

Abstract
Biocompatibility is a major concern for developing biomaterials used in medical devices, tissue engineering and drug delivery. Poly(lactic-co-glycolic acid) (PLGA) is one of the most widely used biodegradable materials, yet still triggers a significant foreign body response that impairs healing. Immune cells including macrophages respond to the implanted biomaterial and mediate the host response, which can eventually lead to device failure. Previously in our laboratory, we found that CD200, an immunomodulatory protein, suppressed macrophage inflammatory activation in vitro and reduced local immune cell infiltration around a biomaterial implant. While in our initial study we used polystyrene as a model material, here we investigate the effect of CD200 on PLGA, a commonly used biomaterial with many potential clinical applications. We fabricated PLGA with varied geometries, modified their surfaces with CD200, and examined macrophage cytokine secretion and phagocytosis. We found that CD200 suppressed secretion of the pro-inflammatory cytokine TNF-α and enhanced secretion of the anti-inflammatory cytokine IL-10, suggesting a role for CD200 in promoting wound healing and tissue remodeling. In addition, we found that CD200 increased phagocytosis in both murine macrophages and human monocytes. Together, these data suggest that modification with CD200 leads to a response that simultaneously prevents inflammation and enhances phagocytosis. This immunomodulatory feature may be used as a strategy to mitigate inflammation or deliver drugs or anti-inflammatory agents targeting macrophages.
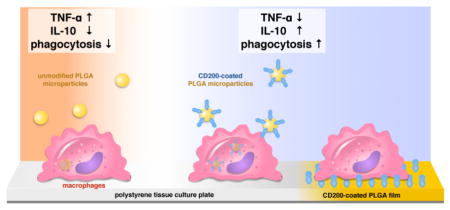
Graphical abstract
CD200 modified PLGA surfaces inhibits inflammatory cytokine (TNF-a) secretion, and enhances anti-inflammatory cytokine secretion (IL-10) and phagocytosis by macrophages.
1. Introduction
The ability to tune the immune response to biomaterials is necessary for many therapeutic applications including vaccines, drug delivery, and tissue engineering. Given the natural host response to foreign materials, vaccine formulations that contain biomaterials have been effective for enhancing the immune response to antigen delivery.1, 2 However, for biomaterial-based drug delivery strategies, activation of the innate immune system by biomaterials can potentially impede the efficacy of the drug, particularly of anti-inflammatory agents.3, 4 Furthermore, in tissue engineering or medical devices, it is desirable to inhibit prolonged inflammatory immune activation in order to enhance tissue regeneration and wound healing around the implanted material.5, 6 Achieving this goal has remained challenging since most materials lead to an inflammatory response when implanted or injected into the body. Therefore, improved strategies to limit inflammation and promote wound healing in response to biomaterials is still a major clinical need.
Among the many different biomaterials, poly(lactic-co-glycolic acid) (PLGA) remains one of the most widely used degradable polymeric materials, likely because of its tunable degradation and non-toxic degradation products.7 PLGA is found in multiple FDA-approved biomedical devices including degradable sutures, screws, and drug delivery carriers, and is also being explored for use in advanced applications including cardiovascular devices and tissue engineering scaffolds.8–12 PLGA micro- and nanoparticles have been explored for delivery of therapeutic agents, and have different potential applications depending on their size. Microscale PLGA particles have been used for site-specific drug delivery including delivering chemotherapeutic agents13,14, or encapsulating anti-inflammatory agent for implantable glucose sensor15, whereas nanoscale of particles are more suitable for system delivery of genes16,17 or vaccines.18 Although considered biocompatible, implantation of PLGA in fact elicits a significant foreign body response, as several studies have shown the formation of dense fibrotic capsule and foreign body giant cells surrounding subcutaneously implanted PLGA materials. 19–21 While this response is more moderate than the response to materials such as polystyrene, it still leads to the failure of many implanted devices since the fibrous capsule isolates the implant and prevents it from interacting with the surrounding tissue. In addition, the use of PLGA for delivery of drugs has been limited by its innate effect on the immune system. For cardiovascular devices, embedding anti-inflammatory agents within the PLGA material has been used as a strategy to inhibit the inflammatory response.20, 22–24 However, anti-inflammatory agents are also known to inhibit endothelialization needed to prevent thrombosis.25, 26 Thus, more specific and strategic methods to modulate the immune response to PLGA without impeding wound healing are necessary.
Our laboratory has previously developed an immunomodulatory strategy based on CD200, a biomolecule that interacts with CD200R expressed on myeloid cells and modulates their activation.27 We generated a recombinant CD200 and tethered it to a polystyrene surface, and examined the effect of CD200-modified surfaces on macrophages, an innate immune cell thought to be important in modulating the inflammatory versus wound healing response to implants. We found that CD200 inhibited macrophage inflammatory activation in vitro, and reduced local immune response towards implanted polystyrene beads in a mouse subcutaneous implant model.28 However, it was unclear whether CD200 only inhibits macrophage inflammatory (also known as M1) activation, or if it in fact could promote an anti-inflammatory (also known as M2) macrophage behavior. Furthermore, it remained unknown whether CD200 could modulate the immune response to a clinically relevant biomaterial.
