: 1μm(A).
AID, Mtr4 proximity is determined by RNA exosome activity in the nucleus of B cells
We wished to determine whether RNA exosome’s RNA processing activity co-localizes Mtr4 and AID in the context of DNA mutagenesis events. It is entirely possible that RNA exosome’s RNase activity is not related with Mtr4 and/or AID. To evaluate the role of functional RNA exosome in promoting the proximity of AID and Mtr4 and eventual complex formation, we performed 3D-STORM analyses in RNA exosome activity proficient and deficient B cells. We used a B cell that has a conditional allele for the Exosc10 gene, a nuclear RNase subunit whose function is important for nuclear RNA exosome function but not for cytoplasmic RNA exosome function (Johnson and Jackson, 2013). As shown in Fig. 4A–C, the distance between AID and Mtr4 in nuclear RNA exosome activity proficient B cells is 145nm in the nucleus and 158 nm in the cytoplasm. However, following inactivation of the nuclear RNA exosome activity (Exosc10COIN/LacZ), the distance of AID and Mtr4 is increased in the nucleus (280 nm) but retained in the cytoplasm (166 nm). As shown in Fig. 4G, from 3D-STORM analyses of 3 individual B cells, the proximity of AID and Mtr4 is perturbed in Exosc10-deficient B cells in the nucleus but not in the cytoplasm. Statistical analyses show that the change in AID-Mtr4 proximity in the nucleus versus the cytoplasm is only significant in the Exosc10-deficient B cells (Fig. 4I) but not in the Exosc10-proficient B cells (Fig. 4H). Moreover, in activity-proficient B cells (Exosc10WT/WT), the proximity of Mtr4 and AID in the nuclear center is 163 nm (Fig. 4J, K, L) and in other parts of the nucleoplasm (labeled as nuclear center-displaced) is 118 nm (Fig. 4J, K, M); the loss of Exosc10 activity leads to an increase in the proximity of Mtr4 and AID, both in the nuclear center and in the other parts of the nucleoplasm (Fig. 4J; see Figs. S4A and S4B for representative examples of nuclear center and nuclear center-displaced regions in Fig. 4J). Finally, in Exosc10COIN/LacZ B cells, cellular expression of both AID and Mtr4 is robust (Fig. S4E). Taken together, these observations unravel the role of nuclear RNA exosome activity in promoting the proximity of Mtr4 and AID in the nucleus, presumably driven by functional requirements to degrade newly transcribed noncoding RNAs expressed from various loci. The small but consistent difference in the proximity of AID-Mtr4 in the nuclear center versus at regions displaced from the center in the nucleoplasm implies subtle changes in the content or interaction-context of the RNA exosome complex in different subnuclear compartments. Microscopy images and movies of Mtr4 and AID localization in RNA exosome nuclear activity proficient (Fig. S4A, S4C) and deficient B cells (Fig. S4B and S4D) are presented.
Figure 4. Analysis of spatial distribution of AID and Mtr4 in the nucleus of B cells isolated from an Exosc10COIN/LacZ mouse.
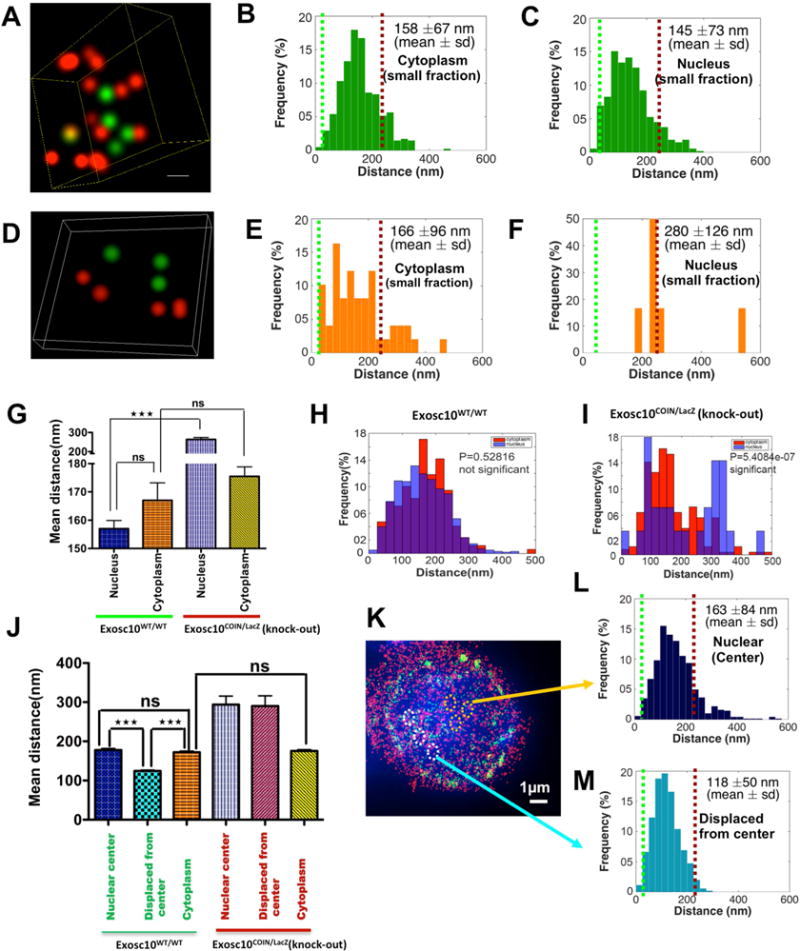
B cells were harvested from the spleen of an Exosc10COIN/LacZ mouse and fixed and prepared for 3D-STORM after 72 hrs of treatment with (1) stimulation cocktail and (2) 4OHT+stimulation cocktail. Reconstructed two color 3D STORM (super-Aresolution) image from a data set of 50,000 frames with Atto488 labeled AID, AlexaFluor647 labeled Mtr4 and DAPI labeled nucleus. Three dimensional views of the boxed region show spatial distribution of AID and Mtr4 molecules inside the nuclei of B cells isolated from (A) wild type cells and (D) Exosc10 knockout cells. Histogram of the distribution of interactions of AID and Mtr4 calculated in the B cell nucleus of (B) wild type & (E) Exosc10 knockout cells and in the corresponding cytoplasms (C) & (F), by using Matlab (2014b, MathWorks) software. (G) Comparison of the distribution of paired interaction of AID and Mtr4 in the nucleus versus cytoplasm by one way ANOVA (Tukey-Kramer test) method in Matlab (2014b, MathWorks) software. Comparison of the distribution of paired interaction of AID and Mtr4 were calculated in the cytoplasm versus nucleus of (H) wild type & (I) Exosc10 knockout B cells using a Student’s t-test in Matlab (2014b, MathWorks) software and P values are noted in the graph. (J) Comparison of the distribution of paired interaction of AID and Mtr4 in the nuclear center versus displaced from center versus cytoplasm by one way ANOVA (Tukey-Kramer test) method in Matlab (2014b, MathWorks) software. (K) Reconstructed two color 3D STORM (super-resolution) image with Alexa488 labeled AID, AlexaFluor647 labeled Mtr4 and DAPI labeled nucleus of wild type Exosc10 cells. Histogram of the distribution of interactions of AID and Mtr4 calculated in (L) nucleus center and (M) displaced from center of wild type cells, by using Matlab (2014b, MathWorks) software. All of the 3D STORM imaging was performed in three different B cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is ×100. Scale bar: 1μm(K). Error bars indicate S.D. (P values: ** <0.01, *** <0.001)
RNA exosome, Mtr4 and Senataxin cooperate to promote DNA antisense strand mutagenesis and DNA double strand break dependent class switch recombination
We generated knockout mutants of AID (AIDmut), Mtr4 (Mtr4mut), and senataxin, a transcription complex-associated RNA/DNA helicase (Setxmut) (Hamperl and Cimprich, 2014). Setx is a well-known RNA/DNA helicase whose function is ubiquitous; this prompted us to evaluate Mtr4’s role in DNA mutagenesis events in conjunction with Setx. The loss-of-function mutants were generated by introducing frameshift mutations in the coding regions of the respective genes in CH12F3 B cell lines. Frameshift mutations were introduced in exon 2 of AID, exon 1 of Setx, and exon 2 of Mtr4 (top, middle, and bottom panels respectively in Fig. S5A). The expression of the AID protein (Fig. S5B) and cellular proliferation (Fig. S6A) of the Setxmut and Mtr4mut cell lines were comparable with control “parental” CH12F3 cells. Following stimulation of parental CH12F3 cells with stimuli cocktail, CSR was observed in approximately 20–30% of the cellular population. As expected, loss of AID expression in AIDmut cells led to complete abolition of CSR (Fig. 5A). Loss of Mtr4 led to a decrease in CSR efficiency whereas loss of Setx had mild effect on CSR efficiency (Fig. 5A and Fig. S5F). The CSR defect in the Mtr4mut CH12F3 cells is partially rescued by the transient transfection of an Mtr4-expressing vector in the cells (Fig. S5D). We generated double mutants of cells that lack both Mtr4 and Setx (Mtr4mut/Setxmut) and found that these cells are remarkably defective in CSR (Fig. 5B and 5C). The lack of Mtr4 protein expression in the Mtr4mut/Setxmut was confirmed by western blotting (Fig. S6C) and the presence of Setx inactivating mutations was detected by sequencing analysis (Fig. S6B). Numerous repeats were performed for each experiment, with 8 independent clones of the Mtr4mut/Setxmut employed in Fig. 5C to generate a comprehensive analysis of the roles of these proteins in catalyzing CSR. In addition, we also demonstrated that at different time points after CSR stimulation, IgM to IgA CSR was affected in the Mtr4mut/Setxmut CH12F3 cells (Fig. S5C). Taken together, we conclude that the RNA helicase activity of Mtr4 and Setx is important for class switch recombination events in the IgH locus of B cells.
Figure 5. Class switch recombination efficiency and DNA-RNA hybrid accumulation and strand specific mutation distribution at IgH 5′ Sμ region in Mtr4 and Setx deficient B cells.
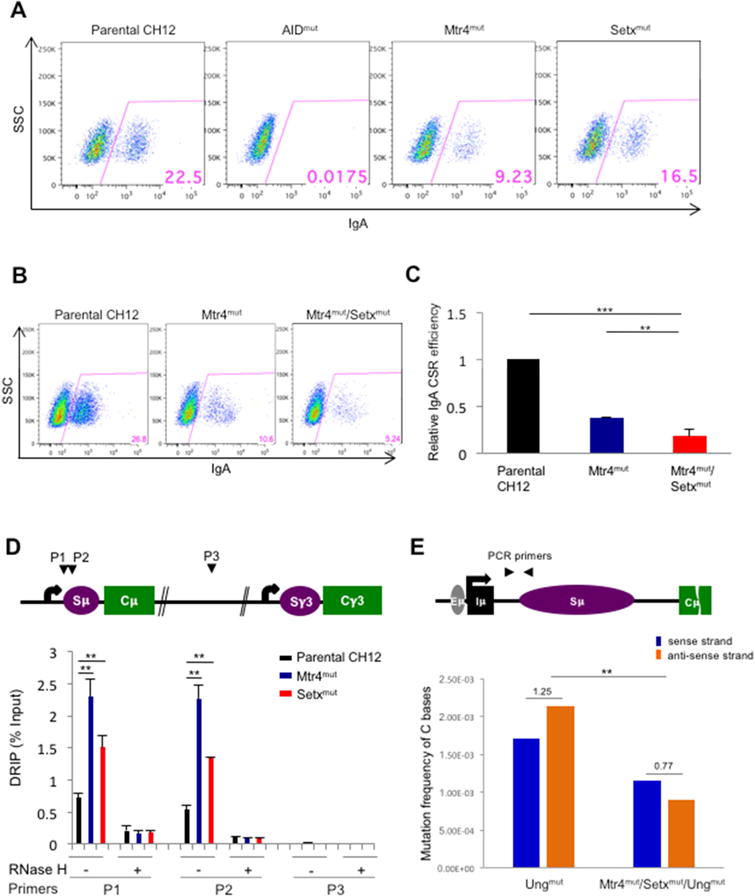
FACS assessment of CSR to IgA of AID, Mtr4 and Setx (single) mutants (A) and Mtr4/Setx double mutant CH12F3 clone (B). Cells were treated for 56h with CSR-stimulating cytokines. (C) IgA CSR efficiency of Mtr4 and Mtr4/Setx mutants was normalized based on CSR levels of parental CH12F3. These results were analyzed from three independently repeated experiments. (D) Schematic map localizing the IgH locus and binding sites of the primer pairs used for DNA-RNA hybrid immunoprecipitation (DRIP) assays. DNA-RNA heteroduplexes were precipitated with the S9.6 antibody and normalized on input. RNase H treated DNA-RNA heteroduplexes were used as a negative control for these experiments. Results were obtained from 4 independent experiments. (E) Schematic map localizing the IgH locus and binding sites of the PCR primer pair for mutation analysis. Mutation frequency of AID-induced somatic hypermutation on sense (non-transcribed) strand and anti-sense (transcribed) strand of IgH 5′Sμ region in Mtr4/Setx/Ung mutant cells compared to Ung mutant CH12F3 cell controls. Ratio of mutation frequency between sense and anti-sense strands is indicated above bar graph. Details of mutations identified by Sanger sequencing in Sup. table 1c.
