

Abstract
Antimalarial drugs have thus far been chiefly derived from two sources—natural products and synthetic drug-like compounds. Here we investigate whether antimalarial agents with novel mechanisms of action could be discovered using a diverse collection of synthetic compounds that have three-dimensional features reminiscent of natural products and are underrepresented in typical screening collections. We report the identification of such compounds with both previously reported and undescribed mechanisms of action, including a series of bicyclic azetidines that inhibit a new antimalarial target, phenylalanyl-tRNA synthetase. These molecules are curative in mice at a single, low dose and show activity against all parasite life stages in multiple in vivo efficacy models. Our findings identify bicyclic azetidines with the potential to both cure and prevent transmission of the disease as well as protect at-risk populations with a single oral dose, highlighting the strength of diversity-oriented synthesis in revealing promising therapeutic targets.
Malaria is a deadly disease caused by protozoan parasites of the genus Plasmodium. Effective eradication strategies have been elusive, primarily owing to the complex life cycle of Plasmodium and the emergence of drug-resistant strains of P. falciparum, the most lethal Plasmodium species in humans1. The majority of the current antimalarial drugs target the asexual blood stage of Plasmodium, in which they parasitize and replicate within erythrocytes2. Even though liver-and transmission-stage parasites do not cause malarial symptoms, prophylaxis and transmission-blocking drugs are essential for the proactive prevention of disease epidemics and to protect vulnerable populations3,4. Unfortunately, the current antimalarial drugs do not address all of the requirements for the targeting of pan-life- cycle activity. Several recent reports have described next-generation drug candidates that may achieve some of these important goals2,5–9. However, eradication will require multiple innovative ways of targeting the parasite10–12. The antimalarial pipeline will therefore benefit from compounds with diverse mechanisms of action, features that should help circumvent the many resistance mechanisms that render existing drugs ineffective.
We identified two key features of a successful strategy for overcoming these challenges. The first of these is the application of modern methods of asymmetric organic synthesis to create unique chemical matter; the second is to test the resulting compounds in a series of phenotype-based screens designed to uncover agents that act on targets essential for several stages of the parasite life cycle (that is, multistage activity). We were encouraged by a small-scale pilot experiment that followed this blueprint and yielded the antimalarial agent ML238 (refs 13–15). The experiments described here excluded this earlier pilot set of compounds.
We tested synthetic compounds with structures that were inspired by the structural complexity and diversity of the entire ensemble of natural products, rather than by specific natural products. In this way, we deliberately break the link to natural selection and the limitations it provides in terms of target diversity16. A high-throughput P. falciparum phenotypic screen of infected erythrocytes was used to detect inhibitors of parasite growth, with counter-screens using parasites that are resistant to approved or developmental drugs, and with liver- and transmission-stage parasites used to facilitate the discovery of compounds that act through novel mechanisms of action and target multiple stages of malarial infection.
Approximately 100,000 compounds, synthesized at the Broad Institute using the build/couple/pair strategy17,18 of diversity-oriented synthesis (DOS), were screened against a multi-drug-resistant strain (P. falciparum strain Dd2) using a phenotypic blood-stage growth-inhibition assay, which models a human blood-stage infection. Compounds scored as positives were counter-screened in parallel against a panel of parasite isolates and diverse drug-resistant clones to deprioritize compounds with previously identified mechanisms of action (Fig. 1a and Supplementary Tables 1, 2). After evaluating results from assays against the liver-stage (Plasmodium berghei strain ANKA) and transmission-stage (P. falciparum strain 3D7) parasites, four chemical series with additional liver-stage and/or transmission-blocking activities (BRD0026, BRD7539, BRD73842 and BRD3444; Fig. 1b–e, Extended Data Table 1 and Supplementary Tables 1, 2) were selected. This layered screening process also yielded other series not described here that may merit attention in the future (available at the Malaria Therapeutics Response Portal, http://portals.broadinstitute.org/mtrp/). Underlying features of DOS helped to guide the selection and development of the four nominated series. The compound collection includes stereoisomeric families that yield stereochemistry-based structure–activity relationships (SSAR); their inclusion indicated the possibility of selective interactions with targets. The short, modular pathways, entailing inter- and intramolecular coupling reactions, facilitate medicinal chemistry optimization. Three of the four series yielded new compound scaffolds against known targets. These include: (i) disruptors of sodium ion regulation mediated by P. falciparum ATPase4 (ref. 9; BRD0026 is active against asexual and late sexual blood stages of parasites, Fig. 1b and Extended Data Fig. 1a – d); (ii) potent and selective inhibitors of P. falciparum dihydroorotate dehydrogenase (pf DHODH)19 (BRD7539 is active against liver-stage and asexual blood-stage parasites; Fig. 1c and Extended Data Fig. 1e – h); and (iii) potent and selective inhibitors of P. falciparum phosphatidylinositol-4-kinase (pf PI4K)20,21 (BRD73842 is active against liver-stage, asexual and late sexual blood-stage parasites; Fig. 1d, Extended Data Figs 1i–m, 2a and Supplementary Table 3). The fourth series was found to inhibit a previously unknown antimalarial target and is characterized in detail below.
Figure 1. Cascading triage strategy reveals targets for some of the hit compounds and highlights potential novel mechanisms of action for others.

a–e, A total of 468 compounds (‘positives’ in the growth inhibition primary assay) were tested in dose against P. falciparum Dd2, a transgenic P. falciparum line expressing Saccharomyces cerevisiae DHODH (PfscDHODH), a P. falciparum strain resistant to NITD609 (Pf NITD609R) and a mammalian cell line (HepG2). P. falciparum ATPase4 is the presumed molecular target of NITD609 (ref. 9). a, Compounds were clustered across the horizontal axis by structural similarity. Colours represent compound potency (EC50). Two compound clusters, exemplified by BRD0026 (b) and BRD7539 (c), showed selectively reduced potency against the Pf NITD609R and PfscDHODH strains, respectively, while BRD73842 (d) and BRD3444 (e) were equipotent across the three P. falciparum strains. Pb, P. berghei; Pf, P. falciparum; Pv, P, vivax; PheRS, phenylalanyl-tRNA synthetase.
Bicyclic azetidines inhibit cytosolic Pf PheRS
The bicyclic azetidine BRD3444 showed multistage activity in vitro (P. falciparum Dd2, blood stage, half-maximal effective concentration (EC50) = 9 nM; P. falciparum 3D7, transmission stage, gametocyte IV–V, EC50 = 663 nM; P. berghei strain ANKA, liver stage, EC50 = 140 nM; Fig. 1e, Extended Data Table 1 and Supplementary Table 1). To elucidate the mechanism of action of the bicyclic azetidine series, three resistant lines were evolved against BRD1095 (Fig. 2a and Extended Data Fig. 2b), a derivative of BRD3444 with increased aqueous solubility, from eight independent cultures (> 8 × 109 inocula). After more than 3 months of drug pressure, EC50 values were increased by 4–84-fold. Two clones were obtained from each culture and genomic DNA from each clone was analysed via whole-genome sequencing (Fig. 3a, b and Supplementary Table 4). Analysis of resistant clones revealed that each had at least one non-synonymous single- nucleotide variant (SNV) in the PF3D7_0109800 locus, which is predicted to encode the alpha subunit of the cytosolic phenylalanyl-tRNA synthetase (Pf PheRS) of P. falciparum (ref. 22). Examination of more than 100 drug-resistant P. falciparum clones failed to reveal even a single SNV in the PF3D7_0109800 locus, indicating that the probability of Pf PheRS having three independent mutations by chance is very low. To confirm that cytosolic PheRS is the molecular target of BRD1095, the compound was assayed against purified recombinant proteins. BRD1095 inhibited the aminoacylation activity of recombinant Pf PheRS in a concentration-dependent manner (half-maximal inhibitory concentration (IC50) = 46 nM; Fig. 3c). We also reasoned that if the primary antiplasmodial mechanism of the bicyclic azetidine series was via inhibition of Pf PheRS activity, then IC50 values for the amino acylation activity of purified recombinant Pf PheRS proteins should correlate with EC50 values obtained in parasite growth inhibition assays. Indeed, a high correlation between the two parameters (r2 = 0.89) was observed using 16 synthetic analogues of BRD1095 covering a range of activities (Fig. 3d and Extended Data Table 2). This notable correlation, together with the aforementioned genetic evidence, indicates that cytosolic Pf PheRS is the relevant molecular target of the bicyclic azetidine series. In addition, supplementation with exogenous l-phenylalanine (but not d-phenylalanine, l-aspartic acid, l-threonine or l-tyrosine) to the in vitro culture medium increased the EC50 value of BRD1095 in a concentration-dependent manner (Supplementary Table 5).
