

Abstract
Aptamers, that exist naturally in living cells as functional elements and can switch nonfluorescent natural targets to fluorophores, are very useful in developing highly sensitive and selective biosensors and screening functional agents. This work demonstrates that human telomeric G-quadruplex (HTG) can serve as a potential fluorophore-switching aptamer (FSA) to target a natural isoquinoline alkaloid. We found that, among the G-quadruplexes studied here and the various structurally similar alkaloids including epiberberine (EPI), berberine (BER), palmatine (PAL), jatrorrhizine (JAT), coptisine (COP), worenine (WOR), sanguinarine (SAN), chelerythrine (CHE), and nitidine (NIT), only the HTG DNA, especially with a 5′-TA-3′ residue at the 5′ end of the G-quadruplex tetrad (5′-TAG3(TTAG3)3-3′, TA[Q]) as the minimal sequence, is the most efficient FSA to selectively light up the EPI fluorescence. Compared to the 5′ end flanking sequences, the 3′ end flanking sequences of the tetrad contribute significantly less to the recognition of EPI. The binding affinity of EPI to TA[Q] (Kd = 37 nM) is at least 20 times tighter than those of the other alkaloids. The steady-state absorption, steady-state/time-resolved fluorescence, and NMR studies demonstrate that EPI most likely interact with the 5′ end flanking sequence substructure beyond the core [Q] and the G-quadruplex tetrad in a much more specific manner than the other alkaloids. The highly selective and tight binding of EPI with the FSA and significantly enhanced fluorescence suggest the potential development of a selective EPI sensor (detection limit of 10 nM). More importantly, EPI, as the brightest FSA emitter among the alkaloids, can also serve as an efficient conformation probe for HTG DNA and discriminate the DNA G-quadruplex from the RNA counterpart. Furthermore, EPI can bind stoichiometrically to each G-quadruplex unit of long HTG DNA multimer with the most significant fluorescence enhancement, which has not been achieved by the previously reported probes. Our work suggests the potential use of EPI as a bioimaging probe and a therapeutic DNA binder.
Graphical Abstract
Besides serving as templates for genetic coding, nucleic acids also play a vital role in other biologically important molecular recognition events. Aptamers are a special group of nucleic acids that display high binding specificity and affinity to their targets. Developed for over two decades, aptamers have received much attention in many fields including sensors, clinical diagnostics, drug delivery, therapeutics, and gene regulation.1–5 The targets of aptamers range from small molecules to proteins, cells, and viruses.6–13
Because of enrichment in functional groups and structural motifs, proteins and cells have an increasing probability to discover nucleic acid sequences as aptamers to target. However, less than a quarter of existing aptamers have been identified for small molecules due to the limited number of functional groups available for binding between them.14 Many techniques, including colorimetric assay (mainly based on gold nanoparticles2,15,16), capillary electrophoresis,17–20 affinity chromatography, 8,11 gradient micro free flow electrophoresis,21 SPR,22 and electrochemistry,23–25 have been utilized as tools to explore the aptamers’ potential to target small molecules. Metal ions,6,7 adenosine,8,15,24 ATP,9,10,23 cocaine,15,25 and thrombin11 have been usually selected as the proof-of-concept representatives for validating the feasibility of these techniques. The fluorescence technique has been identified as a reliable and inexpensive method for aptamer screening and applications.26–29 Advances in fluorescence techniques have made it possible to even visualize single aptamer molecules.28,29 However, labeling the aptamers with fluorophore tags is usually necessary for nonfluorescent targets. The position of the tags in the aptamer and conformation changes caused by such labeling should be carefully optimized in order not to compromise the aptamer binding.30 For fluorescent targets, a more straightforward and label-free method can be established through aptamer bindinginitiated fluorescence changes. Label-free fluorescence technique has been expanded to nonfluorophore targets by conjugating the aptamer (recognition unit) with a second fluorophore-targeting aptamer (signal unit).31 Apparently, there is still a great demand in developing various aptamers to target biologically important small molecules that are more straightforward in design and have a more functional expectation.
The conformational changes of aptamers upon target binding have been extensively studied.7,32,33 However, relatively less attention has been paid for alteration of the small molecule targets’ physicochemical properties upon such binding. This can be directly explored by fluorescence response of the small molecule targets. Small molecule–aptamer complexes have a great potential in the development of unique diagnostics and therapeutics.34 In this respect, a small molecule target that is nonfluorescent alone in solution but is fluorescently lighted up by an aptamer is a promising candidate. In this work, we term this type of aptamer as fluorophore-switching aptamer (FSA). The sensors based on the FSA rationale benefit a low fluorescence background and are label free. A few small molecules with the fluorescence switched on by FSAs have been developed including malachite green,35 GFP-like dyes,36 Rhodamine,37 and Hoechst 3325838 analogs. However, FSAs that exist naturally in living cells and can selectively target natural small molecules, and thus have potentially therapeutic and diagnostic functions, are still lacking if considering the intrinsic functional applications.
Natural isoquinoline alkaloids (IAs, Scheme S1, Supporting Information), especially in the group of protoberberine alkaloids (PA) and benzophenanthridine alkaloids (BA), are abundant in medicinal plant species and have important biological and therapeutic activities including antimicrobial, anti-inflammatory, antioxidation, and anticancer.39,40 Their activities are believed to partially originate from their binding with nucleic acids.39 Although the alkaloid structure-dependent interactions with DNAs (ds-DNA,41,42 G-quadruplex,43–47 and triplex48) and RNAs49 have been thoroughly investigated, no nucleic acid has been found with a highly specific and tight binding to these biologically important IAs. Since these IAs (especially PAs) are almost nonfluorescent and share similar molecular weight and structural frame (Scheme S1, Supporting Information), identifying an aptamer that can selectively target one of these IAs, especially by a label-free fluorescence technique, is very important to expand the IAs’ functionality. Previously, we found that the presence of an abasic site in DNA can significantly enhance IAs’ fluorescence response,50 but the binding selectivity for the alkaloids is far from ideal according to the requirements of an aptamer.