In this study, we examined the effect of CD200 on macrophage behavior in response to PLGA. We fabricated CD200-modified PLGA materials in two different geometries – microparticles and films, which can potentially be used in local drug delivery or tissue engineering applications. Activation and polarization of macrophages were examined by culturing mouse bone marrow-derived macrophages (BMDM) with unmodified and modified PLGA. In addition, we evaluated the effect of CD200 on the phagocytosis of PLGA particles by phagocytes. Our studies show that CD200 inhibits macrophage secretion of the inflammatory cytokine TNF-α, while enhancing secretion of the anti-inflammatory cytokine IL-10. Interestingly, CD200 coating of PLGA particles enhanced their phagocytosis by both mouse macrophages and human monocytes. Moreover, human monocytes and macrophages cultured on CD200-coated surfaces exhibited enhanced phagocytosis of opsonized red blood cells. Together, these data suggest that modification of PLGA materials with CD200 leads to an anti-inflammatory, “eat me” signal.
2. Experimental
2.1 Fabrication of PLGA microparticles and films
Poly(lactic-co-glycolic acid) (PLGA) microparticles were prepared by a water-in-oil single emulsion-solvent evaporation method. PLGA solution was prepared by dissolving 30 mg PLGA (lactide:glycolide=50:50, acid terminated, MW = 38,000–54,000, Sigma-Aldrich) in 3 mL dimethyl carbonate (DMC) (Sigma-Aldrich). Fluorescein (Fisher Scientific) was added to the PLGA solution for microparticles used in the phagocytosis experiment. A single emulsion was prepared by homogenizing the PLGA solution with 1% w/v polyvinyl alcohol (PVA) (Sigma-Aldrich) for 5 min., followed by an extra 30 min. of stirring for solvent evaporation. Resulting PLGA microparticles were washed with phosphate-buffered saline (PBS) (Lonza) and collected using cell strainers with 1, 6, 10, 20, 30, and 40 μm pore sizes (pluriSelect). Microparticle size was determined by microscopic images. The histogram of size distribution was generated using RStudio software. PLGA films were prepared by a solvent evaporation technique. 5% PLGA solution in DMC was dispensed on a 12 mm cover glass (VWR) and the solvent was allowed to evaporate in a desiccator overnight. For scanning electron microscopy (SEM) images, microparticles were transferred to a glass slide and dried overnight in the desiccator. Subsequently, dried samples were sputter-coated with iridium (South Bay Technology Ion Beam Sputtering System) prior to imaging by SEM (FEI Quanta 3D FED Dual Beam). All images were taken at 15kV accelerating voltage and high vacuum.
2.2 Conjugation of CD200 to PLGA microparticles and films
The extracellular domain of CD200 was produced and further biotinylated (herein referred to as CD200) as previously reported.28,29 To prepare streptavidin-conjugated PLGA surfaces, acid-terminated PLGA microparticles or films were placed in 100 mM MES, pH5.5. 100 mg of 1-ethyl-aminopropyl-carbodiimide (EDC) (Thermo Scientific) was then added to react with carboxyl groups for 5 min before 100 mg of N-hydroxysuccinimide (NHS) (Thermo Scientific) was added. The particles or films were incubated with EDC/NHS for 25 min at room temperature. Residual EDC and NHS were washed three times using the reaction buffer, each followed by centrifugation. To conjugate streptavidin or phycoerythrin (PE)-conjugated streptavidin (BioLegend) onto PLGA surfaces, NHS-modified PLGA microparticles and films were incubated in 0.5 mg/mL streptavidin (in PBS) for 2 h. Unreacted streptavidin was removed by washing with PBS. Finally, CD200 was incubated with the streptavidin-modified PLGA in PBS for 2 h. CD200-coated PLGA materials were washed three times prior to use in experiments.
PLGA microparticles coated with or without CD200 were incubated with 20 μg/mL PE monoclonal anti-mouse CD200 antibody (BioLegend) for 2 h on a rotator, and then washed 3 times with PBS. PE signal was detected using an Olympus inverted microscope. To quantify the amount of CD200 conjugated on the microparticles, a Quantibrite™ PE kit (BD Biosciences) was utilized, which contains a set of beads conjugated with four different levels of PE molecules/bead to generate a standard curve. The amount of PE anti-CD200 antibody conjugated on the surfaces was determined using the standard curve of known levels of PE molecules per bead, and the amount of CD200 per particle was calculated assuming a ratio of one antibody bound to one CD200 molecule.
2.3 Cell isolation and culture
All protocols involving animals were approved by the Institutional Animal Care and Use Committee at the University of California Irvine prior to initiation of the study. Bone marrow-derived macrophages (BMDM) were harvested from the femurs of 6–8 week-old C57BL/6 mice (Jackson Laboratories). To isolate and differentiate BMDM, cells were treated with ACK lysis buffer (Invitrogen), centrifuged, and resuspended in D-10 media, which consists of Dulbecco’s modified Eagle medium (DMEM) supplemented with 2mM L-glutamine, 100 units /mL penicillin-streptomycin, 10% heat-inactivated FBS, and 10% media collected from the supernatant of CMG 12–14 cells ectopically expressing recombinant mouse macrophage colony stimulating factor (M-CSF).30 BMDM were cultured at 37°C in a humidified incubator with 5% CO2. BMDM were dissociated using cell dissociation buffer (Invitrogen) on day 7, and seeded on the tissue culture plates (Olympus Plastics) in fresh D-10 culture media.