Figure 2. Structures of key compounds, SSAR study of BRD3444 and X-ray crystal structure of BRD7929.

a, Structures of four bicyclic azetidine compounds. b, SSAR of BRD3444 showing that stereoisomers at the C2 position are equipotent, which suggests that this position is not necessary for activity. c, X-ray crystal structure of BRD7929 showing 3D conformation (BRD7929 was crystallized as a salt with two equivalents of l-tartaric acid; only the structure of BRD7929 is shown for clarity).
Figure 3. The bicyclic azetidine series targets the cytoplasmic Pf PheRS.

a, P. falciparum Dd2 clones resistant to BRD1095, a derivative of BRD3444 with increased aqueous solubility, were selected in vitro and non-synonymous SNVs were identified via whole-genome sequencing. All clones from three individual flasks contained non-synonymous SNVs within the PF3D7_0109800 locus, which encodes the alpha subunit of the cytoplasmic PheRS. b, The non-synonymous SNVs identified in clones from flask 1 (red), flask 2 (blue), and flask 3 (green) are shown overlaid on a homology model based on the human cytoplasmic PheRS (PDB accession 3L4G) generated in PyMol. c, BRD1095 was assayed against purified recombinant proteins of wild-type cytosolic Pf PheRS and a mutant containing a SNV (giving a L550V substitution), identified from the resistant strain. IC50 value of the wild-type PheRS was 0.045 µM, whereas the IC50 value for BRD1095L550V was 1.30 µM (data are mean ± s.d. for two biological and two technical replicates). d, The bicyclic azetidine series showed a strong correlation between blood-stage growth inhibition and biochemical inhibition of cytosolic Pf PheRS activity. We assayed 15 bicyclic azetidine analogues with varying potency against blood-stage parasites (Dd2 strain) against purified recombinant Pf PheRS. The biochemically derived IC50 values correlate strongly (r2 = 0.89) with the EC50 values determined using the blood-stage growth inhibition assay (see Extended Data Table 2 for structure-activity relationship study and chemical structures).
Owing to its newfound susceptibility to inhibition, Pf PheRS joins the aminoacyl-tRNA synthetase class of emerging targets for antimalarial agents23–29. Although they share common tRNA esterification catalytic activities, these proteins are structurally diverse and physiologically distinct enzymes. The target described here (P. falciparum cytosolic PheRS) is unique as it is the first member of the class in which inhibition, as we will describe, results in elimination of asexual blood-, liver- and transmission-stage parasites, preventing disease transmission, ensuring prophylaxis and providing single-dose cures of the disease in mouse models of malaria.
Optimization of the bicyclic azetidine series
BRD3444 exhibited poor solubility (< 1 µM in PBS), high intrinsic clearance in human and mouse microsomes (Clint = 142 and 248 µl min−1 mg−1, respectively) and a high volume of distribution (Vss = 12 l kg−1; all data found in Extended Data Table 3). These results translated to a half-life of 3.7 h in an intravenous pharmacokinetic study in CD-1 mice. Analysis of all eight stereoisomers of BRD3444 included in the primary screen revealed that activity against P. falciparum Dd2 parasites was predominantly found among two isomers differing in stereochemistry at the C2 position (Fig. 2a, b). Therefore, we postulated that the C2 position could be manipulated without loss of in vitro potency and could be used to improve the physicochemical and pharmacokinetic properties of the series. The modular synthetic pathway facilitated the synthesis of advanced analogues that included BRD1095 and BRD7929, in which the hydroxymethyl group at position C2 is replaced with aminomethyl and dimethylaminomethyl substituents, respectively. These bicyclic azetidines showed improved solubility (25 and 15 µ M in PBS, respectively) and greatly improved intrinsic clearance in mouse microsomes (< 20 and 21 µl min−1 mg−1, respectively), while retaining in vitro potency. In an intravenous and oral pharmacokinetic study in mice, both BRD1095 and BRD7929 displayed greatly improved blood clearance relative to BRD3444. BRD7929 also displayed good bioavailability (80%), superior to that of BRD1095 (50%), and improved in vitro potency against P. cynomolgi and P. falciparum liver-stage and P. falciparum transmission-stage parasites (Extended Data Table 1). BRD7929 showed a high Vss of 24 lkg−1 (Extended Data Table 3), which, together with a low blood clearance, translated to a long half-life (32 h), making this compound suitable for single-dose oral treatments. The synthesis pathway enabled the laboratory preparation of 7.5 g of BRD7929 for further testing.
BRD7929 shows in vivo efficacy against all life stages
We evaluated the multistage activity of BRD7929 using mouse malaria models. When BRD7929 activity was evaluated in the blood-stage model with the rodent malaria parasite P. berghei using a luciferase reporter, all infected CD-1 mice treated with a single oral 25 mg kg−1 or 50 mg kg−1 dose became parasite-free and remained so up to the 30-day end-point based on bioluminescent imaging (Extended Data Fig. 3a, b). To evaluate the therapeutic potential of this series, the in vivo efficacy of BRD7929 against the human malaria parasite P. falciparum was determined. Approximately 48 h after inoculation with the blood-stage P. falciparum 3D7HLH/BRD (expressing firefly luciferase), non-obese diabetic/severe combined immunodeficiency (NOD/ SCID) Il2rγ−/− mice engrafted with human erythrocytes (huRBC NSG) were treated with a single dose of BRD7929 and monitored for 30 days (Fig. 4a and Extended Data Fig. 3c). At 25 mg kg−1 (area under curve (AUC) = 62.8 µM h) and 50 mg kg−1 (AUC = 125.6 µM h), a rapid decrease in parasite-associated bioluminescence was observed, while at 6.25 mg kg−1 (AUC = 15.7 µM h) the rate of the loss of bioluminescence was slower. All huRBC NSG mice treated with single oral 12.5 mg kg−1 (AUC = 31.4 µM h), 25 mg kg−1 or 50 mg kg−1 doses were parasite-free for 30 days based on bioluminescent imaging. The AUC in the 25 mg kg−1 single-dose cure observed in the model with P. berghei is estimated to be 27.5 µM h based on pharmacokinetic studies with CD-1 mice. Thus, single-dose cures were observed in the P. berghei CD-1 and P. falciparum huRBC NSG mouse models at similar drug exposure levels (AUC=27.5 and 31.4 µMh, respectively), suggesting that the efficacy against the two Plasmodium species is comparable.