Herein, we found that human telomeric G-quadruplexes (HTGs) can serve as potential aptamers to specifically target epiberberine (EPI, Scheme S1, Supporting Information) with a much higher fluorescence response than the other structurally similar alkaloids including berberine (BER), palmatine (PAL), jatrorrhizine (JAT), coptisine (COP), worenine (WOR), sanguinarine (SAN), chelerythrine (CHE), and nitidine (NIT). It is well-known that HTGs as functional nucleic acids can inhibit the activity of telomerase and thus reduce the risk of cancer evolution.51,52 Thus, in the aspect of diagnostic and therapeutic applications, it is very important to develop the HTG as an FSA for a natural small molecule.4,5 To our knowledge, this is the first report on a natural FSA that can target a therapeutic and natural small molecule using the labelfree fluorescence strategy. Besides serving as the specific FSA’s target, EPI screened here also exhibits very promising applications in recognizing the multimeric HTG, which has not been achieved by the previously reported probes.
EXPERIMENTAL SECTION
Materials and Reagents
DNA species (Table S1, Supporting Information) were synthesized by TaKaRa Biotechnology Co., Ltd. (Dalian, China) and purified by HPLC. For NMR measurements, the 5′-TAG3(TTAG3)3-3′ DNA oligonucleotide (TA[Q], [Q] = 5′-G3(TTAG3)3-3′) was also synthesized (1 μmol scale) using b-cyanoethylphosphoramidite solid-phase chemistry on an Expedite 8909 Nucleic Acid Synthesizer (Applied Biosystem, Inc.) in a DMT-on mode. The DNA sample was purified using MicroPure II Columns from BioSearch Technologies (Novato, CA) as previously described.53 The DNA concentrations were measured by first dissolving DNA in pure water and detecting the UV absorbance at 260 nm using extinction coefficients calculated by the nearest neighbor analysis. BER, SAN, PAL, CHE (Sigma Chemical Co., St. Louis, USA), COP, NIT, JAT (Aladdin Reagent Co., Shanghai, China), EPI (Huicheng Biological Technology Co., Shanghai, China), and WOR (Sichuan Weikeqi Biological Technology Co., Chengdu, China) were obtained in their highest commercially available purities (>98%). Milli-Q water (18.2 MΩ·cm; Millipore Co., Billerica, USA) was used in all experiments. All other chemicals were of analytical-reagent grade (Sigma Chemical Co., St. Louis, USA) and used without further purification.
Fluorescence Measurements
Fluorescence spectra were acquired with a FLSP920 spectrofluorometer (Edinburgh Instruments Ltd., Livingston, UK) at 18 ± 1 °C, which was equipped with a temperature-controlled circulator (Julabo Labortechnik GmbH, Seelbach, Germany). To prepare the G-quadruplex solution, the strand was annealed in a thermocycler (first at 92 °C, then slowly cooled to room temperature) in 25 mM Tris-HCl buffer (pH 7.5) containing 1 mM ethylenediaminetetraacetic acid disodium salt (EDTA) and 100 mM KCl or NaCl. The alkaloid at the specified concentration was added into the DNA solution, and the resulting solution was allowed to incubate for 15 min before fluorescence measurement. The resulting solution was examined within 2 h. No time-dependent fluorescence response was observed after mixing, showing a rapid association between them. Time-resolved fluorescence decay was recorded on a time-correlated single photon counting FLSP920 system. The time-resolved measurement was carried out under the corresponding excitation and emission wavelengths. A Ludox solution was used as scatter for the instrument response. The data were fitted with a multiexponential decay, and χ2 was less than 1.1.
The stoichiometry of EPI binding to DNA was determined by Job’s plot analysis. The total concentration of EPI and the corresponding DNA G-quadruplex unit was maintained at 2 μM, and the EPI-to-DNA concentration ratio was sequentially varied.
Alkaloids were titrated with DNAs for measurement of the binding constants, and the fluorescence intensity was plotted as a function of the aptamer concentration. The data were fitted by KaleidaGraph (Synergy Software) according to a 1:1 binding model.54 The titration experiments were carried out by keeping the alkaloid concentrations at 0.5 μM or 20 nM.
To investigate the effect of ionic strength on the binding of alkaloids to the aptamer, the fluorescence intensities of 1 μM alkaloids in the presence of 1 μM aptamer were measured on addition of increasing Tris-HCl (pH 7.5) concentration by keeping the K+ concentration at 0.1 M.
UV/Vis Absorption Spectra
UV/vis absorption spectra were determined with a UV2550 spectrophotometer (Shimadzu Corp., Kyoto, Japan) at room temperature using a quartz cell with a path length of 1 cm.
1H NMR Measurements
The DNA oligonucleotide was dissolved in 25 mM potassium phosphate buffer (pH 7.0) and 75 mM KCl in 10%/90% D2O/H2O solution. The final NMR sample contained 0.2 mM DNA. The DNA–EPI complexes were prepared by adding an appropriate amount of EPI to the DNA sample. 1D 1H NMR experiments were carried out on a Bruker Avance 600 MHz spectrometer at 25 °C with the WATERGATE water suppression technique. High-resolution 1H NMR spectra were acquired with the following acquisition parameters: time domain, 32 K; 90° pulse width, 11.0 μs; spectral width, 16 ppm; relaxation delay, 1.0 s; and acquisition time, 3.2 s. 128 scans were accumulated. Fourth-order polynomial function was applied for the baseline correction.