All blood samples were collected into CPDA1 at the UCI Institute for Clinical and Translational Science in accordance with guidelines and approval of the University of California, Irvine (UCI) Institutional Review Board. Human peripheral blood lymphocytes and monocytes were isolated from peripheral blood mononuclear cells (PBMCs) by countercurrent elutriation. All experimental samples were 90 – 98% CD11b + in the monocyte fraction by flow cytometry. Human monocyte derived macrophages were generated from monocytes by culture at 5 × 105 cells/ml for 7 days in RPMI1640 (Invitrogen), 10% FBS (HyClone), 2 mM L-Glutamine (Invitrogen) and 1% penicillin/streptomycin (complete media) containing 25 ng/ml recombinant human M-CSF (rhM-CSF) (PeproTech) with addition of fresh media + rhM-CSF at day 3. The adherent human monocyte derived macrophages were harvested at day 7–8 by washing twice with 1xPBS and incubating with non-enzymatic CellStripper (CellGro) for 20 – 30 min. After washing twice with 1x PBS, viable cells were counted using 0.4% Trypan blue solution, a hemocytometer chamber and an optic microscope.
2.4 Viability assay
BMDM were seeded on tissue culture plates at 100,000 cells/cm2. At 6 h after cell seeding, the culture media was replaced with D-10 media containing different combinations of the following stimuli: 0.3 ng/mL lipopolysaccharide (LPS), 1.0 ng/mL interferon-gamma (IFN-γ), 20 ng/mL interleukin-4 (IL-4), and 20 ng/mL interleukin-13 (IL-13). PLGA microparticles with different surface modifications were introduced to the cells together with culture media containing different stimuli. After 36 h of incubation, the viability of BMDM was analyzed by MTT cell proliferation assay kit (ThermoFisher) following the manufacturer’s protocol and reading absorbance at 570 nm.
2.5 Cytokine secretion
BMDM were seeded on the tissue culture plates or on PLGA films with different surface modifications at a cell density of 100,000 cells/cm2. At 6 h after cell seeding, the culture media was replaced with D-10 media containing different combinations of the following stimuli as desribed in section 2.5. For particle experiments, the PLGA microparticles with different surface modifications were introduced to the cells at 8% projective surface coverage together with culture media of different stimuli. After 36 h of incubation, the supernatants were collected and analyzed for TNF-α and IL-10 secretion by enzyme-linked immunosorbent assay (ELISA) following the manufacturer’s instructions (BioLegend).
2.6 Phagocytosis of microparticles
BMDM or human monocytes were seeded in 8-well Lab-Tek Chamber slides (Thermo Scientific) at 62,500 cells/well in D-10 media or RPMI, respectively. PLGA microparticles were seeded at a ratio of 10 particles to 1 cell. After 12 h incubation, the cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 5 min, and washed carefully with PBS. To permeabilize cells, samples were treated with 0.1% Triton X-100 (Sigma-Aldrich) (in PBS) for 5 min. BMDM were then stained with Alexa Fluor-594 conjugated phalloidin (Invitrogen) and Hoechst 33342 dye (Invitrogen) for 1 h, followed by thorough washes with PBS. Human monocytes were stained with Vybrant DiI cell labeling solution (Thermo Scientific) and Hoechst. Finally, the chamber slide was mounted using Fluoromount-G (Southern Biotech). Fluorescence images were obtained using an Olympus inverted microscope. For flow cytometric analysis, cells with phagocytosed fluorescein-containing PLGA microparticles were collected after 12 h (for BMDM) or 3 h (for human monocytes) of particle incubation. The un-phagocytosed particles were washed out with PBS. The cells were then collected using a cell scraper and suspended in PBS. Phagocytosed cell acquisition and analysis were performed using a BD LSR II flow cytometer and FlowJo software.
2.7 Phagocytosis of opsonized erythrocytes
Sheep erythrocytes (Colorado Serum Co., Denver, CO) were sub-optimally opsonized using rabbit anti-sheep erythrocyte immunoglobulin (EA) and the phagocytosis assays were performed as described.31, 32 Briefly, 8-well LabTek chambers (Nunc) were UV ozone treated for 15 min and then silanized with a 4% (3-mercaptopropyl)trimethoxysilane solution (Sigma Aldrich) at RT for 1 h. Meanwhile, streptavidin was crosslinked using sulfo-SMCC (Life Technologies) at RT for 30 min, and unreacted sulfo-SMCC was removed using a 7 kDa Zeba desalting column (Thermo Scientific). Silanized chambers were washed thoroughly with 100% ethanol and dried with a N2 stream. The maleimide-activated streptavidin was diluted to 0.1 mg/mL and incubated on the silanized chambers at RT for 1 h. Streptavidin-modified chambers were then coated with different concentrations of CD200-biotin in PBS and incubated at room temperature for 2 h. After washing chambers twice with PBS, 250 μl of monocytes or human monocyte derived macrophages (0.25 × 106 cells/ml) suspended in phagocytosis buffer (RPMI with 2 mM of glutamine, Pen/Strep and 5 mM of MgCl2), were added to chambers. After spinning down at 700 rpm for 3 min, chambers were placed at 37°C in 5% CO2 air for 30 min. Target particles (EA) were then added (100ul/ well, 1 × 108 EA/ml) and spun down at 700 rpm for 3 min and incubated for 30 min at 37°C in 5% CO2. After removing erythrocytes that were not cell-associated by washing, noningested erythrocytes were further removed by hypotonic lysis. Slides were then fixed in 1% glutaraldehyde (Sigma-Aldrich), stained with Giemsa, and scored by microscopic examination. A minimum of 200 cells per experimental condition in duplicate were quantified by individuals blinded to the experimental design. Percent phagocytosis was defined as the number of monocytes/macrophages ingesting at least one target divided by the total number of monocytes/macrophages counted, multiplied by 100. The phagocytic index was defined as the number of ingested targets per 100 cells counted.