Figure 4. In vivo efficacy studies of BRD7929 using P. falciparum and humanized mouse models.

a, huRBC NSG mice were inoculated with P. falciparum (3D7HLH/BRD) blood-stage parasites 48 h before treatment and BRD7929 was administered as a single 50, 25, 12.5 or 6.25 mg kg−1 oral dose at 0 h (n = 2 for each group, this study was conducted once). Infections were monitored using the in vivo imaging system (IVIS). Bioluminescent intensity was quantified from each mouse and plotted against time. The dotted horizontal line represents the mean bioluminescence intensity level obtained from all the animals before the parasite inoculation. No recrudescence was observed as low as a single 25 mg kg−1 dose of BRD7929 in the infected animals (see Extended Data Fig. 3b). b, huHep FRG-knockout mice were inoculated intravenously with P. falciparum (NF54HT-GFP-luc) sporozoites. BRD7929 was administered as a single 10 mg kg−1 oral dose 1 day after inoculation, and daily engraftment of human erythrocytes was initiated 5 days after inoculation (n = 2 for each group, this study was conducted once). Infections were monitored using IVIS. The dotted horizontal line represents the mean bioluminescence intensity level obtained from all the animals before the sporozoite inoculation. No increase in bioluminescence intensity level was observed from the mice treated with BRD7929 (see Extended Data Fig. 5a). c, huRBC NSG mice were infected with blood-stage P. falciparum (3D7HLH/BRD) parasites for 2 weeks (allowing the gametocytes to mature fully) and were treated with a single oral dose of BRD7929 (12.5 mg kg−1). Blood samples were collected for 11 days and analysed for the presence of the asexual marker SBP1 and the mature gametocyte marker Pfs25 using qRT-PCR (n = 2 for each group, this study was conducted once). The transcription of both SBP1 and Pfs25 decreased to undetectable levels 7 days after treatment, strongly suggesting that BRD7929 eliminates both asexual and gametocyte stages and is capable of preventing parasite transmission to the mosquito (data are mean ± s.d. for three technical replicates for each biological sample).
In a P. berghei liver-stage model, none of the CD-1 mice that were treated with a single dose of 5 or 25 mg kg−1 BRD7929 developed blood-stage parasitaemia within a 30-day period following P. berghei sporozoite inoculation (Extended Data Fig. 4a, b). Furthermore, mice were treated with a single dose of 10 mg kg−1 BRD7929 at various time points before sporozoite inoculation and during liver-stage infection (Extended Data Fig. 4c). All mice treated within the 3 days before inoculation and during liver-stage infection were completely free of blood-stage parasites for the duration of the experiment (32 days), indicating that BRD7929 has potent causal prophylaxis activity. Next, 1 day after inoculation with P. falciparum (NF54HT-GFP-luc)30 sporozoites, FRG knockout (Fah−/−Rag2−/−Il2rg−/−, heavily immunosuppressed) C57BL/6 mice transplanted with human hepatocytes (huHep FRG knockout)31 were treated with a single oral dose of BRD7929 (10 mg kg−1). Human erythrocytes were intraperitoneally injected daily from 5 to 7 days after inoculation. A gradual increase was detected in parasite liver-stage-associated bioluminescence signals from the lower pectoral and upper abdominal regions of the control (vehicle-treated) mice, whereas no increase in bioluminescence signals was observed from the BRD7929-treated mice (Fig. 4b and Extended Data Fig. 5a). For quantitative reverse transcription PCR (qRT-PCR) analysis32, blood samples were also collected 7 days after inoculation (the first day of the blood stage)31 and evaluated for the presence of the blood-stage transcript PF3D7_1120200 (expressing the P. falciparum ubiquitin-conjugating enzyme, UCE) (Extended Data Fig. 5b). The presence of the blood-stage marker was not detected in samples from the BRD7929-treated mice, indicating that BRD7929 eliminated the liver-stage parasites.
Finally, to examine whether BRD7929 has activity against mature gametocytes and prevents parasite transmission to mosquitoes in vivo, CD-1 mice infected with P. berghei were treated with a single oral dose of BRD7929 2 days before exposure to female Anopheles stephensi mosquitoes. One week later, the midguts of the blood-fed mosquitoes were dissected and the number of oocysts was counted (Extended Data Fig. 6a–c). No oocysts were detected in midguts dissected from mosquitoes fed on mice treated with 5 or 20 mg kg−1 BRD7929, concentrations below those found to be efficacious against asexual blood-stage parasites. To determine whether BRD7929 showed in vivo efficacy against P. falciparum in humanized mouse models, huRBC NSG mice were infected with blood-stage P. falciparum 3D7HLH/BRD parasites for 2 weeks to allow the development of mature gametocytes. Subsequently, mice were treated with a single oral dose of BRD7929 (12.5 mg kg−1, AUC = 31.4 µM h). Blood samples were collected for 11 days after treatment and analysed for the presence of the late-sexual−stage-specific transcript of Pfs25 (expressing P. falciparum 25 kDa ookinete surface-antigen precursor, PF3D7_1031000) using qRT–PCR32 (Fig. 4c and Extended Data Fig. 6d−f). The transcription of Pfs25 decreased to undetectable levels 7 days after treatment. Previous literature reports of in vitro cellular sensitivity showed that the Pfs25 marker had a detection limit of 0.02–0.05 gametocytes µl−1 (ref. 33), strongly suggesting that BRD7929 has late-stage gametocidal activity and is capable of preventing the transmission of parasites to the mosquito vector at the same level of exposure as that achieves a single-dose cure in the blood stage.
Safety optimization of the bicyclic azetidine series
While no significant cytotoxicity was observed with BRD3444 and BRD3316, moderate cytotoxicity was observed for bicyclic azetidines BRD7929 (half-maximal cytotoxic concentration (CC50) = 9 µM) and BRD1095 (CC50 = 16 µM) in the HepG2 cell line (Extended Data Fig. 7a). Both BRD1095 and BRD7929 showed inhibition of IKr (encoded by KCNH2, also known as hERG) (IC50 = 5.1 and 2.1 µM, respectively; Extended Data Table 3). Medicinal chemistry efforts have shown that mitigation of ion-channel toxicity is possible while maintaining biological activity; for example, BRD3316 shows no significant inhibition of IKr at > 10 µM, indicating that cardiotoxicity is not intrinsically linked to this series. While BRD3444 showed time-dependent inhibition of CYP3A4, BRD7929 showed no inhibition of any of the major human cytochrome P450 (CYP) isoforms (Extended Data Fig. 7a). No phototoxicity was observed with this series in BALB/c 3T3 mouse fibroblasts following exposure to UVA light. BRD7929 and BRD3316 show desirable pharmacokinetic properties, including good oral bioavailability (F = 80 and 63%, respectively). In addition, BRD7929 has a long half-life that enables single-dose treatment. Based on in vitro microsomal stability data, BRD7929 and advanced analogues in this series are likely to have a similar profile in humans, as metabolic clearance was low for both mouse and human species (Extended Data Table 3). BRD7929 was determined to be non-mutagenic using an Ames test in the presence or absence of S9 mix using the Salmonella typhimurium strains TA100, TA1535, TA98, TA1537 and Escherichia coli strain WP2uvrA (Supplementary Table 6). Histopathological analysis of mice treated at a high dose (100 mg kg−1, estimated Cmax and AUC are 5.4 µM and 110 µMh, respectively) showed no adverse findings in the limited number of organs examined (Extended Data Fig. 7b). Additional studies involving a wider range of organs, doses and compounds will be needed to assess the toxicity of these and related compounds more thoroughly. In NSG mice the estimated Cmax and AUC of the single-dose cure are 833 nM and 31.4 µMh, respectively, affording a 6.5-fold safety margin with respect to Cmax.