RESULTS AND DISCUSSION
HTG in K+ Solution as an Effective FSA for Selectively Targeting and Fluorescently Lighting up Epiberberine
As shown in Scheme S1, Supporting Information, PAs and BAs share a similar heterocyclic structure. These alkaloids are reported to be weakly fluorescent in aqueous solution, with moderate fluorescence enhancement or fluorescence decrease upon binding to nucleic acids, dependent on the nucleic acid sequences and identity of the alkaloids.39,40,45 We aim to develop an FSA that has a tight and selective binding to a natural IA target and, at the same time, functions as a critical transducer element to significantly light up the IA’s fluorescence. Because of their crescent shapes and almost planar rigid structures, IAs seem to exhibit higher binding capacity to G-quadruplexes than to ss- and ds-DNAs/RNAs.39–49 These reports thus help to narrow the selection of the sequence pool for aptamer identification. In this work, considering the potentially practical application of the screened aptamer, G-quadruplexes derived from natural sequences were thus selected as the aptamer candidates to target specifically one of these alkaloids. The G-quadruplex sequences studied here (Table S1, Supporting Information) are mainly derived from human (entries from 1 to 15, Table S1, Supporting Information) and oxytricha (T3TT, T3TT3, T3T3T3) telomeres, oncogene c-myc (mPu22, 1XAV) and c-kit (2O3M) promoters, an HIV integrase inhibitor (T30695), and a G-quadruplex DNAzyme (PS2.M). Their folding conformations in K+ solution have been well documented.55–58 Due to the low abundance of EPI in plants compared to the other alkaloids, little attention has been paid for EPI interaction with nucleic acid. As shown in Figure 1 (inset), EPI alone in solution is nearly nonfluorescent (curve a), while upon binding to TA[Q], EPI becomes a fluorophore with its fluorescence increasing up to 45 times (curve b). The excitation and emission bands of EPI appear at 377 and 540 nm, respectively. The fluorescence quantum yield of TA[Q]-bound EPI is about 0.41, at least 27 times higher than that of EPI alone in aqueous solution (see the Supporting Information).
Figure 1.

Dependence of EPI (1 μM) fluorescence intensity at 540 nm on the G-quadruplex sequences (1 μM) in 0.1 M K+ solution (pH 7.5): [Q] (1), A[Q] (2), T[Q] (3), TA[Q] (4), AT[Q] (5), AA[Q] (6), TT[Q] (7), TA[Q]A (8), TA[Q]T (9), TTA[Q] (10), TTA[Q]TT (11), AAA[Q]AA (12), TA[Q]TT (13), [Q]T (14), [Q]TA (15), mPu22 (16), 1XAV (17), PS2.M (18), T30695 (19), 2O3M (20), T3TT (21), T3TT3 (22), and T3T3T3 (23). Inset: Excitation and emission spectra of EPI in the absence (a) and presence (b) of TA[Q].
We then tested the DNA sequence dependence of EPI fluorescence enhancement in order to screen an optimal FSA. The FSA based on a G-quadruplex structure must have a strong binding with EPI and at the same time have a relatively weak fluorescence response to the other IAs. As shown in Figure 1, the EPI fluorescence enhancement is strongly dependent on the G-quadruplex DNA sequences. Most of the HTGs are more efficient than the others in lighting up the EPI fluorescence in K+ solution. On the basis of the sequence dependence results, we can make the following conclusions for the HTG sequences. First, variation in the flanking sequences at the 5′ end of the HTG core ([Q]) has a larger effect on the EPI emission than the flanking sequences at the 3′ end. G-quadruplexes without any residues at the 5′ end of the core [Q] induce small fluorescence response. Second, at the 5′ end, at least two residues with an adenosine residue immediately adjacent to the core [Q] are required to significantly enhance the EPI fluorescence (for example, TA[Q] with the required least sequence as an efficient FSA). Further lengthening of a residue at the 5′ side (e.g., TA[Q] versus TTA[Q] and TA[Q]TT versus TTA[Q]TT) does not contribute the fluorescence enhancement significantly. Third, varying the sequences at the 3′ end of the core [Q] alone (e.g., TA[Q] versus TA[Q]A, TA[Q]T, and TA[Q]TT) has no significant effect on the EPI fluorescence. Thus, our results indicate that EPI binds to the K+-form telomeric G-quadruplexes most likely at the 5′ end.
The stacking interaction of PAs and BAs with HTGs has been proposed to mainly occur at the 5′ end by structural modeling.43,44 The 3′ end may be the second possible binding site.43,46 The preferable ligand binding at the 5′ end has also been reported for other G-quadruplexes.53 It is well-known that HTGs adopt dominantly hybrid conformations in K+ solution.55–61 The results that only the HTG sequences with appropriate 5′ flanking sequences, i.e., at least two residues with an adenosine residue, can efficiently enhance the EPI fluorescence in K+ solution (Figure 1), indicate that a 5′ flanking adenine followed by additional residue(s) is critical to form the EPI binding pocket at the 5′ end of the hybrid-form telomeric G-quadruplexes. Accordingly, it has been reported that the structured 5′ end sequences is critical for the formation of this hybrid form, as evidenced by a T:A:A triad substructure that is formed with the 5′-TA-3′ at the 5′ end of the core [Q] and the A base from the loop linking the tetrads.59 This may suggest a mode of binding related to this triad, such as “intercalating triad”, which has been reported for the c-myc promoter G-quadruplex recognition.53 Structural studies are underway to determine the exact binding mode of EPI.
We further investigated the dependence of fluorescence responses upon TA[Q] binding on the IA structures. As shown in Figure 2, in the presence of TA[Q] and in 0.1 M K+ condition, PAs (EPI, BER, COP, PAL, and WOR) generally emit higher fluorescence than BAs (JAT, SAN, CHE, and NIT). More importantly, the EPI emission is 18, 5, 13, 14, 68, 87, 135, and 293 times higher than BER, COP, PAL, WOR, JAT, SAN, CHE, and NIT, respectively. The difference in fluorescence resulting from the alkaloid structures can even be observed by the naked eye under UV illumination (Inset of Figure 2). The other G-quadruplexes derived from human telomeric sequence having at least two residues at the 5′ end and a base A immediately adjacent to the core [Q] also have a higher EPI fluorescence response than the other alkaloids (Figure S1, Supporting Information), while the discrimination of EPI from the other alkaloids is slightly less than TA[Q]. Thus, TA[Q] should be the minimal FSA sequence in lighting up EPI. The sequence variation at the 3′ end, however, has no significant effect on the selectivity of EPI binding. Thus, the selective binding to EPI over the other alkaloids is also strongly dependent on the 5′ end flanking sequences. Note that these alkaloids share a similarly heterocyclic structure and all exhibit weak fluorescence especially for EPI, BER, COP, and PAL in solution in the absence of a G-quadruplex. Up to now, no DNA sequence has been reported to selectively discriminate one alkaloid over the others by fluorescence method.39–49 In this work, we demonstrate that HTG, especially with TA[Q] as an efficient and minimal FSA, can target EPI with a high selectivity.