2.8 Statistical Analysis
Statistical analysis was performed using ordinary two-way ANOVA, followed by Tukey’s post hoc test, and p < 0.05 was considered statistically significant.
3. Results and discussion
3.1 Fabrication and characterization of PLGA microparticles and films
PLGA microparticles were generated by single emulsion followed by solvent evaporation. Dimethyl carbonate (DMC) was used as a solvent because of the large range of microparticle sizes produced (Figure S1). To separate particles of different sizes, we size-selected using cell strainers with 1, 6, 10, 20, 30, and 40 μm pore sizes. After filtration, the size distribution was narrowed, as illustrated by scanning electron microscopy imaging (Figure 1A). Quantification of microparticle diameters revealed that particles that passed through the 6 μm cell strainer were 2 μm to 13 μm in diameter, with a median diameter of 7 μm. The particles collected between the 10 μm and 20 μm strainers had a median diameter of 21 μm, and between the 20 μm and 30 μm strainers had a median size of 28 μm. Microparticles collected between the 30 μm and 40 μm strainers exhibited some imperfections and depressions on the particle surface, with a median particle diameter of 35 μm (Figure 1B). Many studies have shown the importance of particle size on immune cell activity.33, 34 In this study, we investigated two particle sizes, 7 μm particles that can potentially be phagocytosed, and 28 μm particles that will not be phagocytosed by immune cells.35, 36 In addition, we fabricated films by dispensing PLGA on a glass slide and allowing the solvent to evaporate in a desiccator overnight. Examination of the surface by SEM showed that the PLGA film had a relatively smooth surface (Figure 1A right).
Figure 1.

characterization of plga microparticles and films. (a) representative scanning electron micrographs of size-selected plga microparticles and film. (b) histograms depicting size distributions of plga microparticles after filtration using 1–6 μm, 10–20 μm, 20–30 μm, and 30–40 μm strainers.
3.2 Modification of PLGA microparticles and films with CD200
Surfaces of PLGA particles and films were modified with CD200 using a biotin-streptavidin binding interaction. Acid-terminated PLGA microparticles or films were reacted with N-hydroxysuccinimide (NHS) via a carbodiimide crosslinker, and then streptavidin was further immobilized on the NHS-modified PLGA surfaces. Using PE-conjugated streptavidin, we observed higher levels after conjugation through amide bond when compared to non-specific adsorption to PLGA particle surfaces (Figure S3). Biotinylated CD200 was then bound to streptavidin-modified PLGA through biotin-streptavidin interaction. The presence of CD200 on PLGA surfaces was confirmed using PE-conjugated anti-CD200 antibody, which detected CD200 on the modified PLGA microparticles and films but not on unmodified surfaces (Figure 2A). Conjugation of CD200 appeared to be highly heterogeneous and large particle-to-particle variations were observed. We quantified the amount of CD200 on 7 μm particles by flow cytometry, which confirmed the heterogeneous levels of CD200 bound to each particle that were observed by microscopy (Figure 2B). Using QuantiBRITE PE beads to calibrate the PE signal, we estimated approximately 1750 CD200 molecules on 2–6 μm-sized particle and 7700 molecules on 7–13 μm-sized particle, with the average density of 30 molecules/μm2. On PLGA films, we conjugated CD200 to surfaces at varying coating concentrations, and evaluated CD200 binding on the surface. Measuring the fluorescent intensity of PE-conjugated anti-CD200 antibodies indicates that the amount of bound CD200 is proportional to the coating concentration, reaching the highest levels at 50 μg/mL (Figure 2C).
Figure 2.

modification of plga microparticles and surfaces with cd200. (a) representative bright field (left) and fluorescent (right) images of different sized unmodified plga or cd200-plga microparticles and films. (b) representative flow cytometry histogram of unmodified plga and cd200-plga 7 μm microparticles (left) and quantification of cd200 (right). data represents mean ± sem of 10,000 particles. (c) mean fluorescence intensity of cd200 bound on plga films with different coating concentration.
3.3 Cell viability in response to incubation with cytokines and microparticles
Viability of BMDM upon stimulation with different cytokines was measured by MTT assay. BMDM were less viable with stimulation of LPS/IFNγ or LPS/IL-4/IL-13 when compared to without stimulation (Figure 3A). When incubated with PLGA microparticles, increasing the amount of PLGA microparticles moderately decreased cell viability (Figure 3B). Different surface modifications of PLGA microparticles did not show any effect on cell viability within same cytokine stimulation condition (Figure 3C). Interestingly, addition of CD200-modified 28μm PLGA particles to LPS/IFN-γ treated cells somewhat enhanced their viability (Figure 3D), suggesting that ligation of CD200 protects macrophages from the toxic effects of LPS/IFN-γ.
Figure 3.

viability of bmdm with cytokine and microparticle treatment. (a) viability of bmdm as measured by mtt assay upon stimulation with 0.3 ng/ml lps and 1 ng/ ml ifnγ, 0.3ng/ml lps, 20 ng/ml il-4 and 20 ng/ml il-13, or left untreated. (b) viability of bmdm treated with different amounts (% surface coverage) of plga or cd200-plga microparticles. viability of bmdm incubated with 7μm (c) or 28μm (d) plga microparticles with different surface modifications and stimulated with cytokines at the same concentrations described in (a). data represents mean ± sem of n=3. * denotes p<0.05.