Although the emergence of resistance in vitro does not necessarily imply that it will happen in vivo, it is indicative of any mechanisms of resistance that could arise in the future. To examine the propensity of de novo resistance selection, P. falciparum Dd2 cultures with initial inocula ranging from 105 to 109 parasites were maintained in medium supplemented with 20 nM BRD7929 (the EC90 of strain Dd2) and monitored for 60 days to identify recrudescent parasitaemia (Extended Data Fig. 7c, d). No recrudescence was observed in Dd2 cultures exposed to a constant pressure of BRD7929, whereas the minimum inoculum of resistance for atovaquone (EC90 = 2 nM) was 107, consistent with previous reports34.
Discussion
Malaria remains one of the deadliest infectious diseases. Available therapeutic agents are already limited in their efficacy, and drug resistance threatens to diminish our ability to prevent and treat the disease further. Despite a renewed effort to identify compounds with antimalarial activity, the drug discovery and development pipeline lacks target diversity and most malaria drugs are only efficacious during the asexual blood stage of parasite infection.
In these studies, we attempted to identify new antimalarial targets by screening a diverse collection of 100,000 compounds with three-dimensional topographic features derived from stereochemical and skeletal elements that are common in natural products but underrepresented in typical screening collections—compounds now accessible using DOS. The compounds are formed in short, modular syntheses that facilitate chemical optimization and manufacturing35,36 and have computed physical properties aimed at accelerating drug discovery37. We used a primary phenotypic screen to identify a subset of compounds that inhibits parasite growth, counter-screens to prioritize molecules with both novel mechanisms of action and activity at multiple stages of the parasite life cycle, and genetic and biochemical studies to illuminate mechanisms of action. These efforts yielded several series of multiple-stage antimalarial compounds with unique scaffolds that modulate both recently described and established molecular targets.
An earlier pilot study tested key elements of the process above using a distinct 8,000-member DOS library, leading to the discovery of ML238 (refs 13, 14), a molecule that inhibits parasite growth with nanomolar potency by targeting the reductase domain of P. falciparum cytochrome b (the Qi site), in contrast to the antimalarial agent atovaquone, which targets the oxidase domain of P. falciparum cytochrome b (the Qo site). The study presented here led to many candidate antimalarial agents. We have, thus far, characterized four of these compound series, namely BRD0026 (targeting P. falciparum ATPase4), BRD7539 (targeting P. falciparum DHODH), BRD73842 (targeting P. falciparum PI4K) and BRD3444 (targeting P. falciparum cytoplasmic PheRS). These series were prioritized as they showed in vitro activity against multiple stages of the P. falciparum life cycle, and this was subsequently confirmed in vivo. We anticipate that additional compound series uncovered by these experiments, made available via the Malaria Therapeutics Response Portal (http://portals.broadinstitute.org/mtrp/), will target additional proteins that function as multiple-stage vulnerabilities in Plasmodium and other Apicomplexa pathogens.
Until now, natural products and synthetic drug-like compounds have served as the primary sources of antimalarial drugs. As parasitic susceptibility to traditional chemotypes decreases, it is becoming increasingly necessary to discover lead compounds that are unaffected by existing mechanisms of resistance. DOS coupled with phenotypic screening offers a systematic means to address this need. The results reported here describe a new target and chemotype—Pf PheRS and bicyclic azetidines such as BRD3316 and BRD7929—that have demonstrated the lowest-concentration single-dose cure of three promising next-generation antimalarials in the pipeline9,38,39 using two mouse models. Single-dose treatments facilitate compliance and overcome cost challenges in resource-deficient regions40. The ability of BRD7929 to eliminate blood-stage (both asexual and sexual) and liver-stage parasites suggests bicyclic azetidines have the potential to cure the disease, provide prophylaxis and prevent disease transmission.
Our findings suggest that DOS-derived compound collections, which comprise three-dimensional structures reminiscent of natural products that have yielded many small-molecule probes of diverse mammalian processes41,42, are also a rich resource for identifying targets and readily optimized chemical scaffolds to supplement the current antimalarial pipeline.
METHODS
In vitro P. falciparum blood-stage culture and assay
Strains of P. falciparum (Dd2, 3D7, D6, K1, NF54, V1/3, HB3, 7G8, FCB and TM90C2B) were obtained from the Malaria Research and Reference Reagent Resource Center (MR4). PfscDHODH, the transgenic P. falciparum line expressing S. cerevisiae DHODH19, was a gift from A. B. Vaidya. P. falciparum isolates were maintained with O-positive human blood in an atmosphere of 93% N2, 4% CO2, 3% O2 at 37 °C in complete culturing medium (10.4 gl−1 RPMI 1640, 5.94 gl−1 HEPES, 5 gl−1 albumax II, 50 mg l−1 hypoxanthine, 2.1 gl−1 sodium bicarbonate, 10% human serum and 43 mgl−1 gentamicin). Parasites were cultured in medium until parasitaemia reached 3–8%. Parasitaemia was determined by checking at least 500 red blood cells from a Giemsa-stained blood smear. For the compound screening, a parasite dilution at 2.0% parasitaemia and 2.0% haematocrit was created with medium. 25 (µl of medium was dispensed into 384-well black, clear-bottom plates and 100 nl of each compound in DMSO was transferred into assay plates along with the control compound (mefloquine). Next, 25 (µl of the parasite suspension in medium was dispensed into the assay plates giving a final parasitaemia of 1% and a final haematocrit of 1%. The assay plates were incubated for 72 h at 37 °C. 10 (µl of detection reagent consisting of 10× SYBR Green I (Invitrogen; supplied in 10,000× concentration) in lysis buffer (20 mM Tris-HCl, 5 mM EDTA, 0.16% (w/v) Saponin, 1.6% (v/v) Triton X-100) was dispensed into the assay plates. For optimal staining, the assay plates were left at room temperature for 24 h in the dark. The assay plates were read with 505 dichroic mirrors with 485 nm excitation and 530 nm emission settings in an Envision (PerkinElmer).
Chemoinformatics clustering
High-throughput screening hits were hierarchically clustered by structural similarity using average linkage on pairwise Jaccard distances43 between ECFP4 fingerprints44. Pipeline Pilot45 was used for fingerprint and distance calculation; clustering and heat-map generation was done in R (ref. 46).
In vitro P. berghei liver-stage assay
HepG2 cells (ATCC) were maintained in DMEM, 10% (v/v) FBS (Sigma), and 1% (v/v) antibiotic–antimycotic in a standard tissue culture incubator (37 °C, 5% CO2). P. berghei (ANKA GFP–luc) infected A. stephensi mosquitoes were obtained from the New York University Langone Medical Center Insectary. For assays, ~ 17,500 HepG2 cells per well were added to a 384-well microtitre plate in duplicate. After 18–24 h at 37 °C the media was exchanged and compounds were added. After 1 h, parasites obtained from freshly dissected mosquitoes were added to the plates (4,000 parasites per well), the plates were spun for 10 min at 1,000 r.p.m. and then incubated at 37 °C. The final assay volume was 30 (µl. After a 48-h incubation at 37 °C, Bright-Glo (Promega) was added to the parasite plate to measure relative luminescence. The relative signal intensity of each plate was evaluated with an EnVision (PerkinElmer) system.