Figure 2.

Fluorescence intensity of the investigated IAs (1 μM) in 0.1 M K+ (red) and Na+ (green) in the presence of TA[Q] (1 μM). Inset: Photographs of these solutions under UV illumination.
A 1:1 binding model has been proposed for the interaction of alkaloids with HTGs.44,45 The formation of 1:1 alkaloid complexes was also confirmed in this work as indicated by the Job’s plot analysis (Figure S2, Supporting Information, with EPI as an example). Fluorescence titration of alkaloids (0.5 μM) by TA[Q] was used here to evaluate the binding strength. The typical titration curves and the fitting results according to the 1:1 binding model are provided in Figure 3. The good fitting to the data also confirms such 1:1 binding model. The binding constants (K11) with TA[Q] for COP, PAL, BER, and WOR are (1.4 ± 0.5) × 106, (4.1 ± 0.4) × 105, (4.1 ± 0.4) × 105, and (5.1 ± 0.4) × 105 M−1, respectively, which are similar to the reported values.44,45 Remarkably, EPI has a much tighter TA[Q] binding with a binding constant of (2.8 ± 0.6) × 107 M−1 (disassociation constant Kd = 37 ± 8 nM). However, JAT, SAN, CHE, and NIT exhibit a negligible fluorescence change upon increasing the TA[Q] concentrations (for clarity, only SAN is shown in Figure 3). Although these alkaloids also bind to G-quadruplex,43,45 no further binding investigation was made here due to the lack of FSA property. To determine the binding constant more accurately,44 we carried out the titration experiments at 20 nM EPI that is comparable with the Kd value, and a similar binding constant was obtained (Figure S3A, Supporting Information). However, the G-quadruplex [Q] without the 5′/3′ flanking sequences has a similar binding affinity to the alkaloids including EPI, PLT, COP, WOR, and BER (Figure S3B, Supporting Information), again suggesting the vital role of the 5′ flanking sequence on the EPI binding. Note that the EPI binding affinity with the HTG is even comparable with the well-evaluated telomerase inhibitors of telomestatin and BRACO-19 (for a review, see ref 30), but these probes do not exhibit the aptamer-switched-on fluorescence properties. Thus, EPI is a promising target for the HTGs having the aptamer-switched-on fluorescence behavior with a high binding affinity and specificity.
Figure 3.

Dependence of fluorescence intensity of IAs (0.5 μM) on gradual addition of TA[Q], showing a 1:1 binding by the curve fitting (solid line). Inset: Photographs of the solutions under UV illumination (from left to right: EPI, BER, COP, PAL, WOR, JAT, SAN, CHE, NIT) with TA[Q] at 0.2 μM.
We also conducted 1D 1H NMR titration experiments to study the binding of EPI with TA[Q]. The TA[Q] alone (EPI free, Figure 4) in 0.1 M K+ solution forms a major hybrid-1 G-quadruplex conformation as indicated by the well-resolved imino proton peaks, as shown previously.59 Upon addition of EPI to the TA[Q] DNA solution, the imino proton peaks of the DNA first broaden at lower EPI equivalence (0.5 N) and then become sharper at higher EPI equivalence (Figure 4), indicating a medium exchange rate of EPI binding to TA[Q] on the NMR time-scale. The observation of a relatively well-resolved imino proton peak suggests a rather specific EPI binding. The upfield-shifting of the DNA imino proton peaks indicates an EPI binding mode of the tetrad stacking.53 Thus, EPI most likely interacts simultaneously with the G-quadruplex tetrad and the triad59 as mentioned above, by intercalating between them. The NMR titration data support the 1:1 binding stoichiometry of EPI with TA[Q], as no further qualitative change is visible in the imino region at the EPI equivalence higher than 1 (Figure 4), also meaning a specific affinity at the intercalating site.
Figure 4.

Imino proton region of the 1D 1H NMR titration spectra of TA[Q] in K+ solution (pH 7.0) at 25 °C. TA[Q] was titrated with the EPI concentration ranging from 0 to 4 equivalence.
In order to verify the role of the G-quadruplex structure-dependent EPI binding on fluorescence enhancement, we carried out the binding experiments in Na+ solution, in which the HTG adopts mainly a basket antiparallel structure.55–61 As shown in Figure 2, the fluorescence intensity of EPI-TA[Q] in Na+ is indeed 12 times lower than in K+. This K+-form-dependent fluorescence enhancement is also visible for the other alkaloids (especially PAs) but with less K+/Na+ factors. Thus, our results suggest that EPI binding and fluorescence enhancement are highly dependent on the G-quadruplex structure.
We compared the absorption spectra using EPI and COP (the second most lighting-up emitter as indicated in Figure 2) as typical examples. As shown in Figure S4A, Supporting Information, TA[Q] binding induces significant red-shifts in the absorption peaks of EPI and COP. However, only EPI exhibits an obvious peak splitting upon binding to TA[Q] with the appearance of four peaks at 359, 377, 465, and 490 nm between 300 and 500 nm (Figure S4A, Supporting Information), indicating a stronger interaction with the G-quadruplex. The ring A moiety of PAs (Scheme S1, Supporting Information) contributes mainly to the absorption peaks between 300 and 400 nm, while the ring C/D moieties contribute mainly to the absorption peaks between 400 and 500 nm.42 Thus, the splitting of absorption peaks at both regions suggests that the substituent pattern in EPI favors a strong binding of both of the moieties to the G-quadruplex. This strong interaction would reduce the chance of excited state intramolecular electron transfer that is responsible for the observed weak fluorescence of alkaloids alone in solution.42 The strong interaction of EPI with TA[Q] was also confirmed by fluorescence decay experiments (Table S2, Supporting Information). The average lifetimes of the EPI-TA[Q] and COP-TA[Q] excited states are 11.26 and 8.53 ns, respectively. Thus, TA[Q] stabilizes the EPI excited state to a much greater degree in comparison with COP, also suggesting that the specific interaction with EPI favors the excited state relaxing via a radiative pathway.