3.4 CD200 inhibits pro-inflammatory and promotes anti-inflammatory activation of macrophages
To assess the effect of CD200 on macrophage activation in response to PLGA particles, we seeded bone marrow-derived macrophages (BMDM) on tissue-culture polystyrene and added unmodified PLGA, streptavidin-PLGA, or CD200-PLGA microparticles to the culture media. Different sized particles were administered at a constant particle projection area in order to minimize the effects of varied surface area on activation of macrophages. We found that without additional cytokine stimulation, all particles had minimal effect on macrophage response and TNF-α and IL-10 secretion levels were undetectable. In the presence of LPS/IFNγ stimulation, macrophages cultured with unmodified PLGA particles secreted higher levels of TNF-α, an inflammatory cytokine, when compared to macrophages without particles (Figure 4A(i) and B(i)). In addition, increasing the amount of particles concomitantly increased TNF-α secretion, suggesting an inflammatory response to the PLGA particles themselves (Figure S2). Modification of PLGA particles with CD200 reduced macrophage TNF-α secretion by approximately fifty percent, whereas coating with streptavidin alone as a control had no effect. Similar results were obtained for both 7 μm and 28 μm microparticles. When stimulated with LPS, IL-4, and IL-13, CD200 coating of 28 μm particles enhanced secretion of IL-10, an anti-inflammatory cytokine, by more than 2-fold, when compared to control microparticles (Figure 4B(ii)). For 7 μm PLGA microparticles, CD200 did not significantly increase IL-10 secretion when compared to unmodified PLGA particles (Figure 4A(ii)).
Figure 4.

cd200 modulates macrophage cytokine secretion in response to plga particles and films. raw levels (left) and levels normalized to unmodified plga (right) of tnf-α (i) and il-10 (ii) secretion by bmdm cultured with 7 μm (a), 28 μm (b) microparticles, or on films (c). for analysis of tnf-α secretion, cells were cultured with 0.3 ng/ml lps and 1 ng/ ml ifnγ. for analysis of il-10 secretion, cells were cultured with 0.3ng/ml lps, 20 ng/ml il-4 and 20 ng/ml il-13. distinct symbols (●,■,▲) represent a separate biological replicate (independent experiment, each performed in duplicate). data represents mean ± sem of n=3. * denotes p<0.05.
On PLGA films, we observed a similar reduction of TNF-α and enhancement of IL-10 secretion. Compared to macrophages seeded on polystyrene, macrophages seeded on unmodified PLGA films secreted approximately two-fold more TNF-α (Figure 4C(i)). Modification of PLGA surfaces with CD200 led to a decrease in TNF-α secretion to baseline levels and increase in IL-10 (Figure 4C(ii)). Interestingly, comparing the effect of CD200 coated on polystyrene, glass, and PLGA had differential results. While CD200 inhibited TNF-α on polystyrene by approximately two-fold similar to that seen on PLGA, no effect was observed on glass (Figure S4A). In addition, CD200 generally inhibited the secretion of IL-10 on polystyrene and glass, in contrast to increased IL-10 when on PLGA (Figure S4B, Figure 4Cii). These data suggest that the biomaterial on which immunomodulatory molecules are presented may play an important role in their effect.
Together, these data suggest that CD200 inhibits pro-inflammatory cytokine secretion in response to both PLGA microparticles and films, consistent with what we have previously observed with CD200-coated polystyrene particles and polystyrene cell culture surfaces.28 In addition, we found that CD200 promotes IL-10 cytokine secretion by macrophages in response to PLGA particles and films, suggesting that this immunomodulatory protein may also enhance alternative activation or pro-healing macrophage function. Interestingly, upregulation of IL-10 was dependent on the biomaterial surface, as CD200-coated polystyrene or glass surfaces did not elicit this effect. These differential effects may be due to different coating densities achieved on each material. Further studies will be needed to elucidate the varied response observed in different material contexts. Nevertheless, our data suggest that CD200-coated PLGA directs macrophages towards an anti-inflammatory phenotype, and can potentially be used for PLGA-based devices to prevent acute inflammation and provide an anti-inflammatory signal that promotes tissue remodeling.
3.5 CD200 enhances phagocytosis of PLGA microparticles
Macrophages are involved not only in secretion of cytokines that help orchestrate inflammatory and wound healing processes, but they are also phagocytes responsible for engulfment of pathogens such as bacteria, foreign microparticles, as well as cellular debris and apoptotic cells during tissue remodeling.37, 38 Therefore, we examined the potential role of CD200 coating on the phagocytosis of PLGA microparticles. In order to detect phagocytosis of particles by microscopy or flow cytometry, 7 μm size fluorescein-containing PLGA microparticles were prepared, modified with CD200, and then introduced to BMDM. After 12 hours, cells were fixed and stained with phalloidin and Hoechst to visualize actin and nuclei respectively by microscopy, which showed particles internalized within cells (Figure 5A and S5). To quantify phagocytic BMDM, cells were collected for measurement of fluorescence intensity by flow cytometry (Figure 5B). We found that coating PLGA microparticles with CD200 led to a significantly higher percentage of phagocytic BMDM when compared to unmodified PLGA microparticles. While the baseline phagocytosis levels were somewhat varied (Figure 5C left), likely because of natural fluctuations in primary cells isolated from individual mice, the % of cells that exhibited positive phagocytosis was approximately 1.7 times higher for cells exposed to CD200-coated particles, when compared to unmodified or streptavidin-coated particles (Figure 5C right). Quantification of the median fluorescein intensity showed that BMDM treated with CD200-PLGA had nearly 3-fold higher fluorescence intensity compared to cells treated with unmodified or streptavidin-coated particles (Figure 5D). Median fluorescein intensity of the phagocytosis-positive BMDM showed no significant differences between PLGA microparticles with different surface modifications, suggesting that the number of particles taken up by phagocytic cells were likely to be similar across the different conditions (Figure 5E).