Error bars indicate S.D. (P values: * <0.05, ** <0.01, *** <0.001)
RNA exosome loss leads to increased ssDNA structures known as R-loops at regions of DNA double strand break formation in the B cell genome (Pefanis and Basu, 2015; Pefanis et al., 2014; Pefanis et al., 2015). In the 5′ regions of the IgH locus switch sequences, we employed the DNA/RNA hybrid immunoprecipitation technique (DRIP) and found enhanced R-loop accumulation following Mtr4 or Setx deletion (Fig. 5D). Concomitantly, following deletion of the UNG gene from these cells to improve mutational detection (details in Fig. S6. D–F), we found an increased ratio of somatic mutation frequency on the sense (nontemplate) DNA strand and a decrease of mutation frequency on the antisense (template) DNA strand in Mtr4 and Setx deficient B cells, and this change of strand mutation ratio was significantly different to what was seen in control Ungmut cells (Fig. 5E). The change in ratio of mutation frequency on sense versus antisense DNA strands between the Ungmut and Mtr4mut/Setxmut/Ungmut was found to be significant, when compared between the hypermutated clones (Fig. 5E). Similar effects on the ratio of DNA strand mutagenesis were also observed in Mtr4mut CH12F3 cells (Fig. S5E), but with reduced overall mutation frequency due to Ung expression in these cells. The details of the mutations analyzed in Figs. 5E and S5E are shown in supplementary table 1c. Collectively, these analyses illustrate that removal of noncoding RNAs associated with the antisense DNA strand by RNA exosome requires the activity of Mtr4 and Setx, without which there is an increased level of ssDNA on the DNA sense strand (non-transcribed/non-template DNA). As would be expected in the case of bidirectional transcription or convergent transcription at switch sequences (Meng et al., 2014; Pefanis et al., 2014; Wang et al., 2016), the possibility exists that ncRNAs associated with the antisense DNA strand are processed by the RNA exosome complex similarly to germline transcripts and provide another mechanism of antisense DNA strand AID access by R-loop formation. However, such an interpretation needs to be approached carefully, as ncRNAs that are associated with the antisense DNA strand are significantly lower in expression level than germline transcripts associated with the sense DNA strand, based on RNA-sequencing data obtained both in the 5′Smu region as well as in the intronic Iμ and Eμ regions (Fig. S6G, H). Both possible mechanisms are outlined in Fig. 7E and various mechanistic possibilities discussed later.
Figure 7. DNA-RNA hybrid and strand specific mutation distribution RNA exosome substrate ncRNA expressing region of Pim1 and CD83 genes in the absence of Mtr4 and Setx.
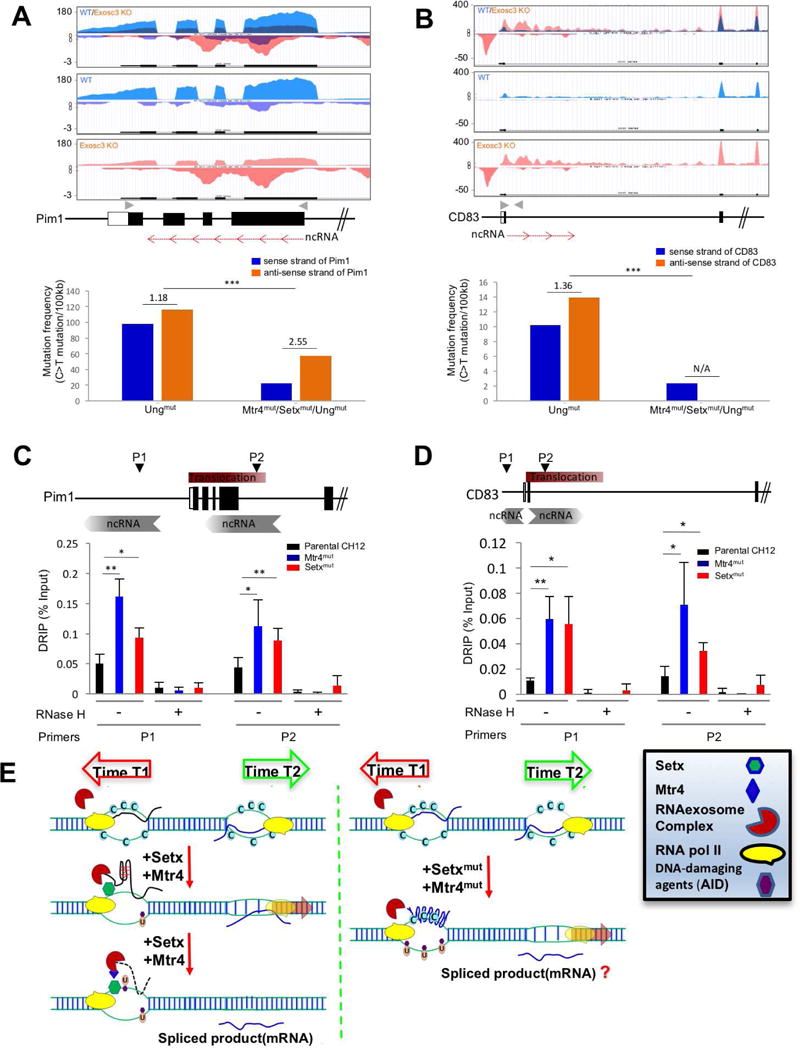
(A, B) RNA exosome substrate ncRNA expression profiles of Pim1 (A) and CD83 (B) loci in WT and Exosc3COIN/COIN mouse B cells (Top). Gray arrowheads indicate primer sites for next generation sequencing (NGS) and red arrows indicate non-coding RNA and its transcription direction on the schematic map of Pim1 and CD83 loci (Middle). Mutation frequency of C bases on sense and anti-sense strands (with respect to Pim1 or CD83 gene transcription in top panel) and ratio of sense strand mutation frequency compare to anti-sense (transcribed) strand mutation frequency in Ung mutant and Mtr4/Setx/Ung mutant CH12F3 cells. Ratio of mutation frequency between sense and anti-sense strand is indicated above bar graph (bottom). (C, D) Schematic map localizing the Pim1 (C) and CD83 (D) loci and binding sites of the primer pairs used for DNA-RNA hybrid immunoprecipitation (DRIP) assays. This experiment was performed as in Fig. 5D. (E) A model elucidating the role of Mtr4 and Setx in restricting ssDNA structure formation at various regions in the genome that have antisense RNA transcription or sense/antisense paired RNA transcription. Time 1 and time 2 represents two separate time points at which two RNAs are expressed inside a genic locus (e.g., Pim1 or CD83). The RNA processing of the two RNAs are potentially through two different mechanisms (e.g., splicing and RNA degradation, respectively). It is to be determined whether the efficient processing of the exosome sensitive ncRNA is important for proper mRNA biogenesis.
Error bars indicate S.D. (P-value: * < 0.05, ** <0.01, *** <0.001); A is obtained from ES cell transcriptome; details of mutations identified by NGS deep sequencing are presented in supplementary table 1c.
c-Myc locus strand specific mutagenesis depends upon the activity of Mtr4 and Setx in unwinding antisense RNAs
B cell lymphomas like DLBCL (diffuse large B cell lymphoma), MM (multiple myeloma) and CLL (chronic lymphocytic leukemia) are caused by inappropriate AID mutagenic activity at various locations in the B cell genome (Basso and Dalla-Favera, 2015). One example of mutagenesis in B cells is in the first intron of the c-Myc locus where a strongly expressed antisense RNA that is a substrate of nuclear RNA exosome activity is found (Fig. 6A). This antisense RNA can be detected easily by RNA-seq in Exosc3-deficient cells (Exosc3COIN/COIN; Fig. 6A bottom panel) but not normal B cells (Exosc3WT/WT; Fig. 6A middle panel). We assessed the ssDNA formation efficiency following Mtr4 and Setx inactivation at this c-Myc intronic region and observed enhanced ssDNA structure formation (Fig. 6B). Strikingly, a comparison of mutations in the antisense versus sense DNA strand of the c-Myc gene corresponding to this intronic sequence demonstrates an Mtr4 and Setx dependent sense/antisense DNA strand mutation mechanism (Fig. 6C). We used Ungmut (base excision repair deficient) and Mtr4mut/Setxmut/Ungmut triple mutant CH12F3 cells to study c-Myc intron associated mutations. In the c-Myc intron there is an increase in the ratio of mutation frequency on the anti-sense DNA strand in comparison to that seen on the sense DNA strand following deletion of Mtr4 and Setx and this change in strand mutation ratio is significantly different to what is seen in the control Ungmut cells. In addition, there is a slight decrease in total mutation frequency in the c-Myc locus in the Mtr4mut/Setxmut/Ungmut cells (Fig. 6C; details of mutational analysis in Sup. Table 1c). Thus, we speculate that the degradation of the exosome sensitive antisense RNA is important for the strand specific distribution of mutations in the c-myc intronic region.
Figure 6. DNA-RNA hybrid and strand specific mutation distribution at the cMyc locus in the absence of Mtr4 and Setx.
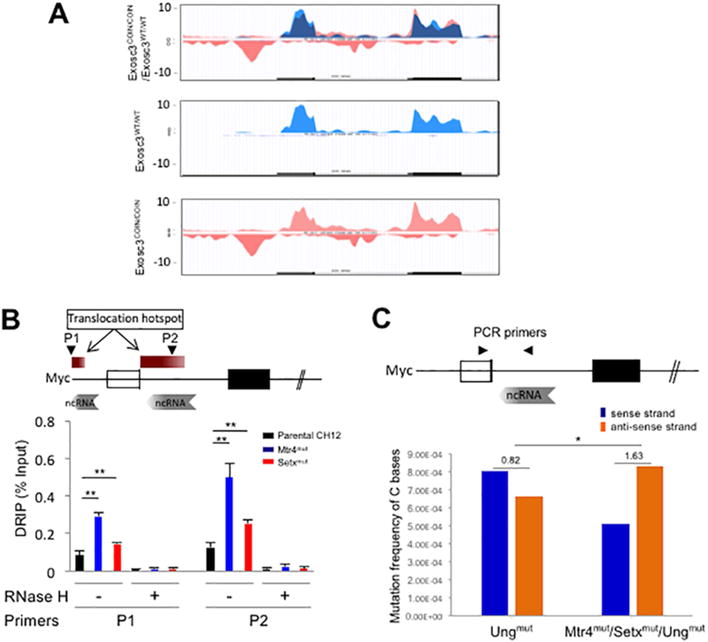
(A) RNA expression profile of the cMyc locus in WT (Exosc3WT/WT; middle panel), Exosc3 k/o (Exosc3COIN/COIN; bottom panel) cells and merged (top panel). RNA exosome substrate antisense RNA shown in pink (bottom panel). (B) Schematic map localizing the cMyc locus and binding sites of the primer pairs used for DRIP assays. DRIP assay to determine single strand DNA structure in the cMyc promoter and intron 1 in Mtr4mut and Setxmut CH12F3 cells. This experiment was performed as described for Fig. 5D. (C) Schematic map localizing the cMyc locus and binding sites of the PCR primer pair for mutation analysis. Mutation frequency of AID-induced somatic hypermutation on sense (non-transcribed) strand and anti-sense (transcribed) strand with respect to cMyc anti-sense RNA transcription (in A) in Mtr4/Setx/Ung mutant CH12F3 cells compared to Ung mutant CH12F3 cell control. Ratio of mutation frequency between sense and anti-sense strand is indicated above bar graph. Details of mutations identified by Sanger sequencing in Sup. table 1c.