Since the alkaloids are positively charged, their binding to G-quadruplex with increasing Tris-HCl concentrations was investigated to reveal the possible contribution of the electrostatic force in the aptamer binding. As shown in Figure S4B, Supporting Information, the EPI fluorescence decays much slower than COP with increasing Tris-HCl concentrations, suggesting that electrostatic force contributes less to the EPI binding to the aptamer. This specific binding of EPI was also confirmed by the Tm experiments (Figure S5, Supporting Information). The EPI binding induces a higher thermostability of the aptamer. Taken together, our experimental results suggest that EPI interacts specifically with the 5′ end structured sequence of HTG, while the binding of the other alkaloids is much less specific.
Selective EPI Sensor
EPI also exhibits wide biological and therapeutic activities including Alzheimer’s disease inhibition,62 cell protection from damage,63 and DNA topoisomerase inhibition.64 The strong aptamer binding of EPI over the other alkaloids provides a possibility in selectively assaying EPI by fluorescence readout. Since BER, PAL, and SAN are abundant in plants and their interactions with DNA have been extensively studied,39–49 in this work, the concentration-dependent fluorescence response of EPI in the presence of the G-quadruplex was directly compared with these alkaloids. As shown in Figure S6A, Supporting Information, in the presence of 1 μM TA[Q], EPI exhibits a significant linear fluorescence response upon increasing its concentration, while the changes in fluorescence for BER, PAL, and SAN are negligible. The detection limit for EPI is about 10 nM by assuming the signal-to-noise ratio of 3, whereas the response of the other alkaloid at this concentration is still at the noise level (Figure S6B, Supporting Information, with PAL as an example). Since the medically important alkaloids are often highly similar in structure, it is valuable to develop a reliable method to identify a special alkaloid. For example, because EPI and BER have an identical molecular weight and the only difference between them is the substituent pattern at the heterocyclic PA backbone, more care should be taken for preparing BER or EPI with a high purity. As shown in Figure S7, Supporting Information, coexistence of 20 times higher BER concentration produces little effect on the EPI fluorescence response, suggesting that TA[Q] binding to EPI is highly selective, and BER does not compete with EPI in binding to the aptamer. There is even no previous report65 on a simple and reliable fluorescence method in identifying one alkaloid from the others with such high selectivity. Thus, our method using the screened aptamer as the selector and fluorescence inducer most likely has the potential in the EPI analysis with a high sensitivity and selectivity.
EPI as an Efficient Conformational Probe for HTG
Although TA[Q] was identified as the minimal FSA in targeting EPI, a significant EPI fluorescence response was also observed for the natural HTGs other than TA[Q], suggesting the potential functional applications of the screened EPI in efficiently exploring the natural HTGs. Because the HTG structure is sequence and environment sensitive,55–61 there is an increasing demand in developing G-quadruplex structure-selective probes.30 It is well-known that the HTG DNA adopts an antiparallel basket structure in Na+ and a hybrid structure in K+.55–61 Thus, EPI could serve as an efficient probe to follow the G-quadruplex conformation changes at different cation solutions. As shown in Figure S8A, Supporting Information, addition of K+ to the EPI-TA[Q] solution containing 0.1 M Na+ induces a prompt increase in EPI fluorescence, in contrast to the small change observed with the identically molecular-weight BER as the fluorescent probe. Up to about 20 times higher fluorescence response of EPI in comparison to BER can be obtained for the K+-induced conformation conversion. Therefore, EPI can be used as a sensitive probe for probing the HTG conformation.
Accumulating evidence suggests that the HTG RNA sequences can also form a G-quadruplex structure.66 The formation of the RNA G-quadruplex in the living cell has been identified.67 Therefore, there is a prompt need to develop selective probes that can discriminate HTG DNA from the RNA counterpart. As shown in Figure S8B, Supporting Information, a much lower fluorescence response for EPI binding to the RNA G-quadruplex of rUA[Q] (Table S1, Supporting Information) is observed with a binding constant of (1.2 ± 0.2) × 106 M−1, about 20 times lower than that with TA[Q]. Thus, EPI could serve as a selective probe to investigate the structure of HTG DNA with a negligible interference from the RNA counterpart. Upon alkaloid binding, the native global structure of HTG DNA in K+ is still maintained.43 Thus, EPI can serve as superior fluorescent probe for G-quadruplex structure over the usually used porphyrin derivatives due to the weakness of their binding-induced structure rearrangement.30
EPI as an Efficient G-Quadruplex Unit-Based Emitter for Long Telomeric DNA G-Quadruplex Multimer
The above experiments were carried out only for the sequences containing a single DNA G-quadruplex unit. However, the length of the HTG 3′ terminal single-stranded overhang is about 150–200 nucleotides.68 Higher-order G-quadruplex structures (beads-on-a-string multimer) with consecutive G-quadruplex units that are connected by TTA linkers can be formed in human telomere.69–71 Thus, there is a great demand to develop a specific ligand that has a similarly strong binding affinity to each individual G-quadruplex unit in the multimer, independent of the G-quadruplex length. The biological applications of the natural IA may be expanded if it can bind to the HTG multimer via a G-quadruplex-unit-based manner. Thus, we further investigated the fluorescence responses of IAs upon binding to the HTGs containing one, two, and three G-quadruplex units (5′-(TTA[Q])nTTA-3′, n = 1, 2, 3), respectively. Among the investigated alkaloids, EPI is still the most efficient emitter upon binding to the long HTGs (Figure S9, Supporting Information). Additionally, EPI also exhibits a G-quadruplex unit-based 1:1 binding (Figure S10, Supporting Information). These results suggest the potential application of EPI as a promising therapeutic candidate targeting the HTGs. Fluorescence titration (with EPI at 0.5 μM) indicates that EPI exhibits a comparable binding constant K11 with these DNAs (Figure 5, in G-quadruplex unit, about 2.6 × 107 M−1 in average), meaning a G-quadruplex length-independent unit binding behavior. Such binding strength was also confirmed with the EPI concentration at 20 nM (best comparable with the Kd value, Figure S11, Supporting Information). Previously, it was reported that some synthetic small molecules can target the G-quadruplex dimer72 and prompt formation of the G-quadruplex sandwich-like assembly.73 Additionally, the quadruplex–quadruplex interface in a multimer has been shown to have a varied effect on the binding of a porphyrin derivative in comparison with the monomer binding.74 We note that EPI screened here is the first reported natural fluorophore that can be used to target independently each of the DNA G-quadruplex units in a long HTG multimer, which occurs in the G-quadruplex monomer.