Figure 5.

phagocytosis of cd200-coated plga microparticles by bmdm. (a) representative fluorescence images of bmdm incubated with unmodified, streptavidin-, and cd200-plga microparticles (green), stained with phalloidin (red), hoechst (blue). scale bar is 20 μm. (b) representative flow cytometry histogram showing fluorescein intensity of bmdm incubated with different plga microparticles. (c) graph of % phagocytosis of particles by bmdm (left). each distinct symbol (●,■,▲) represents a separate biological replicate (independent experiment, each performed in duplicate). graph of phagocytosis of different particles, relative to unmodified plga particles (right). (d) graph of median fluorescein intensity of bmdm treated with different particles, relative to unmodified plga. (e) graph of median fluorescein intensity of phagocytosis+ bmdm in different particle conditions, relative to unmodified plga. data represents mean ± sem. n=3. *p<0.05.
To explore whether enhanced phagocytosis of microparticles by CD200 translates to effects on human cells, we further examined the effects of different PLGA microparticles on primary human peripheral blood monocytes. Similar to what was observed with BMDM, visualization of cells using DiI and Hoechst showed particles taken up within cells (Figure 6A). Analysis by flow cytometry revealed an overall lower percentage of phagocytosis by human monocytes compared to that of BMDM, likely because they were not differentiated to macrophages. CD200 significantly enhanced the phagocytosis of PLGA microparticles. More than 15% of human monocytes were observed to take up CD200-PLGA microparticles, whereas only 8% of cells phagocytosed control PLGA microparticles (Figure 6B and 5C). Overall, human monocytes phagocytosed 2-fold more CD200-coated PLGA microparticles when compared to other microparticles, as shown by the relative phagocytosis and median fluorescein intensity (Figure 6C and 5D). Similar to what was observed for BMDM, individual phagocytic cells did not uptake more CD200-PLGA microparticles than other microparticles, as demonstrated by similar median intensities of phagocytosis-positive cells (Figure 6E).
Figure 6.

phagocytosis of cd200-coated plga microparticles by human monocytes. (a) representative fluorescence images of human monocytes incubated with unmodified, streptavidin-, and cd200-plga microparticles (green), stained with dii (red), and hoechst (blue). scale bar is 20 μm. (b) representative flow cytometry histogram showing fluorescein intensity of human monocytes incubated with different plga microparticles. (c) graph of % phagocytosis of particles by human monocytes. (left). each distinct symbol (●,■,▲) represents a separate biological replicate (independent experiment, each performed in duplicate). graph of monocyte phagocytosis of different particles, relative to unmodified plga condition (right). (d) graph of median fluorescein intensity of human monocytes treated with different particles, relative to unmodified plga. (e) graph of median fluorescein intensity of phagocytosis+ human monocytes treated with different particle conditions, relative to unmodified plga. data represents mean ± sem. n=3. *p<0.05.
Our data demonstrate that CD200 increases the phagocytosis of PLGA particles, and may provide a strategy to enhance delivery of drugs or therapeutics targeting macrophages, while simultaneously preventing inflammation. CD200 has been shown to be highly expressed on apoptotic cell bodies,39 and we hypothesize that CD200-coated PLGA microparticles and apoptotic cell bodies may be taken up by macrophages using a similar mechanisms, during which inflammatory activity is inhibited. Interestingly, CD200 has a remarkably different effect compared to the “don’t eat me” self-molecule, CD47, which has been reported to inhibit phagocytic clearance and enhance nanoparticle delivery to tumor sites.40 These studies suggest that despite their broad classification as tolerizing molecules, CD200 and CD47 have unique roles that may be differentially leveraged for distinct immunomodulatory applications.
3.6 CD200 enhances phagocytosis of opsonized targets by human monocytes and macrophages
We further examined the influence of CD200 on the human phagocyte uptake of opsonized targets. Herein, primary human peripheral blood monocytes or monocyte-derived macrophages were seeded on surfaces coated with different concentrations of CD200, followed by the introduction of opsonized sheep erythrocytes. We found that human monocytes and macrophages cultured on CD200-coated surfaces were significantly more phagocytic than cells on unmodified surfaces, as demonstrated by the increased percentage of cells engulfing erythrocytes (Figure 7A(i) and B(i)). The effect of CD200 on human monocyte phagocytosis was concentration-dependent, with increasing phagocytic activity observed in response to higher CD200 coating concentration. In addition, the phagocytic index or number of targets ingested per monocyte, was also enhanced by CD200 coating (Figure 7A(ii)). Although CD200 enhanced the phagocytic capacity of individual human monocyte, it only had a moderate effect on human macrophages (Figure 7B(ii)), perhaps because the phagocytic activity of these cells is already high. Nevertheless, our data show not only that CD200 enhances the phagocytosis of particles, but also that adhesion of monocyte/macrophages to CD200 surfaces enhances their phagocytosis of opsonized targets. This effect has also been observed with surfaces modified with C1q, a complement protein41 that enhances phagocytosis of pathogens, cellular debris, and apoptotic cells 31, 42 and promotes anti-inflammatory cytokine secretion.43
4. Conclusion
In this study, we demonstrate the effect of the immunomodulatory protein, CD200, on the macrophage response to PLGA. We investigated particles and films, geometries associated with different potential clinical applications including drug delivery and implanted devices, respectively. We found that regardless of geometry, CD200 reduced the secretion of the pro-inflammatory cytokine TNF-α For large particles or surfaces, CD200 enhanced the secretion of the anti-inflammatory cytokine IL-10. Interestingly, although TNF-α was consistently downregulated in the presence of CD200, the increase in IL-10 secretion appeared to be somewhat dependent on the geometry, and the effect was only moderate with smaller particles. It is possible that this observation is limited to the time point of evaluation (24 h of culture) or the stimulation conditions used (LPS, IL-4, and IL-13), and further studies exploring additional time points or cytokines of interest would help provide a deeper understanding of the effect of CD200 on macrophage activation.