A is obtained from ES cell transcriptome; Error bars indicate S.D. (PAvalue: * < 0.05, ** <0.01).
RNA exosome substrate ncRNA transcription associated antisense strand mutations at other regions of the B cell genome as observed in Mtr4, Setx mutant B cells
RNA exosome sensitive RNAs have been found to overlap the sense RNA (i.e., in the same direction as the mRNA inside genic sequences) or span multiple exons in the antisense orientation inside genic sequences. We wondered if the directionality of the RNA exosome sensitive ncRNA transcription is relevant in the determination of strand specific mutagenesis events. In addition, the number of mutations obtained by Sanger sequencing at Sμ or c-Myc intron was not large and alternate assays were required to study the role of Mtr4 and Setx in controlling strand specific mutagenesis with a cohort of a large number of mutations (see. Sup. Table 1c). To address these queries, we selected two different exosome substrate ncRNAs, one in the Pim1 locus and the other in the CD83 locus, and investigated their role in strand mutagenesis using next generation sequencing (NGS). The exosome-sensitive RNA in the Pim1 locus is in the antisense direction to the Pim1 mRNA and spans multiple exons (Fig. 7A; approx. 900 bp in length) whereas the major exosome-sensitive RNA in the CD83 locus is in the same direction as the CD83 mRNA (Fig. 7B). Deep sequencing of either regions unravels a small mutation asymmetry on the sense and antisense strands (with respect to the directionality of the RNA exosome substrate ncRNA) in the control Ungmut cells, perhaps due to the loss of efficient base excision repair or due to inherent replication coupled asymmetric DNA mutagenesis mechanisms that are present in these cells. However, loss of Mtr4 and Setx leads to an increased mutation ratio on the antisense DNA strand relative to the sense DNA strand in the Pim1 locus (Fig. 7A) and this change in strand mutation ratio is significantly different to what is seen in the control Ungmut cells. In contrast, in the CD83 locus where the exosome-sensitive RNA is in the same direction as the CD83 mRNA, mutations are detectable on the DNA sense strand but not on the antisense strand (Fig. 7B). In the Pim1 or the CD83 loci, the overall decrease in the mutation frequency following Mtr4 (and Setx) deletion may be due to inefficient RNA exosome mediated AID targeting, as was shown earlier (Pefanis et al., 2014; Wang et al., 2014). When assayed for RNA/DNA hybrid accumulation, both the relevant region of Pim1 (Fig. 7C) and CD83 (Fig. 7D) accumulate DNA/RNA hybrids in Mtr4mut or Setxmut CH12F3 cells. Thus, it is likely that the RNA exosome substrate RNA unwinding in both the Pim1 and CD83 loci requires the activity of Mtr4 and Setx. One query we wanted to address involves a region normally found unmutated in Ungmut B cells: will Mtr4 and Setx mutations lead to the appearance (or abolition) of specific DNA strand mutations following stabilization of exosome sensitive RNA? To test this possibility, we performed deep sequencing at a region upstream of the CD79b locus (Fig. S7A) and the CD83 locus (Fig. S7B). In the case of CD79b, mutations appeared only in the Ungmut/Mtr4mut/Setxmut cells on the antisense strand (Fig. S7A). On the other hand, detectable mutations on the sense strand at the CD83 upstream region in the Ungmut cells were lost following stabilization of exosome sensitive RNA (Fig. S7B). Taken together, these observations indicate that the activity of RNA exosome to degrade/process nascent ncRNAs with the help of RNA/DNA helicase cofactors Mtr4 and Setx in the nucleus prevents transcription coupled strand asymmetric mutagenesis.
DISCUSSION
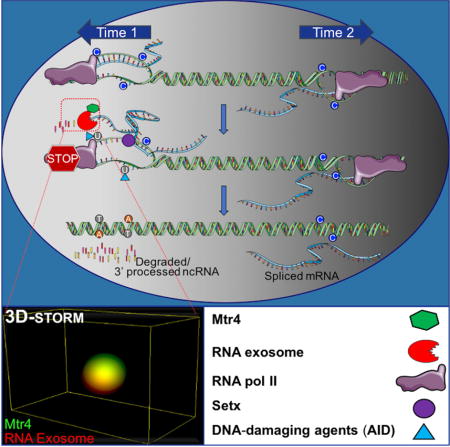
DNA strand asymmetric mutagenesis of mammalian genomes may be modulated by various unknown mechanisms; here we explore the previously unappreciated role of DNA/RNA unwinding followed by 3′-end processing/degradation as a major pathway. We report that the noncoding RNA degradation complex, RNA exosome, utilizes the activities of RNA helicases Mtr4 and Setx to prevent single strand DNA structures like R-loops and to prevent an overwhelming non-template strand DNA mutation burden. The eventual effects of the restriction of strand asymmetric mutagenesis include proper programmed DNA DSB-dependent DNA recombination events as seen for the IgH locus of B cells (failure of which may lead to genomic instability.)
The regulation of many biological processes requires exquisite control over the formation of multi-molecular complexes in specific regions of the cell. Importantly, in these systems, a single protein can evoke different responses depending on its subcellular localization and the interacting partners present in a particular cellular niche. In this context, our study regarding RNA exosome/Mtr4 association and its functional interaction with DNA mutator AID sheds light on time and space restricted protein complex formation in the nucleus. Following stimulation of B cells, RNA exosome undergoes a dramatic migration to the nucleus of B cells (Fig. 1). In the nucleus, it encounters Mtr4 RNA helicase and coordinates efficient strand specific DNA mutagenesis by properly regulating levels of single strand DNA structure formation by post transcriptionally regulating ncRNA levels and associated R-loop formation (Fig. 2 and Fig. 5–7). The IgH locus undergoes DNA mutagenesis and recombination in the nuclear center (Chan et al., 2013) or slightly displaced from the nuclear center of B cells (Holwerda et al., 2013; Yang et al., 2005). Displacement from the nuclear center of the IgH locus could be due to many factors including DNA motion that is associated with passage of time as chromosomes accomplish the long distance DNA interactions required during recombination (Lucas et al., 2014). AID is recruited into the nucleus of B cells during the mitotic phase of the cell cycle (Wang et al., 2016) and mutates DNA predominantly in the early G1 phase (Rush et al., 2004; Wang et al., 2016). We postulate that as B cells recover from M phase and enter G1 phase, chromosome 12 (containing IgH) associates with AID and complexes with RNA exosome, Mtr4, and other relevant factors to orchestrate CSR. It is unlikely that the nuclear lamina associates with the IgH locus, as it would lead to extreme repression of transcription dependent DNA recombination (Zullo et al., 2012). Based on these possibilities, we propose that a localization of Mtr4 in close proximity to RNA exosome and to DNA mutator AID creates an exquisite subnuclear space that orchestrates regulated, restricted, and strand specific DNA mutagenesis in B cells.
Bidirectional transcription (i.e, staggered sense and antisense transcription occurring over a genomic locus) may occur at many regions of the mammalian genome, but the biological function and collateral damage of such transcription on genome integrity is underappreciated. Many possibilities of how bidirectional transcription may determine DNA strand specific mutations are suggested by our study. These include (a) in switch sequences, transcription of exosome sensitive ncRNA occurs both in the sense and antisense orientations and plays a role in mutagenesis on both DNA strands via single-strand DNA structure formation, but this effect should be proportional to the relative levels of transcription on each strand of the template DNA. As is the case for IgSμ, levels of canonical germline transcripts are many folds higher than levels of ncRNAs transcribed in the antisense direction (Fig. S6G, S6H), making the regulation of germline RNA transcription the primary determining factor for switch sequence mutagenesis. Thus, in loci that express RNA exosome sensitive ncRNAs on sense and antisense strands, it is possible that the relative levels of one transcript will determine the frequency of mutation on either DNA strand. (b) As in the c-Myc intronic locus (Fig. 6) or in the Pim 1 locus (Fig. 7A), sense mRNA transcription may play a less significant role in DNA mutagenesis via R-loop formation since mRNAs are rarely transcriptionally stalled and rapidly excluded from the DNA template during transcription by various mechanisms including co-transcriptional splicing (Bentley, 2014; Henriques et al., 2013; Schlackow et al., 2016; Wahba et al., 2011). However, the overlapping antisense c-Myc transcript or Pim1 ncRNA transcript requires unwinding and degradation by the Mtr4/Setx/RNA exosome pathway as otherwise it will increase the asymmetric DNA mutagenesis burden through R-loop formation and stabilization (Fig. 7E, right hand side). Thus, it is possible that at genomic coordinates that have overlapping mRNA and ncRNAs, the metabolism of ncRNAs has a dominant role in determining DNA mutagenesis. (c) RNA exosome substrate ncRNAs are not necessarily expressed in the antisense orientation to genic mRNAs. In the case of the CD83 promoter region (Fig. 7B), it can be seen that a ncRNA in the sense orientation, i.e. in the same direction as the mRNA is transcribed, is responsible for creating DNA mutations and in the absence of Mtr4 and Setx unravels an increased asymmetry. Thus, ncRNAs overlapping mRNAs may have a role in creating genomic instability, if not unwound from DNA by Mtr4/Setx and degraded by RNA exosome. (d) Finally, intergenic regions in the genome that only have antisense RNA expression have very low but detectable mutagenesis levels on the sense DNA strand in Mtr4/Setx deficient cells (Fig. S7A and S7B), thus indicating the role of these RNA helicases in promoting RNA exosome mediated removal of ncRNAs without which strand specific mutations can ensue.
Considering our study as a whole, we propose a new role of RNA exosome, Mtr4, and Setx in controlling the pattern of programmed somatic mutagenesis and preventing enhanced and aberrant strand asymmetric mutagenesis at various other regions which, when not resolved, may create genomic instability. The widespread distribution of mutational asymmetry in cancer genomes originating from various cellular lineages has recently been described (Haradhvala et al., 2016). From a more global perspective, large cohorts of cancer genomes have been shown to demonstrate either transcription associated asymmetric distribution of mutagenesis (T-mutations) or replication associated mutagenesis (R-mutations) (Haradhvala et al., 2016); possibility exists that ncRNA transcription associated single strand DNA structure (shown here) can contribute towards replication asymmetry. More specifically, lung carcinomas harbor an overload of G oxidation driven mutations on the sense strand of various coding genes (Pfeifer and Hainaut, 2003), bladder cancer samples accumulate G/C mutation asymmetry associated with replication (Haradhvala et al., 2016), and liver hepatocellular carcinomas demonstrate sense strand mutations on A residues (Alexandrov et al., 2013a) (Fig. S7C). Some cancer genomes originating from B cells also demonstrate detectable strand asymmetry in the mutagenesis pattern, for example those obtained from Multiple Myeloma patients (Fig. S7D) (Lohr et al., 2014; Lohr et al., 2012). These observations imply that strand asymmetric mutagenesis may play an important role in cancer initiation or progression. Therefore, control of the spreading of strand asymmetric mutagenesis should be managed by overlapping and/or distinct mechanisms and our study provides insight into one such pathway.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Uttiya Basu (ub2121@cumc.columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Exosc3TAPtag/TAPtag, Exosc10COIN/COIN, Rosa26 CreERt2 and Rosa26BirA/BirA mouse models were generated and bred according to the “IACUC” guidelines at Columbia Uniersity, New York.
Cell Culture Conditions
B cells and CH12F3 cells were cultured in RPMI 1640 containing 10% FBS, 2 mM Glutamine, 55μM β-Mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin and 25 mM Hepes. HEK293T cells were cultured in DMEM containing 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. All cells were grown at 37 °C, under an atmosphere containing 5% CO2.