Figure 5.
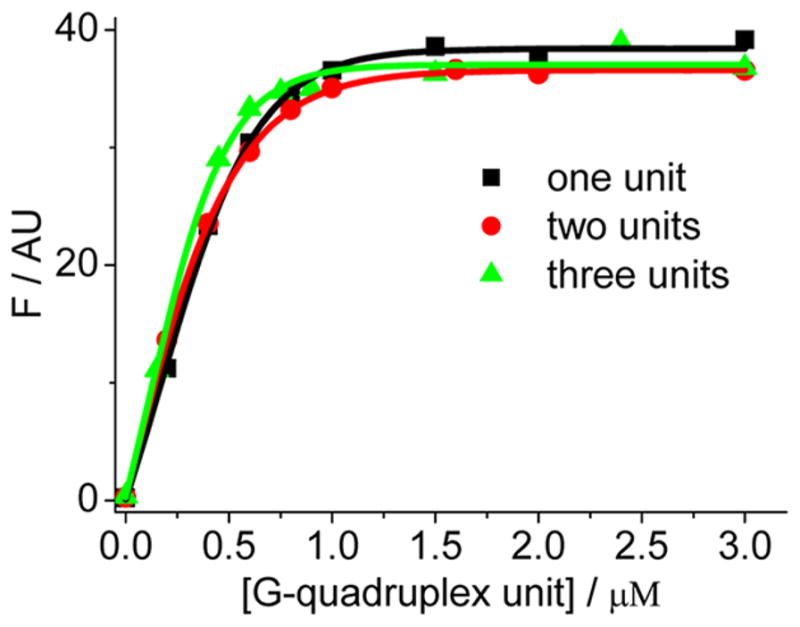
Fluorescence intensity of EPI (0.5 μM) in 0.1 M K+ upon addition of increasing concentrations of 5′-(TTA[Q])nTTA-3′ containing one, two, and three G-quadruplex units with n = 1, 2, 3, respectively, showing a 1:1 binding by the curve fitting (solid line). The X-axis is based on the G-quadruplex unit concentration. The results based on the G-quadruplex strand concentrations are shown in Figure S9, This work was supported by the Zhejiang Provincial Natural Science Foundation of China for Distinguished Young Scholars (Grant No. LR12B05001), the Zhejiang Provincial Public Welfare Project (Grant No. 2014C31150), and the National Natural Science Foundation of China (Grant No. 21075112).
CONCLUSIONS
This work demonstrates that the HTG sequences can be developed as fluorophore-switching aptamers (FSAs) targeting EPI, one of the natural IAs. The natural HTG, especially with 5′-TAG3(TTAG3)3-3′ (TA[Q]) as the minimal sequence, is the most efficient FSA for selectively lighting up the EPI fluorescence, in comparison with BER, PAL, JAT, COP, WOR, SAN, CHE, and NIT. It was found that the 5′ end sequence of the tetrad has a greater effect on the EPI discrimination capacity than the 3′ end sequence, suggesting that the most possible EPI binding site is at the 5′ end. Although these alkaloids have identical parent structures and differ by only the substituent pattern and minor structure modification, at least 20 times tighter binding affinity of the aptamer to EPI was observed. The binding mechanism was briefly discussed. EPI stacking on the 5′ end of the FSA tetrad and interacting with the substructure of the 5′ end flanking sequences are the main driving forces, while the other alkaloids experience more electrostatic force in the G-quadruplex binding. The identified G-quadruplex-based FSA can be used as a switch for developing a highly selective and sensitive EPI sensor (about 10 nM in detection limit) with an enhanced fluorescence response. As an efficient conformation probe, EPI can efficiently differentiate the HTG DNA structure from its RNA counterpart and monitor the DNA G-quadruplex conformation conversion between different topologies. Importantly, EPI exhibits a similar binding capacity to each G-quadruplex unit of the long telomeric DNA G-quadruplex multimer, suggesting that EPI may be used as a functional fluorescence probe for the HTG multimeric recognition. This performance has not been achieved by the previously reported probes. Our work will expand the applications of EPI alkaloid in sensors and therapeutics. Due to the high selectivity and specificity of EPI binding to the HTG, there is further necessity to identify the detailed molecular modeling in order to gain an insight into the structural feature of the alkaloid to target HTG. This will be done in the future by more techniques (for example, 2D NMR and modeling calculation).
Supplementary Material
Footnotes
The authors declare no competing financial interest.