In vitro P. falciparum liver-stage assay
Micropatterned co-culture (MPCC) is an in vitro co-culture system of primary human hepatocytes organized into colonies and surrounded by supportive stromal cells. Hepatocytes in this format maintain a functional phenotype for up to 4–6 weeks without proliferation, as assessed by major liver-specific functions and gene expression47–49. In brief, 96-well plates were coated homogenously with rat-tail type I collagen (50 (µg ml−1) and subjected to soft-lithographic techniques to pattern the collagen into 500-(µm-island microdo-mains that mediate selective hepatocyte adhesion. To create MPCCs, cryopreserved primary human hepatocytes (BioreclamationIVT) were pelleted by centrifugation at 100g for 6 min at 4 °C, assessed for viability using Trypan blue exclusion (typically 70–90%), and seeded on micropatterned collagen plates (each well contained ~ 10,000 hepatocytes organized into colonies of 500 (µM) in serum-free DMEM with 1% penicillin-streptomycin. The cells were washed with serum-free DMEM with 1% penicillin-streptomycin 2–3 h later and replaced with human hepatocyte culture medium48. 3T3-J2 mouse embryonic fibroblasts were seeded (7,000 cells per well) 24 h after hepatocyte seeding. 3T3-J2 fibroblasts were courtesy of H. Green50.
MPCCs were infected with 75,000 sporozoites (NF54) (Johns Hopkins University) 1 day after hepatocytes were seeded48,49. After incubation at 37 °C and 5% CO2 for 3 h, wells were washed once with PBS, and the respective compounds were added. Cultures were dosed daily. Samples were fixed on day 3.5 after infection. For immunofluorescence staining, MPCCs were fixed with − 20 °C methanol for 10 min at 4 °C, washed twice with PBS, blocked with 2% BSA in PBS, and incubated with mouse anti-P. falciparum Hsp70 antibodies (clone 4C9, 2 (µg ml−1) for 1 h at room temperature. Samples were washed with PBS then incubated with Alexa 488-conjugated secondary goat anti-mouse for 1 h at room temperature. Samples were washed with PBS, counterstained with the DNA dye Hoechst 33258 (Invitrogen; 1:1,000), and mounted on glass slides with fluoromount G (Southern Biotech). Images were captured on a Nikon Eclipse Ti fluorescence microscope. Diameters of developing liver stage parasites were measured and used to calculate the corresponding area.
In vitro P. cynomolgi liver-stage assay
All rhesus macaques (Macaca mulatta) used in this study were bred in captivity for research purposes, and were housed at the Biomedical Primate Research Centre (BPRC; AAALAC-certified institute) facilities under compliance with the Dutch law on animal experiments, European directive 86/609/EEC and with the ‘Standard for Humane Care and Use of Laboratory Animals by Foreign Institutions’ identification number A5539-01, provided by the Department of Health and Human Services of the US National Institutes of Health. The local independent ethical committee first approved all protocols. Non-randomized rhesus macaques (male or female; 5– 14 years old; one animal per month) were infected with 1×106 P. cynomolgi (M strain) blood-stage parasites, and bled at peak parasitaemia. Approximately 300 female A. stephensi mosquitoes (Sind-Kasur strain, Nijmegen University Medical Centre St Radboud) were fed with this blood as described previously51.
Rhesus monkey hepatocytes were isolated from liver lobes as described by previously52. Sporozoite infections were performed within 3 days of hepatocyte isolation. Sporozoite inoculation of primary rhesus monkey hepatocytes was performed as described previously53,54. On day 6, intracellular P. cynomolgi malaria parasites were fixed, stained with purified rabbit antiserum reactive against P. cynomolgi Hsp70.1 (ref. 53), and visualized with FITC-labelled goat anti- rabbit IgG antibodies. Quantification of small ‘hypnozoite’ exoerythrocytic forms (1 nucleus, a small round shape, a maximal diameter of 7 (µm) or large ‘developing parasite’ exoerythrocytic forms (more than 1 nucleus, larger than 7 (µm and round or irregular shape) was determined for each well using a high-content imaging system (Operetta, PerkinElmer).
In vitro transmission-blocking assay (gametocyte IV–V)
P. falciparum 3D7 stage IV–V gametocytes were isolated by discontinuous Percoll gradient centrifugation of parasite cultures treated with 50 mM N-acetyl-d-glucosamine for 3 days to kill asexual parasites. Gametocytes (1.0 × 105) were seeded in 96-well plates and incubated with compounds for 72 h. In vitro anti-gametocyte activity was measured using CellTiter-Glo (Promega).
In vitro transmission-blocking imaging assay (early, I–III; and late, IV–V, gametocyte)
A detailed description of the method is published elsewhere55. In brief, NF54pf-LUC-GFP highly synchronous gametocytes were induced from a single intra-erythrocytic asexual replication cycle. On day 0 of gametocyte development, spontaneously generated gametocytes were removed by VarioMACS magnetic column (MAC) technology. Early stage I gametocytes were collected on day 2 of development and late-stage gametocytes (stage IV) on day 8 using MAC columns. Percentage parasitaemia and haematocrit was adjusted to 10 and 0.1, respectively. 45 (µl of parasite sample were added to PerkinElmer Cell carrier poly-d-lysine imaging plates containing 5 (µl of test compound at 16 doses, including control wells containing 4% DMSO and 50 (µM puromycin (0.4% and 5 (µM final concentrations, respectively), the plates sealed with a membrane (Breatheasy or 4ti-05 15/ST) and incubated for 72 h in standard incubation conditions of 5% CO2, 5% O2, 90% N2 and 60% humidity at 37 °C. After incubation, 5 (µl of 0.07 (µg ml−1 MitoTracker Red CM-H2XRos (MTR) (Invitrogen) in PBS was added to each well, and plates were resealed with membranes and incubated overnight under standard conditions. The following day, the plates were brought to room temperature for at least one hour before being measured on the Opera QEHS Instrument. Image analysis was performed using an Acapella (PerkinElmer)-based algorithm that identifies gametocytes of the expected morphological shape with respect to degree of elongation and specifically those parasites that are determined as viable by the MitoTracker Red CM-H2XRos fluorescence size and intensity. IC50 values were determined using GraphPad Prism 4, using a 4-parameter log dose, nonlinear regression analysis, with sigmoidal dose–response (variable slope) curve fit.
P. falciparum standard membrane feeding assay
P. falciparum transmission-blocking activity of BRD7929 was assessed in a standard membrane feeding assay as previously described56. In brief, P. falciparumNF54 hsp70-GFP-luc reporter parasites were cultured up to stage V gametocytes (day 14). Test compounds were serially diluted in DMSO and subsequently in RPMI medium to reach a final DMSO concentration of 0.1%. Diluted compound was either pre-incubated with stage V gametocytes for 24 h (indirect mode) or directly added to the blood meal (direct mode). Gametocytes were adjusted to 50% haematocrit, 50% human serum and fed to A. stephensi mosquitoes. All compound dilutions were tested in duplicate in independent feeders. After 8 days, mosquitoes were collected and the relative decrease in oocysts density in the midgut was determined by measurement of luminescence signals in 24 individual mosquitoes from each cage. For each vehicle (control) cage, an additional 10 mosquitoes were dissected and examined by microscopy to determine the baseline oocyst intensity.