METHOD DETAILS
Exosc3TAPtag allele construction
Mouse BAC library clone bMQ386a13 harboring the Exosc3 locus was modified using bacterial homologous recombination. A TAP tag comprising coding sequences for the FLAG epitope tag, TEV protease cleavage site, and biotin tag (de Boer et al., 2003) was inserted in frame immediately downstream of the Exosc3 ATG initiator codon. A neo selection cassette flanked by FRT sites was inserted into a non-conserved region of the first intron of Exosc3. BAC clones modified through bacterial homologous recombination were screened by junctional PCR. The 3.8 kb TAPtag-neo modification was fully sequence verified for the BAC clone used in mESC targeting. Upstream and downstream homology arms in the Exosc3TAPtag-neo BAC targeting vector were 41kb and 84kb, respectively. Gene targeting was performed in 129S6/SvEvTac × C57BL/6Tac hybrid mESC cells. Targeted clones were identified using loss of allele assay. Exosc3TAPtag-neo/+ mESCs were microinjected into blastocysts to give rise to chimeric mice, which were subsequently crossed with Tg(ACTB:FLPe) female mice (Jackson Laboratory) to remove the neo selection cassette and germline transmit the Exosc3TAPtag allele. Following removal of the FLPe transgene, Exosc3TAPtag/+ mice were crossed with ROSA26BirA/BirA mice (Driegen et al., 2005) to generate experimental cohorts. All mouse experiments were performed in adherence with protocols approved by the Columbia University Institutional Animal Care and Use Committee.
Cell culture and IgH class switch recombination analysis
Splenic B cells of TAPtag mice were isolated using CD43 magnetic beads (Miltenyi Biotec) negative selection and cultured in RPMI1640 containing 15% FBS with 20 μg/ml LPS (Sigma-Aldrich) and/or 20 ng/ml IL4 (R&D) for 48~72 hrs. CH12F3 cells were cultured in RPMI1640 containing 10% FBS and stimulated with 20 μg/ml LPS (Sigma-Aldrich), 20 ng/ml IL4 (R&D) and 1 ng/ml TGFβ (R&D). Stimulated splenic B cells were stained with B220-PE (Biolegend) and IgG1-FITC (BD Bioscience) and stimulated mutant CH12F3 cells were stained with IgA-FITC (BD Bioscience). Cells were analyzed by using LSRII and FlowJo (BD Bioscience).
Purification of RNA exosome complex binding proteins
The biotin and FLAG affinity tags were genetically incorporated in the endogenous locus of the Exosc3 subunit of the mouse RNA exosome (Figure S1). B cells were isolated from two TAP-tagged Exosc3, as well as, two control mouse spleens and grown in culture for 72hours, yielding approximately 100×106 cells per condition. Cells were collected, washed 2X with cold PBS, pelleted and lysed with 300ul per 25–30 million cells of Nuclear Extract Buffer A (20mM HEPES, 10mM KCl, 1mM EDTA, 0.1mM Na3VO4, 0.2%(v/v) NP-40, 10%(v/v) Glycerol, supplemented with 1mM DTT, 1mM PMSF, Protease Inhibitor Cocktail1:1000). Following a 10-minute incubation on ice, cells were slowly homogenized using a glass douncer (100–300 times), to achieve at least 80% cell lysis, and subsequently pelleted at 4300g for 15 minutes at 4 °C. The protein lysate was collected, and the cell pellet was further lysed with equal volume (1:1 volume of Buffer A) of Nuclear Extract Buffer B (20mM HEPES, 10mM KCl, 1mM EDTA, 0.1mM Na3VO4, 250mM NaCl, 20%(v/v) Glycerol, supplemented with 1mM DTT, 1mM PMSF, Protease Inhibitor Cocktail1:1000), homogenized slowly using a glass douncer (30 times), incubated with rotation for 30 minutes at 4 °C and pelleted at 20000g for 20 minutes at 4 °C. The second fraction of the protein lysate was collected. In case of purification of core RNA exosome complex, 100 mM KCl was used in Buffers A and B. To increase the yield, this procedure was repeated using the Buffer A isolated protein fraction and the remaining cell pellet. The combined lysate fractions were precleared for 2 hours with 80μl of mouse IgG beads per sample. Immunoprecipitation was carried out using a total of 120μl of pre-equilibrated mouse FLAG-M2 agarose per sample. Extracts were incubated overnight at 4 °C, with rotation. Subsequently, agarose was pelleted and washed 3X using Buffer A/B (1:1) – 15-minute ration at 4 °C, and pelleted at 300g for 4 minutes. Proteins were eluted 2X – 3-hrs elution with rotation each time, using 320μl of FLAG peptide (100ug/ul) per elution. Supernatant containing the RNA exosome complex and its interacting proteins was collected and further purified using 200μl of pre-equilibrated Streptavidin agarose – overnight incubation at 4 °C with rotation. Subsequently, agarose was pelleted, washed 3X with Buffer A/B, and bound proteins were eluted in 400μl of 0.3% SDS buffer by boiling – 10 minutes at 95 °C.
Sample Preparation for Mass Spectrometry
Mass spectrometry of core RNA exosome complex
Tryptic peptides from digested proteins were trapped on a Symmetry C18 Trap column and analyzed in 120-minute chromatograms with a 75 μm ID × 25 cm HSS T3, 1.8 μm particle diameter reverse phase C18 column at a flowrate of 300 nL/min with an acetonitrile/formic acid gradient (NanoAcquity UPLC, Waters Corp.). Spectra were recorded in resolution ion mobility positive ion mode with a Synapt G2 quadrupole-time-of-flight HDMS mass spectrometer (Waters Corp.). Data analysis was performed with ProteinLynx Global Server (PLGS) (Vers. 2.5, RC9, Waters Corp.) with search against a reference database of mouse protein sequences (Uniprot). Post-processing of PLGS data by accurate mass and retention time-based data mining was done Elucidator Protein Expression Data Analysis System, Version 3.3 (3.3.0.1.SP3_CRE52.21) (PerkinElmer/Rosetta Biosoftware). Cluster analysis in Fig. 1A was performed with the Elucidator software. This section is relevant to Fig. 1A.
Mass spectrometry of RNA exosome megacomplex with associated proteins
The protein complexes recovered from TAP purification were fractionated on a 4–20 % SDS-PAGE gradient gel. A zinc staining protocol was used to stain the proteins. Unknown proteins were directly excised from the gel and destained in 2% citric acid (9). In-gel digestions were performed as described previously, and modified porcine trypsin (Promega) was employed (100–200 ng/digestion) to digest the sample overnight at 37 °C (10). Formic acid was added to terminate the reaction (1 μl of 88% formic acid for every 50 μl of digestion sample), and the gel slices were sonicated for 20 min to recover the peptides. This aqueous sample was concentrated in a speed-Vac and further purified by a C18 reverse phase Ziptip (Whatman). The peptides bound to the column were eluted in 50% acetonitrile/0.1% trifluoroacetic acid. LC-MS/MS (Thermo/LTQ-Orbitrap Velos) was used to identify the unknown proteins. The MS/MS spectra from each LC-MS/MS run were searched against the Uniprot Mouse database using SEQUEST search engine in Proteome Discoverer 1.3 software. This section is relevant to Figure 1A.
IP/Western
Cultured splenic B cells were lysed in IP buffer (50 mM Tris [7.4], 150 mM NaCl, 1 mM EDTA, 0.1 % Triton X-100) containing proteinase inhibitor (11836170001 Roche) on the ice for 20 min. Extracted protein was precipitated with FlagM2-magnetic beads (Sigma-Aldrich) or Streptavidin agarose beads (Thermo scientific) at 4 °C for 16 hrs. Beads were washed 3 times with IP lysis buffer and eluted. IP products were separated on the 12 % SDS gel and transferred on the PVDF membrane. IP products were analyzed using FlagM2-HRP (Sigma-Aldrich), Streptavidin-HRP (Thermo Scientific) and Exosc3 antibody (Genway).
Cloning
The full length human AID and Mtr4 genes were PCR amplified using primers listed in key resource table and inserted into mammalian expression pCDNA3.1 vector (Invitrogen), which was previously linearized by the NheI restriction enzyme. HA-tag was introduced at the N-terminus of the AID and Mtr4 proteins.
Protein expression and purification
hMtr4 was isolated from HEK-293T cells. Multiple 15cm plates of HEK-293T cells were grown to confluence and maintained in Dulbecco’s Modified Eagle Medium (DMEM medium supplemented with 4 mM L-Glutamine, 10% fetal bovine serum at 37° C with 5% CO2). HEK-293T cells were transfected with hMtr4 construct with previously described Polyethylenimine (PEI) based transfection protocol. After 72–96 hours of transfection, HEK-293T cells were removed from the plates and dissolved in lysis buffer. Cells were disrupted by a French press and the supernatant was collected after centrifuging at 10,000 g for 1 hr at 4 °C. Affinity purification with agarose anti-HA resin was conducted based on principles originally described (Rigaut et al., 1999), except for solubilization procedure and buffers.
Molecular beacon-based helicase assay for Mtr4
DNA and RNA oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA) in purified form as lyophilized solids. They were then dissolved in DNase/RNase-free water (Qiagen, USA), and concentrations were determined from the extinction coefficients provided. The reporter RNA oligonucleotide(16mer), labeled with 6-FAM at its 5′ end and Iwova black at its 3′end was annealed to the bottom strand of a 22 nucleotide ssRNAs and ssDNAs at a 1:1 molar ratio in 10 mM Tris-HCl pH 8.5, placing in 95 °C, and then allowing to cool to room temperature for ∼1 h to anneal. The sequences of the oligonucleotides used in the study are listed in key resources table. The Mtr4 helicase enzyme used for this study was a recombinant protein with N-terminal HA-tag. Cloning, expression, and purification of this Mtr4 protein has been described in early section of methods. Each reaction contained 40 mM MOPS pH 6.5, 100 mM NaCl, 4 mM MgCl2, 5% glycerol, 0.01% NP-40 substitute, 4 mM DTT, 2μM substrate nucleic acids (RNA-RNA &RNA-DNA), 10μM enzyme(Mtr4), 4mM ATP and 1 U/μl of Ribolock (Thermo Fisher). Reactions were carried out in 100 μL, in triplicate, in white half-volume 96-well polystyrene plates at room temp. Fluorescence was measured as arbitrary units (a.u.) in each well every 5min. using a Tecan infinite 500 fluorescence spectrophotometer [excitation/emission at 485/535 nm (5/10 nm slit width)]. Three replicates of data were analyzed using Matlab software, and a first-order exponential decay model was used to determine the pseudo-first order rate constant.