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Liu J, Cao Z, Lu Y. Chem Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li D, Song S, Fan C. Acc Chem Res. 2010;18:631–641. doi: 10.1021/ar900245u. [DOI] [PubMed] [Google Scholar]
- 3.Iliuk AB, Hu L, Tao WA. Anal Chem. 2011;83:4440–4452. doi: 10.1021/ac201057w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu G, Ye M, Donovan MJ, Song E, Zhao Z, Tan W. Chem Commun. 2012;48:10472–10480. doi: 10.1039/c2cc35042d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deigan KE, Ferré-D’Amaré AR. Acc Chem Res. 2011;44:1329–1338. doi: 10.1021/ar200039b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiang Y, Lu Y. Inorg Chem. 2014;53:1925–1942. doi: 10.1021/ic4019103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng D, Zou R, Lou X. Anal Chem. 2012;84:3554–3560. doi: 10.1021/ac300690r. [DOI] [PubMed] [Google Scholar]
- 8.Deng Q, German I, Buchanan D, Kennedy RT. Anal Chem. 2001;73:5415–5421. doi: 10.1021/ac0105437. [DOI] [PubMed] [Google Scholar]
- 9.Sazani PL, Larralde R, Szostak JW. J Am Chem Soc. 2004;126:8370–8371. doi: 10.1021/ja049171k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng D, Seferos DS, Giljohann DA, Patel PC, Mirkin CA. Nano Lett. 2009;9:3258–3261. doi: 10.1021/nl901517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Q, Li XF, Shao Y, Le XC. Anal Chem. 2008;80:7586–7593. doi: 10.1021/ac801206s. [DOI] [PubMed] [Google Scholar]
- 12.Keefe AD, Pai S, Ellington A. Nat Rev Drug Discovery. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balogh Z, Lautner G, Bardáczy V, Komorowska B, Gyurcsányi RE, Mészáros T. FASEB J. 2010;24:4187–4195. doi: 10.1096/fj.09-144246. [DOI] [PubMed] [Google Scholar]
- 14.McKeague M, DeRosa MC. J Nucleic Acids. 2012;2012:748913. doi: 10.1155/2012/748913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Lu Y. Angew Chem, Int Ed. 2006;45:90–94. [Google Scholar]
- 16.Wei H, Li B, Li J, Wang E, Dong S. Chem Commun. 2007:3735–3737. doi: 10.1039/b707642h. [DOI] [PubMed] [Google Scholar]
- 17.Mendonsa SD, Bowser MT. Anal Chem. 2004;76:5387–5392. doi: 10.1021/ac049857v. [DOI] [PubMed] [Google Scholar]
- 18.Drabovich AP, Berezovski M, Okhonin V, Krylov SN. Anal Chem. 2006;78:3171–3178. doi: 10.1021/ac060144h. [DOI] [PubMed] [Google Scholar]
- 19.Bao J, Krylova SM, Reinstein O, Johnson PE, Krylov SN. Anal Chem. 2011;83:8387–8390. doi: 10.1021/ac2026699. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Z, Ravelet C, Perrier S, Guieu V, Roy B, Perigaud C, Peyrin E. Anal Chem. 2010;82:4613–4620. doi: 10.1021/ac100755q. [DOI] [PubMed] [Google Scholar]
- 21.Turgeon RT, Fonslow BR, Jing M, Bowser MT. Anal Chem. 2010;82:3636–3641. doi: 10.1021/ac902877v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang AL, McKeague M, Liang JC, Smolke CD. Anal Chem. 2014;86:3273–3278. doi: 10.1021/ac5001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuo X, Song S, Zhang J, Pan D, Wang L, Fan C. J Am Chem Soc. 2007;129:1042–1043. doi: 10.1021/ja067024b. [DOI] [PubMed] [Google Scholar]
- 24.Zhang S, Hu R, Hu P, Wu ZS, Shen GL, Yu RQ. Nucleic Acids Res. 2010;38:e185. doi: 10.1093/nar/gkq728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Qi H, Shen L, Gao Q, Zhang C. Electroanalysis. 2008;20:1475–1482. [Google Scholar]
- 26.Zhang CY, Johnson LW. Anal Chem. 2009;81:3051–3055. doi: 10.1021/ac802737b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cruz-Aguado JA, Penner G. Anal Chem. 2008;80:8853–8855. doi: 10.1021/ac8017058. [DOI] [PubMed] [Google Scholar]
- 28.Pitchiaya S, Heinicke LA, Custer TC, Walter NG. Chem Rev. 2014;114:3224–3265. doi: 10.1021/cr400496q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song W, Strack RL, Svensen N, Jaffrey SR. J Am Chem Soc. 2014;136:1198–120. doi: 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vummidi BR, Alzeer J, Luedtke NW. Chem Bio Chem. 2013;14:540–558. doi: 10.1002/cbic.201200612. [DOI] [PubMed] [Google Scholar]
- 31.Stojanovic MN, Kolpashchikov DM. J Am Chem Soc. 2004;126:9266–9270. doi: 10.1021/ja032013t. [DOI] [PubMed] [Google Scholar]
- 32.Carrasquilla C, Lau PS, Li Y, Brennan JD. J Am Chem Soc. 2012;134:10998–11005. doi: 10.1021/ja304064a. [DOI] [PubMed] [Google Scholar]
- 33.Gopinath SCB, Misono TS, Kumar PKR. Crit Rev Anal Chem. 2008;38:34–47. [Google Scholar]
- 34.Famulok M, Hartig JS, Mayer G. Chem Rev. 2007;107:3715–3743. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- 35.Stead SL, Ashwin H, Johnston B, Tarbin JA, Sharman M, Kay J, Keely BJ. Anal Chem. 2010;82:2652–2660. doi: 10.1021/ac902226v. [DOI] [PubMed] [Google Scholar]
- 36.Paige JS, Wu KY, Jaffrey SR. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sunbul M, Jaschke A. Angew Chem, Int Ed. 2013;52:13401–13404. doi: 10.1002/anie.201306622. [DOI] [PubMed] [Google Scholar]
- 38.Sando S, Narita A, Hayami M, Aoyama Y. Chem Commun. 2008;33:3858–3860. doi: 10.1039/b808449a. [DOI] [PubMed] [Google Scholar]
- 39.Bhadra K, Kumar GS. Med Res Rev. 2011;31:821–862. doi: 10.1002/med.20202. [DOI] [PubMed] [Google Scholar]