In vitro resistance selections
In vitro resistance selections were performed as previously described15. In brief, approximately 1 × 109 P. falciparum Dd2 parasites were treated with 60 nM (EC99.9) or 150 nM (10 × EC50) of BRD1095 in each of four independent flasks for 3–4 days. After the compounds were removed, the cultures were maintained in compound-free complete RPMI growth medium with regular media exchange until healthy parasites reappeared. Once parasitaemia reached 2–4%, compound pressure was repeated and these steps were executed for about 2 months until the initial EC50 shift was observed. Three out of four independent selections pressured at 60 nM developed a phenotypic EC50 shift. None of the selections pressured at 150 nM resulted in resistant parasites. After an initial shift in the dose–response phenotype was observed, selection at an increased concentration was repeated in the same manner until at least a threefold shift in EC50 was observed. Selected parasites were then cloned by limiting dilution.
BRD73842-resistant selections were conducted in a similar manner except that parasites were initially treated at 0.5 (µM (10 × EC50) for 4 days or 150 nM (EC99.9) for 2 days in each of two independent flasks. The Y1356N mutant was derived from a flask pressured at 0.5 (µM and the L1418F mutant was developed from one of the flasks exposed to the 150 nM.
Whole-genome sequencing and target identification
DNA libraries were prepared for sequencing using the Illumina Nextera XT kit (Illumina), and quality-checked before sequencing on a Tapestation. Libraries were clustered and run as 100-bp paired-end reads on an Illumina HiSeq 2000 in RapidRun mode, according to the manufacturer’s instructions. Samples were analysed by aligning to the P. falciparum 3D7 reference genome (PlasmoDB v. 11.1). To identify SNVs and CNVs, a sequencing pipeline developed for P. falciparum (Plasmodium Type Uncovering Software, Platypus) was used as previously described, with the exception of an increase in the base quality filter from 196.5 to 1,000 (ref. 57).
P. falciparum DHODH biochemical assay
Substrate-dependent inhibition of recombinant P. falciparum DHODH protein was assessed in an in vitro assay in 384-well clear plates (Corning 3640) as described previously58. A 20-point dilution series of inhibitor concentrations were assayed against 2–10 nM protein with 500 µM l-dihydroorotate substrate (excess), 18 µM dodecylubiquinone electron acceptor (~Km), and 100 µM 2,6-dichloroindophenol indicator dye in assay buffer (100 mM HEPES pH 8.0, 150 mM NaCl, 5% glycerol, 0.5% Triton X-100). Assays were incubated at 25 °C for 20 min and then assessed via OD600. Data were normalized to 1% DMSO and excess inhibitor (25 µM DSM265; ref. 7).
Human DHODH biochemical assay
Substrate-dependent inhibition of recombinant human DHODH protein was assessed in an in vitro assay in 384-well clear plates (Corning 3640) as described previously59. A 20-point dilution series of inhibitor concentrations was assayed against 13 nM protein with 1 mM l-dihydroorotate substrate (excess), 100 (µM dodecylubiquinone electron acceptor, and 60 µM 2,6-dichloroindophenol indicator dye in assay buffer (50 mM Tris HCl pH 8.0, 150 mM KCl, 0.1% Triton X-100). Assays were incubated at 25 °C for 20 min and then assessed via OD600. Data were normalized to 1% DMSO and no enzyme.
P. vivax PI4K biochemical assay
The synthetic gene for full-length P. vivax PI4K (PVX098050) was synthesized from GeneArt (ThermoScientific), and was expressed and purified as previously described20. Aliquots of P. vivax PI4K(3 were flash-frozen in liquid nitrogen and stored at − 80°C. Full-length human PI4KB (uniprot gene Q9UBF8-2) was expressed and purified as previously described60. 100 nM extruded lipid vesicles were made to mimic Golgi organelle vesicles (20% phosphatidylinositol, 10% phosphatidylserine, 45% phosphatidylcholine and 25% phosphatidylethanolamine) in lipid buffer (20 mM HEPES pH 7.5 (room temperature), 100 mM KCl, 0.5 mM EDTA). Lipid kinase assays were carried out using the Transcreener ADP2 FI Assay (BellBrook Labs) following the published protocol as previously described61. 4-(µl reactions ran at 21 °C for 30 min in a buffer containing 30 mM HEPES pH 7.5, 100 mM NaCl, 50 mM KCl, 5 mM MgCl2, 0.25 mM EDTA, 0.4% (v/v) Triton X-100, 1 mM TCEP, 0.5 mg ml−1 Golgi-mimic vesicles and 10 (µM ATP. P. vivax PI4K(β was used at 7.5 nM and human PI4KB was used at 200 nM. Fluorescence intensity was measured using a Spectramax M5 plate reader with excitation at 590 nm and emission at 620 nm (20-nm bandwidth). IC50 values were calculated from triplicate inhibitor curves using GraphPad Prism software.
PheRS homology modelling
The model was built using the SWISS-MODEL online resource62–64 and Prime65 (Schrödinger Release 2015-2: Prime, version 4.0, Schrödinger), with human PheRS (PDB accession 3L4G) as a template for P. falciparum PheRS (PlasmoDB Gene ID: PF3D7_0109800). The template was chosen based on highest sequence identity and similarity identified via PSI-BLAST. Target-template alignment was made using ProMod-II and validated with Prime STA. Coordinates from residues that were conserved between the target and the template were copied from the template to the model, and remaining sites were remodelled using segments from known structures. The side chains were then rebuilt, and the model was finally refined using a force field.
P. falciparum cytoplasmic PheRS biochemical assay
Protein sequences of both α- (PF3D_70109800) and β- (PF3D7_1104000) subunits of cytoplasmic P. falciparum PheRS were obtained from PlasmoDB (http://plasmodb.org/plasmo/). Full length α- and (β-subunit gene sequences optimized for expression in E. coli were cloned into pETM11 (Kanamycin resistance) and pETM20 (ampicillin resistance) expression vectors using Nco1 and Kpn1 sites and co-transformed into E. coli B834 cells. Protein expression was induced by addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and cells were grown until an OD600 of 0.6–0.8 was reached at 37 °C. They were then allowed to grow at 18 °C for 20 h after induction. Cells were separated by centrifugation at 5,000g for 20 min and the bacterial pellets were suspended in a buffer consisting of 50 mM Tris–HCl (pH 7.5), 200 mM NaCl, 4 mM (β-mercaptoethanol, 15% (v/v) glycerol, 0.1 mg ml−1 lysozyme and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed by sonication and cleared by centrifugation at 20,000g for 1 h. The supernatant was applied on to prepacked NiNTA column (GE Healthcare), and bound proteins were eluted by gradient-mixing with elution buffer (50 mM Tris–HCl (pH 7.5), 80 mM NaCl, 4 mM (β-mercaptoethanol, 15% (v/v) glycerol, 1 M imidazole). Pure fractions were pooled and loaded on to heparin column for further purification. Again, bound proteins were eluted using gradient of heparin elution buffer 50 mM Tris–HCl (pH 7.5), 1 M NaCl, 4 mM (β-mercaptoethanol, 15% (v/v) glycerol). Pure fractions were again pooled and dialysed overnight into a buffer containing 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 4 mM (β-mercaptoethanol, 1 mM DTT and 0.5 mM EDTA. TEV protease (1:50 ratio of protease:protein) was added to the protein sample and incubated at 20 °C for 24 h to remove the polyhistidine tag. Protein was further purified via gel-filtration chromatography on a GE HiLoad 60/600 Superdex column in 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 4 mM (β-mercaptoethanol, 1 mM MgCl2. The eluted protein (a heterodimer of P. falciparum cPheRS) were collected, assessed for purity via SDS–PAGE and stored at − 80 °C.