3D-STORM super-resolution sample preparation
Splenic B cells from Exosc3 TAP-tagged, Exosc10Wt/Wt and Exosc10COIN/LacZ mouse were prepared using CD43 microbead (Miltenyi Biotec) negative selection and cultured in RPMI 1640 containing 15% FBS, 2 mM Glutamine, 55μM βA Mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin and 25 mM Hepes. Isolated B-cells were cultured with 20 μg ml−1 LPS (Sigma) and 20ng ml−1 IL4(R&D system) for three days. B-Cells from day1 and day3 were immobilized on MatTek1.5mm glass cover-slips by cytospin and subsequently fixed with 3% Paraformaldehyde (Electron Microscopy Sciences) + 0.1% glutaraldehyde (Electron Microscopy Sciences) for 5 min. at room temperature, washed three times in 0.1 M phosphate buffer (PB), permeabilized in 0.5% (v/v) Triton X-100 supplemented with 5% normal goat serum(NGS) in 0.1 M phosphate buffer (PB) for 10 min and immunostained with primary antibodies, anti-Mtr4 (Polyclonal anti Mtr4 antibody produced in mouse-cat.no. ab187884-Abcam), anti-AID (Basu et al., 2011) and anti-Flag-tag (Polyclonal Anti-flag tag antibody produced in Chicken, cat.no. ab1170, Abcam for TAP taged RNA exosome complex) in PBS for 3h. The cells were then washed in PBS three times and stained with photo-switchable secondary antibodies Atto488 (Anti-Rabbit IgG(H+L), Host: Goat antibody, ATTO 488 conjugated, cat.no. 18772-Sigma) and Alexa fluor647 (Anti-Chicken IgG (H+L), Host: Donkey antibody, Alexa Fluor 647 conjugated, cat.no. 703-606-155- Jackson Immuno Research) compatible for STORM imaging for 2 h. Cells were then washed twice each in PBS. Immediately before STORM imaging, PBS was removed and freshly prepared imaging medium buffer (Containing cystamine and glucose oxidase mixture for oxygen scavenging)(Dani et al., 2010). The two colors (Atto488/Alexa488 and Alexa flour 647) were imaged sequentially. Imaging buffer helped to keep dye molecules in a transient dark state. Subsequently, individual dye molecules were excited stochastically with high laser power at their excitation wavelength (488 nm for Atto488/Alexa488 or 647 nm for Alexa flour 647, respectively) to induce blinking on millisecond time scales. STORM images and the correlated high-power confocal stacks were acquired via a CFI Apo TIRF 100× objective (1.49 NA) on a Nikon Ti-E inverted microscope equipped with a Nikon N-STORM system, an Agilent laser launch system, an Andor iXon Ultra 897 EMCCD (with a cylindrical lens for astigmatic 3D-STORM imaging) camera, and an NSTORM Quad cube (Chroma). This setup was controlled by Nikon NIS-Element AR software with N-STORM module. To obtain images, the field of view was selected based on the live EMCCD image under 488-nm illumination. 3D STORM data sets of 20,000 frames for HEK293T and 50,000 frames for B-Cells were collected. Lateral drift between frames were corrected by tracking 488, 561, and 647 fluorescent beads (TetraSpeck, Life Technologies). STORM images were processed to acquire coordinates of localization points using the N-STORM module in NIS-Elements AR software. Identical settings were used for every image. Each localization is depicted in the STORM image as a Gaussian peak, the width of which is determined by the number of photons detected (Betzig et al., 2006). All of the 3D STORM imaging were performed in three different HEK293T/B-Cells (from independent experiments) and repeated three or more times. Details of 3D-STORM microscopy associated statistical analyses, number of biological repeats and number of technical repeats are provided in Supplementary Table 1d.
Controls used to set up 3D-STORM
To evaluate the non-specific binding of secondary antibodies with the cells, we collected 3D STORM images from (A) B cells (isolated from AID K/O mouse) labelled with primary antibody (anti AID) and secondary antibody (Atto 488) (Fig.S3G) (B) B-Cells (isolated from Exosc3TAPtag/TAPtag) labelled with secondary antibody (alexa fluor 647) alone (Fig.S3H).
As a negative control for the localization of RNA exosome complex & its cofactors, we transfected HEK293T cells with RNA exosc3 constructs along with an HA-tagged RFP protein (non-interacting protein with RNA exosome complex) and collected 3D STORM images (Fig. S2D, S2E& S2F). In the conditions of our assays, we set closest proximity of two proteins by labelling B-cells with Exosc3 and Exosc5 subunits of RNA exosome (Fig. S2A, S2B and S2C).
RT-qPCR
RNA was isolated by Trizol reagent (Invitrogen, 15596018) from B cells, and then DNaseI (Qiagen, 79254) treated 1ug RNA was used for cDNA synthesis using superscript IV reverse transcriptase (Life Technology, 18091050). Primers sequences used for PCR are in Supplementary table S2.
DRIP
DRIP was performed following previously used protocol (Pefanis et al., 2014). WT, Setxmut and Mtr4mut CH12F3 cells were stimulated by LPS, IL4 and TGFβ for 24 hrs, and then cells were collected and lysed. DNA of each cell type was purified by phenol/chloroform extraction followed by EtOH precipitation and subsequently digested using restriction enzymes BsoBI, HindIII, NheI, NcoI and StuI (New England Biolabs). DNA/RNA fragments treated with or without RNase H were precipitated by S9.6 antibody (Kerafast) with Protein A/G agarose (Thermo Scientific). Precipitated DNA was eluted and purified by phenol/chloroform extraction and EtOH precipitation. DRIP products were measured using qPCR analysis employing the primers in Supplementary table S2.
CRISPR Cas9
Mutations in each target were generated following previous protocols (Pefanis et al., 2014). Guide RNAs were designed using the online tool (http://tools.genome-engineering.org) and the following guide RNA sequences were used: AID – TGAGACCTACCTCTGCTACG, Setx – AAGCGCTATGCATCTAGCAC, Mtr4 – AATCAGAGTCAGCCAGTGGG and AAATGTGGCCCCAGTTGCCC, Ung- CGCAACGTGCCTGCCGGCTT and TCACGGACGCGGTCGTGTCC. DNA oligos of target guide RNA sequences were inserted into BbsI site of pSpCas9(BB)-2A-GFP (Addgene), and construction of each target was transfected into CH12F3 cells by nucleofector (Lonza). Transfected cells were isolated into 96 well culture plates by cell sorter (BD FACSAria II) after incubation for 24 hrs and incubated for 1 week. Genomic DNA of each clones were used as a template for PCR/sequencing screening to seek mutated clones by insertion or deletion. Primers sequences used for screening are in Supplementary table S2. PCR products of mutated clones were inserted into pGEM-T Easy (Promega) vector and their mutated regions were confirmed by sequencing analysis.
Proliferation assay
Proliferation of each mutant CH12F3 cells was tested using Violet Proliferation Dye 450 (BD Bioscience). Cells were collected and treated at 1μM VPD at 37 °C for 10 min. After washing with PBS, cells were cultured and/or tested by FACS for 4 days.
Somatic mutation experiment and analysis for Sanger sequencing
UNG mutant and Mtr4/Setx mutant CH12F3 cells were stimulated by LPS (Sigma-Aldrich), IL4 (BD Bioscience) and TGFβ (R&D) for 48 hrs, and genomic DNA of each clone was prepared for PCR. Primers (see supplementary table S2) were used for amplifying IgH 5′ Sμ and cMyc intron 1 regions. PCR products were inserted into pGEMT-easy vector (Promega) and transformed into XL1 Blue competent cells. Transformed XL1 blue colonies were submitted for sequencing analysis. Specific DNA regions of Non-transcribed strand were sequenced. Raw sequences were manually reversed and complemented to match the reference if necessary. We used ClustalW to do multiple sequence alignment. Non-relevant ends of each sequence were removed by truncate the primer region include extra 5 bases. Sequence with few bases were removed. Since AID primarily affects C, we focus on mutation frequency of C and G to other bases. G mutation frequency of non-transcribed strand can be used to reflect the C mutation frequency of transcribed strand, as transcribed and non-transcribed strands are complementary. Clones without C or G mutation were excluded from further analysis. For clones that have C or G mutation, we calculate mutation frequency of C and G as follows.
where denominators are counted from reference sequence and numerators are counted from clonal sequence compared to reference sequence.
After the clone specific mutation frequencies are calculated, we use the ratio of C mutation frequency in anti-sense (transcribed) strand over C mutation frequency in sense (non-transcribed) strand to depict the strand mutation bias for case and control group. For example, if (μC1, μC2, …, μCm) and (μ′C1, μ′C2, …, μ′Cm) are C mutation frequencies in anti-sense (transcribed) and sense (non-transcribed) strand respectively, the ratios are
where is the average C mutation frequency in non-transcribed strand. To test whether there is significant difference between strand mutation biases in control samples and in case samples, we employ the Wilcoxon rank sum test to accesses the difference between ratios in case and control samples.
Next Generation Sequencing experiments
Genomic DNA was extracted 6 days (experiment 1) or 7 days (experiment 2) after stimulation with LPS/IL4/TGFβ, from Ung mutant and Mtr4/Setx/Ung mutant CH12F3 cells or unstimulated wt CH12F3 cells as a control. Two independent sets of PCR were performed for each target using high fidelity Taq polymerase (M0491S, NEB) and following primers. 100ng Template genomic DNA was amplified in 25~28 cycle. PCR primers sequences used for amplification of non IgH loci are in Supplementary table S2. PCR products were purified using spin column (28004, Qiagen). Next Generation Sequencing was performed according to the user guide Ion Xpress™ Plus gDNA Fragment Library Preparation (Cat. no. 4471269, Life Technologies). Briefly, PCR products (1μg) were fragmented by enzymatic digestion (Ion Shear™ Plus Reagents Kit, Cat. no. 4471248) and ligated to Barcodes and Adapters (Ion Plus Fragment Library Kit, Cat. no. 4471252). After 200 bp size selection step on E-Gel precast agarose electrophoresis system, final amplification was performed. Libraries were sequenced on an Ion Proton™ System.
Next Generation Sequencing analysis
Detailed pipeline analysis protocol will be published in a method article (François Boyer, Ophélie Martin and Eric Pinaud, manuscript in preparation). Briefly, fastq data files were aligned on reference sequences using BWA-MEM algorithm. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [Q-Bio]). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. (arXiv:1303.3997v1 [q-bio.GN]), and nucleotides were counted position-wise with igvtools (Robinson et al., 2011). Unstimulated Wt CH12F3 samples were used to determine background error frequencies for each possible mutation event. Then, mutations in stimulated samples were called at each site with a nucleotide frequency significantly greater than local background (p<0.001, fold increase >2). AID hotspots were defined as WRCY (or reverse complement RGYW) and C (or G) mutations were studied. Total mutations and mutation frequencies were calculated, as well as mutation ratios between C and G (corresponding to sense or anti-sense DNA strand, depending on genome region). Finally, Fisher’s exact test was used to evaluate mutation differences between Ung mutated cells and Mtr4/Setx/Ung mutated CH12F3 cells.
Analysis of somatic mutation in cancer
We analyzed somatic coding mutations from whole exome data in six different control matched tumors. We considered those with coverage >=20 reads in both tumor and normal, < 2% variant allele frequency in the Normal and >= 5% in the tumor. For variants harbored in genes transcribed on the reverse strand we reverse complemented the variant nucleotides. Mutations were tallied and compared in pairs for differences in abundance. Significance was assessed using a Proportion Test. The following mutations were grouped together A>C/T>G, A>G/T>C, A>T/T>A, C>G/G>C, C>A/G>T, C>T/G>A. Bladder, Liver and Lung whole exome sequencing data were obtained from Broad’s GDAC at gdac.broadinstitue.org. Multiple Myeloma data was obtained from (Lohr et al., 2014).
QUANTIFICATION AND STATISTICAL ANALYSIS
3D-STORM super-resolution data statistical analysis
Proximity of RNA exosome complex with its cofactors (AID and Mtr4) were calculated by using algorithm “Nearest Neighbors Search” in the Matlab (2014b, Math works). The 50nm range was used to maximize detection of co-localization. Distribution of the interaction between RNA exososme complex and its cofactors were represented in the form of frequency histogram plot by using Matlab (2014b, Math works) software. We also compared the distribution of interaction between RNA exosome complex with its cofactors in the nucleus and cytoplasm by using a paired Student’s T-test in Matlab (2014b, Math works) software. Comparison of the distribution of paired interaction of AID and Mtr4 in the nuclear center versus displaced from center versus cytoplasm were performed by one-way ANOVA (Tukey-Kramer test) method in Matlab (2014b, MathWorks) software. All of the 3D STORM imaging were performed in three different B-Cells (from independent experiments) and repeated three or more times. Data are presented as the means ± SEM or SD (indicated in the legend for each figure). Statistical precision measures (mean ± s.e.m/SD) and statistical significance are reported in the Figures and the Figure Legends when necessary. All Error bars indicate S.D. (P values: ** <0.01, *** <0.001).