- 40.Maiti M, Kumar GS. Med Res Rev. 2007;27:649–695. doi: 10.1002/med.20087. [DOI] [PubMed] [Google Scholar]
- 41.Bhadra K, Maiti M, Kumar GS. Biochim Biophys Acta. 2008;1780:1054–1061. doi: 10.1016/j.bbagen.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Hirakawa K, Hirano T. Photochem Photobiol. 2008;84:202–208. doi: 10.1111/j.1751-1097.2007.00220.x. [DOI] [PubMed] [Google Scholar]
- 43.Bessi I, Bazzicalupi C, Richter C, Jonker HRA, Saxena K, Sissi C, Chioccioli M, Bianco S, Bilia AR, Schwalbe H, Gratteri P. ACS Chem Biol. 2012;7:1109–1119. doi: 10.1021/cb300096g. [DOI] [PubMed] [Google Scholar]
- 44.Arora A, Balasubramanian C, Kumar N, Agrawal S, Ojha RP, Maiti S. FEBS J. 2008;275:3971–3983. doi: 10.1111/j.1742-4658.2008.06541.x. [DOI] [PubMed] [Google Scholar]
- 45.Bhadra K, Kumar GS. Biochim Biophys Acta. 2011;1810:485–496. doi: 10.1016/j.bbagen.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 46.Bazzicalupi C, Ferraroni M, Bilia AR, Scheggi F, Gratteri P. Nucleic Acids Res. 2013;41:632–638. doi: 10.1093/nar/gks1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai LP, Hagihara M, Jiang ZH, Nakatani K. Chem Bio Chem. 2008;9:2583–2587. doi: 10.1002/cbic.200800256. [DOI] [PubMed] [Google Scholar]
- 48.Sinha R, Saha I, Kumar GS. Chem Biodiversity. 2011;8:1512–1528. [Google Scholar]
- 49.Çetinkol ÖP, Hud NV. Nucleic Acids Res. 2008;37:611–621. doi: 10.1093/nar/gkn977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu F, Sun Y, Shao Y, Xu S, Liu G, Peng J, Liu L. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048251. No. e48251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 52.Patel DJ, Phan AT, Kuryavyi V. Nucleic Acids Res. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai J, Carver M, Hurley LH, Yang D. J Am Chem Soc. 2011;133:17673–17680. doi: 10.1021/ja205646q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mohanty J, Bhasikuttan AC, Nau WM, Pal H. J Phys Chem B. 2006;110:5132–5138. doi: 10.1021/jp056411p. [DOI] [PubMed] [Google Scholar]
- 55.Chaires JB, Graves D. Quadruplex Nucleic Acids. Springer; Berlin, Heidelberg: 2013. [Google Scholar]
- 56.Liu L, Shao Y, Peng J, Huang C, Liu H, Zhang L. Anal Chem. 2014;86:1622–1631. doi: 10.1021/ac403326m. and references therein. [DOI] [PubMed] [Google Scholar]
- 57.Dai J, Punchihewa C, Ambrus A, Chen D, Jones RA, Yang D. Nucleic Acids Res. 2007;35:2440–2450. doi: 10.1093/nar/gkm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang D, Okamoto K. Future Med Chem. 2010;2:619–646. doi: 10.4155/fmc.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phan AT, Kuryavyi V, Luu KN, Patel DJ. Nucleic Acids Res. 2007;35:6517–6525. doi: 10.1093/nar/gkm706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dai J, Carver M, Punchihewa C, Jones RA, Yang D. Nucleic Acids Res. 2007;35:4927–4940. doi: 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai J, Carver M, Yang D. Biochimie. 2008;90:1172–1183. doi: 10.1016/j.biochi.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jung HA, Min BS, Yokozawa T, Lee JH, Kim YS, Choi JS. Biol Pharm Bull. 2009;32:1433–1438. doi: 10.1248/bpb.32.1433. [DOI] [PubMed] [Google Scholar]
- 63.Yokozawa T, Satoh A, Cho EJ, Kashiwada Y, Ikeshiro Y. J Pharm Pharmacol. 2005;57:367–374. doi: 10.1211/0022357055470. [DOI] [PubMed] [Google Scholar]
- 64.Kobayashi Y, Yamashita Y, Fujii N, Takaboshi K, Kawakami T, Kawamura M, Mizukami T, Nakano H. Planta Med. 1995;61:414–418. doi: 10.1055/s-2006-958127. [DOI] [PubMed] [Google Scholar]
- 65.For a review: Grycová L, Dostál J, Marek R. Phytochemistry. 2007;68:150–175. doi: 10.1016/j.phytochem.2006.10.004.
- 66.Zhang DH, Fujimoto T, Saxena S, Yu HQ, Miyoshi D, Sugimoto N. Biochemistry. 2010;49:4554–4563. doi: 10.1021/bi1002822. [DOI] [PubMed] [Google Scholar]
- 67.Xu Y, Suzuki Y, Ito K, Komiyama M. Proc Natl Acad Sci USA. 2010;107:14579–14584. doi: 10.1073/pnas.1001177107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wright WE, Tesmer VM, Huffman KE, Levene SD, Shay JW. Genes Dev. 1997;11:2801–2809. doi: 10.1101/gad.11.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu H, Gu X, Nakano S, Miyoshi D, Sugimoto N. J Am Chem Soc. 2012;134:20060–20069. doi: 10.1021/ja305384c. [DOI] [PubMed] [Google Scholar]
- 70.Heddi B, Phan AT. J Am Chem Soc. 2011;133:9824–9833. doi: 10.1021/ja200786q. [DOI] [PubMed] [Google Scholar]
- 71.Buscaglia R, Miller MC, Dean WL, Gray RD, Lane AN, Trent JO, Chaires JB. Nucleic Acids Res. 2013;41:7934–7946. doi: 10.1093/nar/gkt440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao C, Wu L, Ren J, Xu Y, Qu X. J Am Chem Soc. 2013;135:18786–18789. doi: 10.1021/ja410723r. [DOI] [PubMed] [Google Scholar]
- 73.Zhu LN, Wu B, Kong DM. Nucleic Acids Res. 2013;41:4324–4335. doi: 10.1093/nar/gkt103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cummaro A, Fottichia I, Franceschin M, Giancola C, Petraccone L. Biochimie. 2011;93:1392–1400. doi: 10.1016/j.biochi.2011.04.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.