Nuclear encoded tRNAPhe from P. falciparum was synthesized using an in vitro transcription method as described earlier22,66. Aminoacylation and enzyme inhibition assays for P. falciparum cytosolic PheRS were performed as described earlier22,67. Enzymatic assays were performed in buffer containing 30 mM HEPES (pH 7.5), 150 mM NaCl, 30 mM KCl, 50 mM MgCl2, 1 mM DTT, 100 (µM ATP, 100 (µM l-phenylalanine, 15 (µM P. falciparum tRNAPhe, 2 U ml−1 E. coli inorganic pyrophosphatase (NEB) and 500 nM recombinant P. falciparum PheRS at 3 °C. Reactions at different time points were stopped by the addition of 40 mM EDTA and subsequent transfer to ice. Recombinant maltose binding protein was used as negative control. The cPheRS inhibition assays were performed using inhibitor concentrations of 0.01 nM, 0.1 nM, 1 nM, 10 nM, 100 nM, 1(µM, 5(µM and 10(µM for strong binders and 1 nM, 10 nM, 100 nM, 1 (µM, 10(µM, 100 (µM and 500 (µM for weaker binders in the assay buffer. Enzymatic and inhibition experiments were performed twice in triplicate.
Mammalian cell cytotoxicity assays
Mammalian cells (HepG2, A549, and HEK293) were obtained from the ATCC and cultured routinely in DMEM with 10% FBS and 1% (v/v) antibiotic–antimycotic. For cytotoxicity assays, 1 × 106 cells were seeded into 384-well plates 1 day before compound treatment. Cells were treated with ascending doses of compound for 72 h, and viability was measured using Cell-Titer Glo (Promega). All cell lines were tested for Mycoplasma contamination using Universal mycoplasma Detection Kit (ATCC).
In vitro ADME/PK and safety assays
In vitro characterization assays (protein binding, microsomal stability, hepatocyte stability, cytochrome P450 (CYP) inhibition, and aqueous solubility) were performed according to industry- standard techniques. Ion channel inhibition studies were performed using the Q-Patch system using standard techniques.
Animal welfare
All animal experiments were conducted in compliance with institutional policies and appropriate regulations and were approved by the institutional animal care and use committees for each of the study sites (the Broad Institute, 0016-09–14; Harvard School of Public Health, 03228; Eisai, 13-05, 13-07, 14-C-0027). No method of randomization or blinding was used in this study.
In vivo P. berghei blood-stage assay
CD-1 mice (n = 4 per experimental group; female; 6–7-week-old; 20–24 g, Charles River) were intravenously inoculated with approximately 1 × 105 P. berghei (ANKA GFP-luc) blood-stage parasites 24 h before treatment and compounds were administered orally (at 0 h). Parasitaemia was monitored by the in vivo imaging system (IVIS SpectrumCT, PerkinElmer) to acquire the bioluminescence signal (150 mg kg−1 of luciferin was intraperitoneally injected approximately 10 min before imaging). In addition, blood smear samples were obtained from each mouse periodically, stained with Giemsa, and viewed under a microscope for visual detection of blood parasitaemia. Animals with parasitaemia exceeding 25% were humanely euthanized.
In vivo P. berghei causal prophylaxis assay
CD-1 mice (n = 4 per experimental group; female; 6–7-week-old; 20–24 g, Charles River) were inoculated intravenously with approximately 1 × 105 P. berghei (ANKA GFP-luc) sporozoites freshly dissected from A. stephensi mosquitoes. Immediately after infection, the mice were treated with single oral doses of BRD7929; infection was monitored as described for the P. berghei erythrocytic-stage assay. For time-course experiments, the time of compound treatment (single oral dose of 10 mg kg−1) was varied from 5 days before infection to 2 days after infection.
In vivo P. berghei transmission-stage assay
CD-1 (n = 3 per experimental group; female; 6–7-week-old; 21–24 g, Charles River) mice were infected with P. berghei (ANKA GFP-luc) for 96 h before treatment with vehicle or BRD7929 (day 0). On day 2, female A. stephensi mosquitoes were allowed to feed on the mice for 20 min. After 1 week (day 9), the midguts of the mosquitoes were dissected out and oocysts were enumerated microscopically (12.5 × magnification).
In vivo P. falciparum blood-stage assay
In vivo adapted P. falciparum (3D7HLH/BRD) were selected as described previously68. In brief, NSG mice (n = 2 per experimental group; female; 4–5-week-old; 19–21 g; The Jackson Laboratory) were intraperitoneally injected with 1 ml of human erythrocytes (O-positive, 50% haematocrit, 50% RPMI 1640 with 5% albumax) daily to generate mice with humanized circulating erythrocytes (huRBC NSG). Approximately 2 × 107 blood-stage P. falciparum 3D7HLH/BRD (ref. 69) were intravenously infected to huRBC NSG mice and > 1% parasitaemia was achieved 5 weeks after infection. After three in vivo passages, the parasites were frozen and used experimentally.
Approximately 48 h after infection with 1 × 107 blood-stage of P. falciparum 3D7HLH/BRD, the mean parasitaemia was approximately 0.4%. huRBC NSG mice were orally treated with a single dose of compound and parasitaemia was monitored for 30 days by IVIS to acquire the bioluminescence signal (150 mg kg−1 of luciferin was intraperitoneally injected approximately 10 min before imaging).
In vivo P. falciparum transmission-stage assay
huRBC NSG mice (n = 2 per experimental group; female; 4–5-week-old; 18–20 g; Jackson Laboratory) were infected with blood-stage P. falciparum 3D7HLH/BRD for 2 weeks to allow the development of mature gametocytes. Subsequently the mice were treated with a single oral dose of BRD7929. Blood samples were collected for 11 days. For molecular detection of parasite stages, 40 µl of blood was obtained from control and treated mice. In brief, total RNA was isolated from blood samples using RNeasy Plus Kit with genomic DNA eliminator columns (Qiagen). First-strand cDNA synthesis was performed from extracted RNA using SuperScript III First-Strand Synthesis System (Life Technologies). Parasite stages were quantified using a stage-specific qRT–PCR assay as described previously33,69. Primers were designed to measure transcript levels of PF3D70501300 (ring stage parasites), PF3D7_1477700 (immature gametocytes) and PF3D7_1031000 (mature gametocytes). Primers for PF3D7_1120200 (P. falciparum UCE) transcript were used as a constitutively expressed parasite marker. The assay was performed using cDNA in a total reaction volume of 20 µl, containing primers for each gene at a final concentration of 250 nM. Amplification was performed on a Viia7 qRT–PCR machine (Life Technologies) using SYBR Green Master Mix (Applied Biosystems) with the following reaction conditions: 1 cycle × 10 min at 95 °C and 40 cycles × 1 s at 95 °C and 20 s at 60 °C. Each cDNA sample was run in triplicate and the mean Ct value was used for the analysis. Ct values obtained above the cut-off (negative control) for each marker were considered negative for the presence of specific transcripts. Blood samples from each mouse before parasite inoculation were also tested for ‘background noise’ using the same primer sets. No amplification was detected from any samples.