In addition to modulation of cytokine secretion, CD200 enhanced monocyte and macrophage phagocytosis of PLGA particles and opsonized targets. When particles were coated with CD200, a higher percentage of BMDM engulfed at least one particle, although the phagocytic capacity of individual cells was similar. The same trend was observed in human monocytes incubated with CD200-PLGA microparticles. Moreover, human monocyte and macrophage phagocytosis of opsonized erythrocytes was also enhanced in the presence of CD200. Together, these data suggest that CD200 promotes phagocytosis without triggering pro-inflammatory activation.
The immunomodulatory activity of CD200, specifically eliciting a decrease in TNF-α and increase in IL-10, suggests that it may be used to mitigate inflammation and promote wound healing in response to implanted biomaterials. In addition, the “eat me” signal may potentially be leveraged as a strategy to improve delivery of therapeutics into macrophages. While biomaterials have been tremendously beneficial in the controlled delivery of drugs or genes, delivery of anti-inflammatory agents has been challenging given the inflammatory response to the biomaterials themselves. Our findings suggest that CD200 may be used enhance delivery of anti-inflammatory agents to treat diseases including atherosclerosis or arthritis, where the phagocytic activity of macrophages to clear cellular debris may be desirable, but inhibition of an inflammatory response and/or preventing the induction of an adaptive response to self proteins is definitely desirable. Exploring the many applications where CD200 may beneficially modulate the host response to biomaterials will be an important area of future work.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH) National institute of Dental and Craniofacial Research (NIDCR) and Office of Director’s Grant DP2DE023319 (to W.F.L), National Institute on Aging (NIA) Grant AG00538 (to A.J.T.), National Institute of Allergy and Infectious Diseases (NIAID) Grant AI120846 (to M.B.L.). We gratefully acknowledge the Laboratory for Electron and X-ray Instrumentation (LEXI), the Stem Cell Research Center, and the Institute for Immunology Flow Cytometry Facilities at the University of California, Irvine. We thank the staff of the University of California, Irvine, Institute for Clinical and Translational Science for obtaining blood and healthy volunteers for donating blood. We thank Xiuhui Gao and Ci Ren for technical assistance in the laboratory.
References
- 1.Acharya AP, Clare-Salzler MJ, Keselowsky BG. Biomaterials. 2009;30:4168–4177. doi: 10.1016/j.biomaterials.2009.04.032. [DOI] [PubMed] [Google Scholar]
- 2.Saroja C, Lakshmi P, Bhaskaran S. Int J Pharm Investig. 2011;1:64–74. doi: 10.4103/2230-973X.82384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naahidi S, Jafari M, Edalat F, Raymond K, Khademhosseini A, Chen P. J Control Release. 2013;166:182–194. doi: 10.1016/j.jconrel.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 4.Kohane DS, Langer R. Chem Sci. 2010;1:441–446. [Google Scholar]
- 5.Vishwakarma A, Bhise NS, Evangelista MB, Rouwkema J, Dokmeci MR, Ghaemmaghami AM, Vrana NE, Khademhosseini A. Trends Biotechnol. 2016;34:470–482. doi: 10.1016/j.tibtech.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Chan G, Mooney DJ. Trends Biotechnol. 2008;26:382–392. doi: 10.1016/j.tibtech.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. Journal of Controlled Release. 2012;161:505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 8.Formiga FR, Pelacho B, Garbayo E, Abizanda G, Gavira JJ, Simon-Yarza T, Mazo M, Tamayo E, Jauquicoa C, Ortiz-de-Solorzano C, Prosper F, Blanco-Prieto MJ. J Control Release. 2010;147:30–37. doi: 10.1016/j.jconrel.2010.07.097. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Venkatraman SS, Boey FY, Loo JS, Tan LP. Biomaterials. 2006;27:5588–5595. doi: 10.1016/j.biomaterials.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 10.Venkatraman SS, Tan LP, Joso JF, Boey YC, Wang X. Biomaterials. 2006;27:1573–1578. doi: 10.1016/j.biomaterials.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Westedt U, Wittmar M, Hellwig M, Hanefeld P, Greiner A, Schaper AK, Kissel T. J Control Release. 2006;111:235–246. doi: 10.1016/j.jconrel.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Karp JM, Shoichet MS, Davies JE. Journal of Biomedical Materials Research Part A. 2003;64A:388–396. doi: 10.1002/jbm.a.10420. [DOI] [PubMed] [Google Scholar]
- 13.Kim I, Byeon HJ, Kim TH, Lee ES, Oh KT, Shin BS, Lee KC, Youn YS. Biomaterials. 2012;33:5574–5583. doi: 10.1016/j.biomaterials.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 14.Sahoo SK, Panda AK, Labhasetwar V. Biomacromolecules. 2005;6:1132–1139. doi: 10.1021/bm0492632. [DOI] [PubMed] [Google Scholar]