ADDITIONAL RESOURCES
The Exosc3TAP mice are available for other investigators following completion of “Uniform Biological Material Transfer Agreement” implemented by Columbia University.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-AID | (Basu et al., 2011) | N/A |
| Goat polyclonal anti AID | Novusbio | NB100-93454 |
| Anti-β actin | Sigma-Aldrich | A1978 |
| Anti-DNA/RNA hybrid (S9.6) | Kerafast | ENH001 |
| Anti-Exosc3 | Genway | GWB-FF795C |
| Anti Exosc3 | Santa Cruz Biotechnology | Sc-98776 |
| Anti-SKIV2L2 (Mtr4) | Abcam | ab70551 |
| B220-PE | Biolegend | 103208 |
| IgG1-FITC | BD Bioscience | 553443 |
| IgA-FITC | BD Bioscience | 559354 |
| Anti-Flag M2 magnetic beads for IP | Sigma-Aldrich | M8823 |
| Anti-Flag M2-HRP for WB | Sigma-Aldrich | A8592 |
| Chicken polyclonal anti Flag-tag | Abcam | Ab1170 |
| Alexa488 (Against-Goat) | Jackson Immuno Research | 805-547-008 |
| Alexa Fluor 647 (Anti-Rabbit) | Jackson Immuno Research | 711-605-152 |
| Goat antibody, ATTO 488 conjugated | Rockland Immunochemical | 610-152-041 |
| Nuclease-Free Water | Qiagen | 129115 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LPS | Sigma-Aldrich | L4130 |
| IL4 | Peprotech | 214-14 |
| TGFβ | R&D | 240-B-010 |
| cOmplete ™ | Roche | 11836170001 |
| Tris base | Fisher | BP152-1 |
| Streptavidin-HRP | Thermo Scientific | PI-21130 |
| VPD450 | BD Bioscience | 562158 |
| Q5 HF-Taq | NEB | M0491S |
| NaCl | Sigma | S5150-1L |
| HA-peptide | Fisher | 26184 |
| Flag-peptide | Sigma | F3290-4MG |
| M-280 Streptavidin Dynabeads | Life Technologies | 11205D |
| Protein G Dynabeads | Life Technologies | 10003D |
| Protein A/G agarose | Pierce | 20423 |
| Anti-HA agarose | Fisher | 26181 |
| M2-agarose anti-FLAG resin | Sigma | A22220-1ml |
| Glycerol | Sigma | G5516-1L |
| Dithiothreitol (DTT) | Gold Biotech | DTT25 |
| MOPS | Sigma | M1442-500ml |
| Polyethylenimine (PEI) | Fisher | NC9197339 |
| Cystamine | Sigma | 30070-10G |
| Protease Inhibitor Cocktail tab. | Sigma | 11-836-170-001 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Life Technology | 10565018 |
| Paraformaldehyde | Electron Microscopy Sciences | 15710 |
| Glutaraldehyde | Fisher | 50-262-10 |
| Normal goat serum(NGS) | Jackson Immuno Research Laboratories, Inc. | 005-000-121 |
| RiboLock | Fisher | EO0381 |
| MgCl2 | Sigma | M1028 |
| MOPS buffer | Sigma | M1442 |
| Critical Commercial Assays | ||
| Ion Xpress™ Plus gDNA Fragment Library Preparation | Life Technologies | 4471269 |
| Ion Shear™ Plus Reagents Kit | Life Technologies | 4471248 |
| Ion Plus Fragment Library Kit | Life Technologies | 4471252 |
| CD43 magnetic beads | Miltenyi biotec | 130-049-801 |
| Mouse B Cell Nucleofector Kit | Lonza | VPA-1010 |
| Deposited Data | ||
| RNA-seq data | (Pefanis et al., 2014; Pefanis et al., 2015) | SRP042355 |
| Experimental Models: Cell Lines | ||
| CH12F3 | (Nakamura et al., 1996) | N/A |
| 129S6/SvEvTac X C57BL/6Tac hybrid mESC | Columbia U. transgenic facility | N/A |
| HEK293T | ATCC | ATCC-CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Exosc3TAPtag/TAPtag mouse | This paper | N/A |
| Exosc10COIN/COIN mouse | (Pefanis et al., 2015) | N/A |
| Rosa26 CreERt2 mouse | Gift from Dr. Piere Chambon (IGBMC) | N/A |
| Rosa26BirA/BirA mouse | (Driegen et al., 2005) | N/A |
| Recombinant DNA | ||
| BAC clone | Source Bioscience | bMQ386a13 |
| pSpCas9(BB)-2A-GFP (pX458) | Addgene | 48138 |
| pGEM-T Easy | Promega | A1360 |
| pcDNA3.1 | Fisher | V79020 |
| Sequence-Based Reagents | ||
| Primers for sanger sequencing (see table S2) | This paper | N/A |
| Primers for NGS (see table S2) | This paper | N/A |
| Primers for DRIP (see table S2) | This paper | N/A |
| Primers for RT-qPCR (see table S2) | This paper | N/A |
| Sequences of AID gRNA: TGAGACCTACCTCTGCTACG | This paper | N/A |
| Sequences for Setx gRNA: AAGCGCTATGCATCTAGCAC | This paper | N/A |
| Sequences for Mtr4 gRNA: AATCAGAGTCAGCCAGTGGG and AAATGTGGCCCCAGTTGCCC | This paper | N/A |
| Sequences for Ung gRNA: CGCAACGTGCCTGCCGGCTT and TCACGGACGCGGTCGTGTCC | This paper | N/A |
| Primers for sub cloning of hMtr4 in to pCDNA3.1- reverse primer: 5′ACGTCAGCTAGCCTATAA ATA CAA AGA3′ | This paper | N/A |
| Primers for sub cloning of hAID in to pCDNA3.1- forward primer: 5′GTGGTCGCTAGCATG TACCCATACGATGTT CCAGATTACGCTATG GACAGCCTCTTG3′ | This paper | N/A |
| Primers for sub cloning of hAID in to pCDNA3.1- reverse primer: 5′ACGTCAGCTAGCTCA AAG TCCCAAAGT3′ | This paper | N/A |
| Primers for unwinding assay for Mtr4- R16- Top strand-reporter: 5′AGCACCGUAAAGACGC3′ | This paper | N/A |
| Primers for unwinding assay for Mtr4- R22- Bottom strand: 5′GCGUCUUUACGGUGCUUAAAAA3′ | This paper | N/A |
| Primers for unwinding assay for Mtr4- D22- Bottom strand: 5′GCGTCTTTACGGTGCTTAAAAA3′ | This paper | N/A |
| Software and Algorithms | ||
| BWA-MEM. arXiv:1303.3997v1 [q-bio.GN] | http://github.com/lh3/bwa | |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/in dex.shtml |
| Samtools | http://samtools.sourceforge.net/ | |
| MATLAB | https://www.mathworks.com | |
| Rstudio | https://www.rstudio.com/ | |
| R | https://www.r-project.org/ | |
| NIS element | NIKON | N/A |
| FlowJo | BD Bioscience | N/A |
| Other | ||
| Bladder, Liver and Lung mutational exosome data | gdac.broadinstitute.org | |
| Multiple Myeloma | (Lohr et al., 2014) | http://www.ncbi.nlm.nih.gov/pubmed/24434212 |
| Illustration image | Servier Medical Art | http://www.servier.com/Powerpoint-image-bank |
Supplementary Material
(A) Schematic representation of strategy showing the TAP-tag Exosc3 allele. The TAP-tag was inserted in the 5′ region of exon1 together with a Neo cassette. The Neo gene was removed via mating to a FLPe recombinase mouse. Orange triangle represents FRT site. This diagram is not drawn to scale. (B) PCR products resulting from genotyping of Exosc3 TAPtag mice. Map shows primers location in Exosc3 gene locus. (C) Resting B cells from TAPtag-Exosc3 mice were treated with LPS and IL4 and assayed for IgG1 expression after 72 hrs. (D) Schematic representation of the steps involved in the purification of the RNA exosome complex from B cells. (E) Flag and Streptavidin IPs were performed and the presence and functionality of tags validated by western blot prior to mass spectrometry. (F) Co-immunoprecipitation of RNA exosome complex with Mtr4 in HEK293T cells. Flag-tagged Exosc3 and HA-tagged Mtr4-expressing plasmids were overexpressed in HEK293T cells. Lysates were co-immunoprecipitated using anti-FLAG-agarose beads, and western blots were probed with indicated antibodies.
Reconstructed two color 3D STORM (super-resolution) video from data set of 50,000 frames of fixed splenic B cells, in which AID & Exosc3 were labeled with atto488 and Alexa Fluor 647 respectively. This video shows 360° rotation in X and Y axis for spatial distribution of AID and RNA exosome in the nucleus of mouse B cells.
Reconstructed two color 3D STORM (super-resolution) video from data set of 20,000 frames of fixed HEK293Tcells, in which AID & Exosc3 were labeled with atto488 and Alexa Fluor 647 respectively. This video shows 360° rotation in X and Y axis for spatial distribution of AID and RNA exosome in the nucleus of non-B cells(HEK293T).
Reconstructed two color 3D STORM (super-resolution) video from a data set of 50,000 frames with Alexa488 labeled AID, AlexaFluor647 labeled Mtr4, show 360° rotation in X and Y axis for spatial distribution of AID and Mtr4 molecules inside the nuclei of B cells isolated from wild type B cells.
Reconstructed two color 3D STORM (super-resolution) video from a data set of 50,000 frames with Alexa488 labeled AID, AlexaFluor647 labeled Mtr4, show 360 ° rotation in X and Y axis for spatial distribution of AID and Mtr4 molecules inside the nuclei of B cells isolated from RNA exosome nuclear activity deficient (Exosc10COIN/LacZ) mouse.
Table S1 (related with Figure 1,2,3,4,5, 6 and 7): (a) RNA exosome complex protein identity for negative control following mass spectrometry. (b) RNA exosome complex protein identity from Exosc3TAP mouse following mass spectrometry. (c) Sanger sequencing and Next generation sequencing (NGS) analysis in Mtr4 and Setx deficient CH12F3. (d) Details of statistical analyses and biological and technical repeats performed for the 3D-STORM experiments presented in Figs. 1–4 and Figs. S2–4.
B cells were harvested from Exosc3 TAP-tagged mice and fixed after 72 hrs of treatment with stimulation cocktail. HEK293T cells were transfected with Red fluorescence protein(RFP) with N-terminus HA-tag and hExosc3 mammalian expression vectors and then fixed similar to B-Cells post 16hrs of transfection. Reconstructed two color 3D STORM (super-resolution) image for (A) Atto488 labeled Exosc5, AlexaFluor647 labeled Exosc3 & DAPI labeled nucleus of B-Cell and (D) for Atto488 labelled RFP and AlexaFluor647 labeled hExosc3 in HEK293T cell. Histogram of the distribution of interactions of Exosc3 and Exosc5 calculated in the B-cell (B) and RFP & Exosome complex in HEK293T cell (E), by using custom written algorithm “Nearest Neighbors Search” in the Matlab (2014b, Math works) software. (C) Two components Exosc3 and Exosc5 of RNA exosome complex were selected since they have closest distance in crystal structure (PDB-2NN6) and used as positive control for the interaction. All of the 3D STORM imaging were performed in three different B-Cells/HEK293T cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is × 100.
B cells were harvested from Exosc3 TAP-tagged mice and fixed after 72 hrs of treatment with stimuli cocktail. (A) Reconstructed two color 3D STORM (super-resolution) image for Atto488 labeled AID, AlexaFluor647 labeled Exosc3 & DAPI labeled nucleus. (B) Three dimensional views of the boxed region. (C) Higher magnification three dimensional [3D] view of the boxed region. Histogram for the distribution of interactions of AID and RNA exosome complex were calculated in the nucleus of (D)B cell & (E) HEK293T cell and cytoplasm of (E) B cell & (L) HEK293T cell, by using Matlab (2014b, Math works) software. Comparison of the distribution of paired interaction of AID and RNA exosome in the nucleus and cytoplasm of (F) B cell & (M) HEK293t cell were calculated by using a paired Student’s T-test in Matlab (2014b, Math works) software and P values are mentioned in the graph. (G) Control experiment for non-specific binding primary and secondary antibodies- (Top panel) B cells were harvested from AID K/O mice & labeled with primary antibody AID, secondary antibody Atto 488& nucleus with DAPI and (Bottom panel) B cells were harvested from Exosc3 TAP-tagged mice & labeled with secondary antibody AlexaFluor647& nucleus with DAPI. (H) Comparison of the inter molecular distance between AID molecules in the nucleus and cytoplasm of a activated B-cell, calculated by using Matlab (2014b, Math works) software. Reconstructed two color 3D STORM (super-resolution) image from (I) B cell and (J) HEK293T cell, in which AID & Exosc3 were labeled with atto488 and Alexa Fluor 647 respectively. All of the 3D STORM imaging were performed in three different B-Cells/HEK293T cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is × 100. Scale bars: 1μm(A) and 2μm(G). (P value: ** <0.01).
B cells were harvested from the spleen of Exosc10CC/LacZ mice and fixed after 72 hrs of treatment with (1) stimulation cocktail and (2) 4OHT with stimulation cocktail. Reconstructed two color 3D STORM (super-resolution) (A and C) image from a data set of 50,000 frames with Alexa488 labeled AID, AlexaFluor647 labeled Mtr4 and DAPI labeled nucleus, show spatial distribution of AID and Mtr4 molecules inside the nuclei of B cells isolated from wild type cells, and (B and D) image from Exosc10 knockout cells. (E) Conditional inversion allele of Exosc10 in 4OHT and stimuli cocktail treated Exosc10 deficient (Exosc10COIN/LacZ) cells was validated by flow cytometry analysis (upper panel). RNA expression level of Exosc10, AID and Mtr4 genes in Exosc10 deficient (Exosc10COIN/LacZ) cells was tested by RT-qPCR after 72 hrs of 4OHT and stimulation cocktail treatment. RNA level (lower panel). Results were obtained from 4 experimental repeats. Error bars indicate S.D. (P value: *** <0.001). All of the 3D STORM imaging were performed in three different B-Cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is ×100. Scale bars: 1μm(A) &(B).
(A) Brief map showing gRNA target regions and sequencing results of target region in AID mutant, Setx mutant and Mtr4 mutant. Red arrows indicate insertion or deletion junction of each allele. (B) Western blot analysis shows AID expression in stimulated AID, Setx and Mtr4 mutant CH12F3. (C) IgM to IgA CSR in Mtr4/Setx mutant CH12F3 cells was tested every day for 72h. (D) IgA CSR was assayed to test Mtr4 rescue effect after transfecting Mtr4 expressing plasmid DNA. (E) Schematic map localizing the IgH locus and binding sites of the PCR primer pair for mutation analysis. Mutation frequency of AID-induced somatic hypermutation on sense (non-transcribed) strand and anti-sense (transcribed) strand of IgH 5′Sμ region in Mtr4 mutant CH12F3 cells compared to WT CH12F3 cell controls. Ratio of mutation frequency between sense and anti-sense strands is indicated above bar graph. Details of mutations identified by Sanger sequencing in Sup. table 1c. (P values: ** <0.01) (F) IgA efficiency of two different Setx clones was normalized based on CSR level of parental cells. Error bars indicate S.D. (P values: ** <0.01, *** <0.001)
(A) Proliferation test of mutant CH12F3 clones using VPD450. (B) Sequencing analysis of gRNA target region on the Setx gene locus in Setx/Mtr4 double mutant clone. Setx gRNA was transfected into Mtr4 mutant cells. Red arrows indicate the junction of deleted region. Proliferation test of Setx/Mtr4 double mutant using VPD450 (bottom panel). (C) Western analysis of Mtr4 expression in Mtr4 mutant CH12F3 cells. (D) Sequencing analysis of gRNA target region at the Ung gene locus in Ung mutant CH12f3 clone and Mtr4/Setx/Ung mutant CH12F3 clone. (E) Validation of Ung gene expression using qRT-PCR in Ung mutant CH12F3 cells (F) Class switch recombination efficiency in Ung mutant Ch12F3 cells (G) RNA-seq tracks from IGV browser for WT B cells (Exosc3WT/WT; middle panel) RNA exosome mutant B Cells (Exosc3COIN/COIN; bottom panel) and overlap (top panel) for sense (blue) and antisense (red) RNAs expressed in 5′ and 3′ IgH Sμ regions (H) and intronic enhancer Eμ, Iμ and Sμ region.
(A, B) RNA exosome substrate ncRNA expression profiles of CD79b (C) and CD83 (D) TSS locus in WT and Exosc3coin/coin mouse B cells (Top). Gray arrow heads indicate primer sites for next generation sequencing (NGS) and red arrows indicate non-coding RNA and its transcription direction on the schematic map (Middle). Mutation frequency of C bases on transcribed and non-transcribed strands and ratio of non-transcribed strand mutation frequency compare to transcribed strand mutation frequency in Ung mutant and Mtr4/Setx/Ung mutant CH12F3 cells (Bottom). The number above bar graph means C to T mutation count on AID mutation hotspot motif. Details of mutations identified by NGS deep sequencing in Sup. table 1c.
(C, D) Mutational counts representing somatic coding variants across 4 different tumors in patients with coding mutations in analyzed genes. A red bar highlights specific transcriptional, replicative or mutational nucleotide changes. Indicating asymmetry in Liver, Lung and Bladder cancer, respectively (E). The largest number of mutations concurrent with Multiple Myeloma (F). P-value is indicated above each bar graph.
Reconstructed single color 3D-STORM video from a data set of 50,000 frames of fixed splenic B cells before CSR stimulation, in which Exosc3 was labeled with AlexaFluor647. This video shows 360° rotation in X and Y axis for RNA exosome.
Reconstructed single color 3D-STORM video from a data set of 50,000 frames of fixed splenic B cells after CSR stimulation, in which Exosc3 was labeled with AlexaFluor647. This video shows 360° rotation in X and Y axis for RNA exosome.
HIGHLIGHTS.
Mtr4 and Setx cooperatively unwind RNA exosome sensitive ncRNA from DNA/RNA hybrids.
Physical proximity of Mtr4 and AID is a function of nuclear RNA exosome activity.
Mtr4 and Setx restricts transcription coupled asymmetric DNA mutagenesis by AID.
Acknowledgments
We thank Drs. David Schatz (Yale University) for insightful comments; Victor Lin (Herbert Irving Comprehensive Cancer Center transgenic facility) for targeting and generation of Exosc3TAP allele ES cells and mouse models; Dr. Tasuku Honjo (Kyoto University) for providing CH12-F3 cells; and, Dr. Pierre Chambon (IGBMC and Université de Strasbourg) for ROSA:Ert2-Cre allele. This work was supported by grants to U.B. (NIAID, (1R01AI099195), NIH (DP2OD008651), Leukemia & Lymphoma Society, and the Pershing Square Sohn Cancer Research Alliance); D.K. (NIAID (F31AI098411-01A1)); B.L. (EMBO fellowship ALTF 906-2015); Evangelos Pefanis (Regeneron Pharmaceuticals), O.M. (M.E.N.S.E.R. PhD fellowship of the French government); F.B. (Fondation Partenariale de l’Université de Limoges and ALURAD); E.P. (Comité d’Orientation Recherche Cancer en Limousin (FJA/NP-2014-126), La Ligue contre le Cancer-Institut National du Cancer (INCa grant #9363; PLBIO #15-256)); M.C. (Association pour la Recherche contre le Cancer (PGA120150202338) and Agence Nationale de la Recherche (16-CE15-0019-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONTRIBUTIONS
J.L. generated CRISPR/Cas9 genome edited clones, performed DRIP, I.P., mutation analyses studies and mass spec data analyses; P.G. performed, analyzed and stored 3D-STORM data, I.P. and assessed Mtr4’s DNA/RNA unwinding activity; D.K. studies Setx mutants; V.G. and E.Pefanis generated Exosc3TAP mouse; E.L. and RR performed analyses for Fig. S 7C, D; Y.C. and H.D. performed mass spectrometry on Exosc3TAP complex; O.M., F.B., M.C., and E.P., performed NGS assays and subsequent strand mutation analyses in Fig. 7., U.B., conceived, directed and analyzed experiments; and U.B. (with J.L.) wrote the manuscript.
References
- Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nature reviews Genetics. 2015;16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013a;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell reports. 2013b;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nature reviews Immunology. 2015;15:172–184. doi: 10.1038/nri3814. [DOI] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, et al. The RNA Exosome Targets the AID Cytidine Deaminase to Both Strands of Transcribed Duplex DNA Substrates. Cell. 2011a;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL. Coupling mRNA processing with transcription in time and space. Nature reviews Genetics. 2014;15:163–175. doi: 10.1038/nrg3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Chan EA, Teng G, Corbett E, Choudhury KR, Bassing CH, Schatz DG, Krangel MS. Peripheral subnuclear positioning suppresses Tcrb recombination and segregates Tcrb alleles from RAG2. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4628–4637. doi: 10.1073/pnas.1310846110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution Imaging of Chemical Synapses in the Brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer E, Rodriguez P, Bonte E, Krijgsveld J, Katsantoni E, Heck A, Grosveld F, Strouboulis J. Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7480–7485. doi: 10.1073/pnas.1332608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driegen S, Ferreira R, van Zon A, Strouboulis J, Jaegle M, Grosveld F, Philipsen S, Meijer D. A generic tool for biotinylation of tagged proteins in transgenic mice. Transgenic Res. 2005;14:477–482. doi: 10.1007/s11248-005-7220-2. [DOI] [PubMed] [Google Scholar]
- Goodman MF, Scharff MD, Romesberg FE. AID-initiated purposeful mutations in immunoglobulin genes. Advances in immunology. 2007;94:127–155. doi: 10.1016/S0065-2776(06)94005-X. [DOI] [PubMed] [Google Scholar]
- Hamperl S, Cimprich KA. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA repair. 2014;19:84–94. doi: 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, et al. Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell. 2016;164:538–549. doi: 10.1016/j.cell.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, Adelman K. Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Molecular cell. 2013;52:517–528. doi: 10.1016/j.molcel.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holwerda SJ, van de Werken HJ, Ribeiro de Almeida C, Bergen IM, de Bruijn MJ, Verstegen MJ, Simonis M, Splinter E, Wijchers PJ, Hendriks RW, et al. Allelic exclusion of the immunoglobulin heavy chain locus is independent of its nuclear localization in mature B cells. Nucleic acids research. 2013;41:6905–6916. doi: 10.1093/nar/gkt491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januszyk K, Lima CD. The eukaryotic RNA exosome. Curr Opin Struct Biol. 2014;24:132–140. doi: 10.1016/j.sbi.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SJ, Jackson RN. Ski2-like RNA helicase structures: common themes and complex assemblies. RNA biology. 2013;10:33–43. doi: 10.4161/rna.22101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim C, Kazadi D, Rothschild G, Basu U. Regulation of AID, the B-cell genome mutator. Genes Dev. 2013;27:1–17. doi: 10.1101/gad.200014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nature reviews Genetics. 2012;13:204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, Lee S, Chittenden TW, D’Gama AM, Cai X, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94–98. doi: 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, Sougnez C, Knoechel B, Gould J, Saksena G, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell. 2011;43:624–637. doi: 10.1016/j.molcel.2011.06.028. [DOI] [PubMed] [Google Scholar]
- Lucas JS, Zhang Y, Dudko OK, Murre C. 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell. 2014;158:339–52. doi: 10.1016/j.cell.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015;161:1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, Meyers RM, Amor C, Wasserman CR, Neuberg D, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014;159:1538–1548. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S, Honjo T. High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int Immunol. 1996;8:193–201. doi: 10.1093/intimm/8.2.193. [DOI] [PubMed] [Google Scholar]
- Pefanis E, Basu U. RNA Exosome Regulates AID DNA Mutator Activity in the B Cell Genome. Advances in immunology. 2015;127:257–308. doi: 10.1016/bs.ai.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, Economides AN, Basu U. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature. 2014b;514:389–393. doi: 10.1038/nature13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu ZP, et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell. 2015;161:774–789. doi: 10.1016/j.cell.2015.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP, Hainaut P. On the origin of G –> T transversions in lung cancer. Mutation research. 2003;526:39–43. doi: 10.1016/s0027-5107(03)00013-7. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotech. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush JS, Fugmann SD, Schatz DG. Staggered AID-dependent DNA double strand breaks are the predominant DNA lesions targeted to S mu in Ig class switch recombination. Int Immunol. 2004;16:549–557. doi: 10.1093/intimm/dxh057. [DOI] [PubMed] [Google Scholar]
- Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nature reviews Genetics. 2015;16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- Schlackow M, Nojima T, Gomes T, Dhir A, Carmo-Fonseca M, Proudfoot NJ. Distinctive Patterns of Transcription and RNA Processing for Human lincRNAs. Molecular cell. 2016 doi: 10.1016/j.molcel.2016.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uson ML, Ordonez H, Shuman S. Mycobacterium smegmatis HelY Is an RNA-Activated ATPase/dATPase and 3′-to-5′ Helicase That Unwinds 3′-Tailed RNA Duplexes and RNA:DNA Hybrids. Journal of bacteriology. 2015;197:3057–3065. doi: 10.1128/JB.00418-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, Vuica-Ross M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Molecular cell. 2011;44:978–988. doi: 10.1016/j.molcel.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Kieffer-Kwon KR, Oliveira TY, Mayer CT, Yao K, Pai J, Cao Z, Dose M, Casellas R, Jankovic M, et al. The cell cycle restricts activation-induced cytidine deaminase activity to early G1. The Journal of experimental medicine. 2016 doi: 10.1084/jem.20161649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Fan M, Kalis S, Wei L, Scharff MD. A source of the single-stranded DNA substrate for activation-induced deaminase during somatic hypermutation. Nature communications. 2014;5:4137. doi: 10.1038/ncomms5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei PC, Chang AN, Kao J, Du Z, Meyers RM, Alt FW, Schwer B. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell. 2016;164:644–655. doi: 10.1016/j.cell.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Riblet R, Schildkraut CL. Sites that direct nuclear compartmentalization are near the 5′ end of the mouse immunoglobulin heavy-chain locus. Molecular and cellular biology. 2005;25:6021–6030. doi: 10.1128/MCB.25.14.6021-6030.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullo JM, Demarco IA, Pique-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012;149:1474–1487. doi: 10.1016/j.cell.2012.04.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic representation of strategy showing the TAP-tag Exosc3 allele. The TAP-tag was inserted in the 5′ region of exon1 together with a Neo cassette. The Neo gene was removed via mating to a FLPe recombinase mouse. Orange triangle represents FRT site. This diagram is not drawn to scale. (B) PCR products resulting from genotyping of Exosc3 TAPtag mice. Map shows primers location in Exosc3 gene locus. (C) Resting B cells from TAPtag-Exosc3 mice were treated with LPS and IL4 and assayed for IgG1 expression after 72 hrs. (D) Schematic representation of the steps involved in the purification of the RNA exosome complex from B cells. (E) Flag and Streptavidin IPs were performed and the presence and functionality of tags validated by western blot prior to mass spectrometry. (F) Co-immunoprecipitation of RNA exosome complex with Mtr4 in HEK293T cells. Flag-tagged Exosc3 and HA-tagged Mtr4-expressing plasmids were overexpressed in HEK293T cells. Lysates were co-immunoprecipitated using anti-FLAG-agarose beads, and western blots were probed with indicated antibodies.