- 15.Ju YM, Yu B, West L, Moussy Y, Moussy F. Journal of Biomedical Materials Research Part A. 2010;93:200–210. doi: 10.1002/jbm.a.32512. [DOI] [PubMed] [Google Scholar]
- 16.Bivas-Benita M, Romeijn S, Junginger HE, Borchard G. European Journal of Pharmaceutics and Biopharmaceutics. 2004;58:1–6. doi: 10.1016/j.ejpb.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soderquist RG, Sloane EM, Loram LC, Harrison JA, Dengler EC, Johnson SM, Amer LD, Young CS, Lewis MT, Poole S. Pharmaceutical research. 2010;27:841–854. doi: 10.1007/s11095-010-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, Torensma R, Figdor CG. Journal of Controlled Release. 2010;144:118–126. doi: 10.1016/j.jconrel.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Yoon SJ, Kim SH, Ha HJ, Ko YK, So JW, Kim MS, Yang YI, Khang G, Rhee JM, Lee HB. Tissue Eng Part A. 2008;14:539–547. doi: 10.1089/tea.2007.0129. [DOI] [PubMed] [Google Scholar]
- 20.Patil SD, Papadmitrakopoulos F, Burgess DJ. J Control Release. 2007;117:68–79. doi: 10.1016/j.jconrel.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 21.Hickey T, Kreutzer D, Burgess DJ, Moussy F. Journal of Biomedical Materials Research. 2002;61:180–187. doi: 10.1002/jbm.10016. [DOI] [PubMed] [Google Scholar]
- 22.Dang TT, Bratlie KM, Bogatyrev SR, Chen XY, Langer R, Anderson DG. Biomaterials. 2011;32:4464–4470. doi: 10.1016/j.biomaterials.2011.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhardwaj U, Sura R, Papadimitrakopoulos F, Burgess DJ. J Diabetes Sci Technol. 2007;1:8–17. doi: 10.1177/193229680700100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patil SD, Papadimitrakopoulos F, Burgess DJ. Diabetes Technol Ther. 2004;6:887–897. doi: 10.1089/dia.2004.6.887. [DOI] [PubMed] [Google Scholar]
- 25.Wolff JEA, Guerin C, Laterra J, Bressler J, Indurti RR, Brem H, Goldstein GW. Brain Research. 1993;604:79–85. doi: 10.1016/0006-8993(93)90354-p. [DOI] [PubMed] [Google Scholar]
- 26.Chen WL, Lin CT, Yao CC, Huang YH, Chou YB, Yin HS, Hu FR. Ocul Immunol Inflamm. 2006;14:215–223. doi: 10.1080/09273940600732380. [DOI] [PubMed] [Google Scholar]
- 27.Kim YK, Chen EY, Liu WF. J Mater Chem B. 2016;4:1600–1609. doi: 10.1039/c5tb01605c. [DOI] [PubMed] [Google Scholar]
- 28.Kim YK, Que R, Wang SW, Liu WF. Advanced Healthcare Materials. 2014;3:989–994. doi: 10.1002/adhm.201300532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kingston RE, Kaufman RJ, Bebbington CR, Rolfe MR. Current Protocols in Molecular Biology. 2001 doi: 10.1002/0471142727.mb1623s60. [DOI] [PubMed] [Google Scholar]
- 30.Morgado P, Ong YC, Boothroyd JC, Lodoen MB. Infection and Immunity. 2011;79:4401–4412. doi: 10.1128/IAI.05562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bobak DA, Gaither TA, Frank MM, Tenner AJ. The Journal of Immunology. 1987;138:1150–1156. [PubMed] [Google Scholar]
- 32.Nepomuceno RR, Ruiz S, Park M, Tenner AJ. The Journal of Immunology. 1999;162:3583–3589. [PubMed] [Google Scholar]
- 33.Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, Jhunjhunwala S, Chiu A, Siebert S, Tang K, Hollister-Lock J, Aresta-Dasilva S, Bochenek M, Mendoza-Elias J, Wang Y, Qi M, Lavin DM, Chen M, Dholakia N, Thakrar R, Lacik I, Weir GC, Oberholzer J, Greiner DL, Langer R, Anderson DG. Nat Mater. 2015;14:643–651. doi: 10.1038/nmat4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Takashi N, Akasaka T, Roska ID, Uo M, Totsuka Y, Watari F. Key Engineering Materials. 2004;254–256:919–922. [Google Scholar]
- 35.Tabata Y, Ikada Y. Biomaterials. 1988;9:356–362. doi: 10.1016/0142-9612(88)90033-6. [DOI] [PubMed] [Google Scholar]
- 36.Champion JA, Walker A, Mitragotri S. Pharmaceutical Research. 2008;25:1815–1821. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Journal of Clinical Investigation. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 39.Rosenblum MD, Olasz E, Woodliff JE, Johnson BD, Konkol MC, Gerber KA, Orentas RJ, Sandford G, Truitt RL. Blood. 2004;103:2691–2698. doi: 10.1182/blood-2003-09-3184. [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraser DA, Laust AK, Nelson EL, Tenner AJ. J Immunol. 2009;183:6175–6185. doi: 10.4049/jimmunol.0902232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Webster SD, Galvan MD, Ferran E, Garzon-Rodriguez W, Glabe CG, Tenner AJ. The Journal of Immunology. 2001;166:7496–7503. doi: 10.4049/jimmunol.166.12.7496. [DOI] [PubMed] [Google Scholar]
- 43.Fraser DA, Bohlson SS, Jasinskiene N, Rawal N, Palmarini G, Ruiz S, Rochford R, Tenner AJ. J Leukoc Biol. 2006;80:107–116. doi: 10.1189/jlb.1105683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.