Reconstructed two color 3D STORM (super-resolution) video from data set of 50,000 frames of fixed splenic B cells, in which AID & Exosc3 were labeled with atto488 and Alexa Fluor 647 respectively. This video shows 360° rotation in X and Y axis for spatial distribution of AID and RNA exosome in the nucleus of mouse B cells.
Reconstructed two color 3D STORM (super-resolution) video from data set of 20,000 frames of fixed HEK293Tcells, in which AID & Exosc3 were labeled with atto488 and Alexa Fluor 647 respectively. This video shows 360° rotation in X and Y axis for spatial distribution of AID and RNA exosome in the nucleus of non-B cells(HEK293T).
Reconstructed two color 3D STORM (super-resolution) video from a data set of 50,000 frames with Alexa488 labeled AID, AlexaFluor647 labeled Mtr4, show 360° rotation in X and Y axis for spatial distribution of AID and Mtr4 molecules inside the nuclei of B cells isolated from wild type B cells.
Reconstructed two color 3D STORM (super-resolution) video from a data set of 50,000 frames with Alexa488 labeled AID, AlexaFluor647 labeled Mtr4, show 360 ° rotation in X and Y axis for spatial distribution of AID and Mtr4 molecules inside the nuclei of B cells isolated from RNA exosome nuclear activity deficient (Exosc10COIN/LacZ) mouse.
Table S1 (related with Figure 1,2,3,4,5, 6 and 7): (a) RNA exosome complex protein identity for negative control following mass spectrometry. (b) RNA exosome complex protein identity from Exosc3TAP mouse following mass spectrometry. (c) Sanger sequencing and Next generation sequencing (NGS) analysis in Mtr4 and Setx deficient CH12F3. (d) Details of statistical analyses and biological and technical repeats performed for the 3D-STORM experiments presented in Figs. 1–4 and Figs. S2–4.
B cells were harvested from Exosc3 TAP-tagged mice and fixed after 72 hrs of treatment with stimulation cocktail. HEK293T cells were transfected with Red fluorescence protein(RFP) with N-terminus HA-tag and hExosc3 mammalian expression vectors and then fixed similar to B-Cells post 16hrs of transfection. Reconstructed two color 3D STORM (super-resolution) image for (A) Atto488 labeled Exosc5, AlexaFluor647 labeled Exosc3 & DAPI labeled nucleus of B-Cell and (D) for Atto488 labelled RFP and AlexaFluor647 labeled hExosc3 in HEK293T cell. Histogram of the distribution of interactions of Exosc3 and Exosc5 calculated in the B-cell (B) and RFP & Exosome complex in HEK293T cell (E), by using custom written algorithm “Nearest Neighbors Search” in the Matlab (2014b, Math works) software. (C) Two components Exosc3 and Exosc5 of RNA exosome complex were selected since they have closest distance in crystal structure (PDB-2NN6) and used as positive control for the interaction. All of the 3D STORM imaging were performed in three different B-Cells/HEK293T cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is × 100.
B cells were harvested from Exosc3 TAP-tagged mice and fixed after 72 hrs of treatment with stimuli cocktail. (A) Reconstructed two color 3D STORM (super-resolution) image for Atto488 labeled AID, AlexaFluor647 labeled Exosc3 & DAPI labeled nucleus. (B) Three dimensional views of the boxed region. (C) Higher magnification three dimensional [3D] view of the boxed region. Histogram for the distribution of interactions of AID and RNA exosome complex were calculated in the nucleus of (D)B cell & (E) HEK293T cell and cytoplasm of (E) B cell & (L) HEK293T cell, by using Matlab (2014b, Math works) software. Comparison of the distribution of paired interaction of AID and RNA exosome in the nucleus and cytoplasm of (F) B cell & (M) HEK293t cell were calculated by using a paired Student’s T-test in Matlab (2014b, Math works) software and P values are mentioned in the graph. (G) Control experiment for non-specific binding primary and secondary antibodies- (Top panel) B cells were harvested from AID K/O mice & labeled with primary antibody AID, secondary antibody Atto 488& nucleus with DAPI and (Bottom panel) B cells were harvested from Exosc3 TAP-tagged mice & labeled with secondary antibody AlexaFluor647& nucleus with DAPI. (H) Comparison of the inter molecular distance between AID molecules in the nucleus and cytoplasm of a activated B-cell, calculated by using Matlab (2014b, Math works) software. Reconstructed two color 3D STORM (super-resolution) image from (I) B cell and (J) HEK293T cell, in which AID & Exosc3 were labeled with atto488 and Alexa Fluor 647 respectively. All of the 3D STORM imaging were performed in three different B-Cells/HEK293T cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is × 100. Scale bars: 1μm(A) and 2μm(G). (P value: ** <0.01).
B cells were harvested from the spleen of Exosc10CC/LacZ mice and fixed after 72 hrs of treatment with (1) stimulation cocktail and (2) 4OHT with stimulation cocktail. Reconstructed two color 3D STORM (super-resolution) (A and C) image from a data set of 50,000 frames with Alexa488 labeled AID, AlexaFluor647 labeled Mtr4 and DAPI labeled nucleus, show spatial distribution of AID and Mtr4 molecules inside the nuclei of B cells isolated from wild type cells, and (B and D) image from Exosc10 knockout cells. (E) Conditional inversion allele of Exosc10 in 4OHT and stimuli cocktail treated Exosc10 deficient (Exosc10COIN/LacZ) cells was validated by flow cytometry analysis (upper panel). RNA expression level of Exosc10, AID and Mtr4 genes in Exosc10 deficient (Exosc10COIN/LacZ) cells was tested by RT-qPCR after 72 hrs of 4OHT and stimulation cocktail treatment. RNA level (lower panel). Results were obtained from 4 experimental repeats. Error bars indicate S.D. (P value: *** <0.001). All of the 3D STORM imaging were performed in three different B-Cells (from independent experiments) and repeated three or more times. 3D STORM super resolution image magnification is ×100. Scale bars: 1μm(A) &(B).
(A) Brief map showing gRNA target regions and sequencing results of target region in AID mutant, Setx mutant and Mtr4 mutant. Red arrows indicate insertion or deletion junction of each allele. (B) Western blot analysis shows AID expression in stimulated AID, Setx and Mtr4 mutant CH12F3. (C) IgM to IgA CSR in Mtr4/Setx mutant CH12F3 cells was tested every day for 72h. (D) IgA CSR was assayed to test Mtr4 rescue effect after transfecting Mtr4 expressing plasmid DNA. (E) Schematic map localizing the IgH locus and binding sites of the PCR primer pair for mutation analysis. Mutation frequency of AID-induced somatic hypermutation on sense (non-transcribed) strand and anti-sense (transcribed) strand of IgH 5′Sμ region in Mtr4 mutant CH12F3 cells compared to WT CH12F3 cell controls. Ratio of mutation frequency between sense and anti-sense strands is indicated above bar graph. Details of mutations identified by Sanger sequencing in Sup. table 1c. (P values: ** <0.01) (F) IgA efficiency of two different Setx clones was normalized based on CSR level of parental cells. Error bars indicate S.D. (P values: ** <0.01, *** <0.001)
(A) Proliferation test of mutant CH12F3 clones using VPD450. (B) Sequencing analysis of gRNA target region on the Setx gene locus in Setx/Mtr4 double mutant clone. Setx gRNA was transfected into Mtr4 mutant cells. Red arrows indicate the junction of deleted region. Proliferation test of Setx/Mtr4 double mutant using VPD450 (bottom panel). (C) Western analysis of Mtr4 expression in Mtr4 mutant CH12F3 cells. (D) Sequencing analysis of gRNA target region at the Ung gene locus in Ung mutant CH12f3 clone and Mtr4/Setx/Ung mutant CH12F3 clone. (E) Validation of Ung gene expression using qRT-PCR in Ung mutant CH12F3 cells (F) Class switch recombination efficiency in Ung mutant Ch12F3 cells (G) RNA-seq tracks from IGV browser for WT B cells (Exosc3WT/WT; middle panel) RNA exosome mutant B Cells (Exosc3COIN/COIN; bottom panel) and overlap (top panel) for sense (blue) and antisense (red) RNAs expressed in 5′ and 3′ IgH Sμ regions (H) and intronic enhancer Eμ, Iμ and Sμ region.
(A, B) RNA exosome substrate ncRNA expression profiles of CD79b (C) and CD83 (D) TSS locus in WT and Exosc3coin/coin mouse B cells (Top). Gray arrow heads indicate primer sites for next generation sequencing (NGS) and red arrows indicate non-coding RNA and its transcription direction on the schematic map (Middle). Mutation frequency of C bases on transcribed and non-transcribed strands and ratio of non-transcribed strand mutation frequency compare to transcribed strand mutation frequency in Ung mutant and Mtr4/Setx/Ung mutant CH12F3 cells (Bottom). The number above bar graph means C to T mutation count on AID mutation hotspot motif. Details of mutations identified by NGS deep sequencing in Sup. table 1c.
(C, D) Mutational counts representing somatic coding variants across 4 different tumors in patients with coding mutations in analyzed genes. A red bar highlights specific transcriptional, replicative or mutational nucleotide changes. Indicating asymmetry in Liver, Lung and Bladder cancer, respectively (E). The largest number of mutations concurrent with Multiple Myeloma (F). P-value is indicated above each bar graph.
Reconstructed single color 3D-STORM video from a data set of 50,000 frames of fixed splenic B cells before CSR stimulation, in which Exosc3 was labeled with AlexaFluor647. This video shows 360° rotation in X and Y axis for RNA exosome.
Reconstructed single color 3D-STORM video from a data set of 50,000 frames of fixed splenic B cells after CSR stimulation, in which Exosc3 was labeled with AlexaFluor647. This video shows 360° rotation in X and Y axis for RNA exosome.