In vivo P. falciparum liver-stage assay
FRG knockout on C57BL/6 (human repopulated, > 70%) mice (huHep FRG knockout; n = 2 per experimental group; female; 5.5–6-month-old; 19–21 g; Yecuris) were inoculated intravenously with approximately 1 × 105 P. falciparum (NF54HT-GFP-luc) sporozoites and BRD7929 was administered as a single 10 mg kg−1 oral dose one day after inoculation31. Infection was monitored daily by IVIS. Daily engraftment of human erythrocytes (0.4 ml, O-positive, 50% haematocrit, 50% RPMI 1640 with 5% albumax) was initiated 5 days after inoculation. For qPCR analysis, blood samples (40 µl) were collected 7 days after inoculation. For molecular detection of the blood-stage parasite, 40 µl of blood was obtained from control and treated mice. In brief, total RNA was isolated from blood samples using RNeasy Plus Kit with genomic DNA eliminator columns (Qiagen). First-strand cDNA synthesis was performed from extracted RNA using SuperScript III First-Strand Synthesis System (Life Technologies). The presence of the blood-stage parasites was quantified using a highly stage-specific qRT–PCR assay as described previously33,70. Primers were designed to measure transcript levels of PF3D7_1120200 (P. falciparum UCE). The assay was performed using cDNA in a 20 µl total reaction volume containing primers for each gene at a final concentration of 250 nM. Amplification was performed on a Viia7 qRT–PCR machine (Life Technologies) using SYBR Green Master Mix (Applied Biosystems) and the reaction conditions are as follows: 1 cycle × 10 min at 95 °C and 40 cycles × 1 s at 95 °C and 20 s at 60 °C. Each cDNA sample was run in triplicate and the mean Ct value was used for the analysis. Ct values obtained above the cut-off (negative control) for each marker were considered negative for presence of specific transcripts. Blood samples from each mouse were also tested for background noise using the same primer sets before parasite inoculation. No amplification was detected from any samples.
Resistance propensity determination assay
In vitro cultures of P. falciparum Dd2, with the initial inocula ranging from 105 to 109 parasites, were maintained in complete medium supplemented with 20 nM of BRD7929 (EC90 against Dd2). Media was replaced with fresh compound added daily and cultures monitored for 60 days to identify propensity for recrudescent parasitaemia as described34. Atovaquone was used as a control (EC90 = 2 nM).
Solubility assay
Solubility was determined in PBS pH 7.4 with 1% DMSO. Each compound was prepared in triplicate at 100 µM in both 100% DMSO and PBS with 1% DMSO. Compounds were allowed to equilibrate at room temperature with a 750 r.p.m. vortex shake for 18 h. After equilibration, samples were analysed by UPLC–MS (Waters) with compounds detected by single-ion reaction detection on a single quadrupole mass spectrometer. The DMSO samples were used to create a two-point calibration curve to which the response in PBS was fit.
Plasma protein binding assay
Plasma protein binding was determined by equilibrium dialysis using the Rapid Equilibrium Dialysis (RED) device (Pierce Biotechnology) for both human and mouse plasma. Each compound was prepared in duplicate at 5 µM in plasma (0.95% acetonitrile, 0.05% DMSO) and added to one side of the membrane (200 µl) with PBS pH 7.4 added to the other side (350 µl). Compounds were incubated at 37 °C for 5 h with 350 r.p.m. orbital shaking. After incubation, samples were analysed by UPLC–MS (Waters) with compounds detected by SIR detection on a single quadrupole mass spectrometer.
hERG channel inhibition assay
The required potency to inhibit the hERG channel in expressed cell lines were evaluated using an automated patch-clamp system (QPatch-HTX).
Mouse pharmacokinetics assay
Pharmacokinetics of BRD3444 and BRD1095 were performed by Shanghai ChemPartner Co. Ltd., following single intravenous and oral administrations to female CD-1 mice. BRD3444 and BRD1095 were formulated in 70% PEG400 and 30% aqueous glucose (5% in H2O) for intravenous and oral dosing. Test compounds were dosed as a bolus solution intravenously at 0.6 mg kg−1 (dosing solution; 70% PEG400 and 30% aqueous glucose, 5% in H2O) or dosed orally by gavage as a solution at 1 mg kg−1 (dosing solution; 70% PEG400 and 30% aqueous glucose, 5% in H2O) to female CD-1 mice (n = 9 per dose route). Pharmacokinetic parameters of BRD7929 and BRD3316 were determined in CD-1 mice. BRD7929 and BRD3316 were formulated in 10% ethanol, 4% Tween, 86% saline for both intravenous and oral dosing. Pharmacokinetic parameters were estimated by non-compartmental model using WinNonlin 6.2. Pharmacokinetic parameters for BRD7929 and BRD3316 were estimated by a non-compartmental model using proprietary Eisai software. Pharmacokinetic parameters of BRD7539 and BRD9185 were determined in CD-1 mice. Compounds were formulated in 70% PEG300 and 30% (5% glucose in H2O) at 0.5 mg ml−1 for oral dosing, and 5% DMSO, 10% cremophor, and 85% H2O at 0.25 mg ml−1 for intravenous formulation. Pharmacokinetic parameters were estimated by non-compartmental model using WinNonlin 6.2. Pharmacokinetics of BRD7539 and BRD9185 were performed by WuXi AppTec. The protocol was approved by Eisai IACUC, 13-07, 13, 05, and 14-c-0027.
Metabolic stability assay
Compounds were evaluated in vitro to determine their metabolic stability in incubations containing liver microsomes or hepatocytes of mouse and human. In the presence of NADPH, liver microsomes (0.2 mg ml−1) from mouse (CD-1) and human were incubated with compounds (0.5 and 5 µM) for 0, 10 and 90 min. The depletion of compounds in the incubation mixtures, determined using liquid chromatography tandem mass spectromety LC–MS/MS, was used to estimate Km and Vmax values and determine half-lives for both mouse and human microsomes.
CYP inhibition assay
Compounds were evaluated in vitro for the potential inhibition of human cytochrome P450 (CYP) isoforms using human liver microsomes. Two concentrations (1 and 10 µM) of compound were incubated with pooled liver microsomes (0.2 mg ml−1) and a cocktail mixture of probe substrates for selective CYP isoform. The selective activities tested were CYP1A2-mediated phenacetin O-demethylation, CYP2C8-mediated rosiglitazone para-hydroxylation, CYP2C9-mediated tolbutamide 4′ -hydroxylation, CYP2C19-mediated (S)-mephenytoin 4′ -hydroxylation, CYP2D6-mediated (±)-bufuralol 1′ -hydroxylation and, CYP3A4/5-mediated midazolam 1′ -hydroxylation. The positive controls tested were α -naphthoflavone for CYP1A2, montelukast for CYP2C8, sulfaphenazole for CYP2C9, tranylcypromine for CYP2C19, quinidine for CYP2D6, and k etoconazole for CYP3A4/5. The samples were analysed by LC–MS/MS. IC50 values were estimated using nonlinear regression.
Time-dependent inactivation assay
The time-dependent inactivation potential of compounds were assessed in human liver microsomes for CYP2C9, CYP2D6, and CYP3A4/5 by determining KI and kinact values when appropriate. Two concentrations (6 and 30 µM) of compound were incubated in primary reaction mixtures containing phosphate buffer and 0.2 mg ml−1 human liver microsomes for 0, 5, and 30 min in a 37 °C water bath. The reactions were initiated by the addition of NADPH. Phosphate buffer was substituted for NADPH solution for control. At the respective times, 25 µl of primary incubation was diluted tenfold into pre- incubated secondary incubation mixture containing each CYP-selective probe substrate in order to assess residual activity. The second incubation time was 10 min. The probe substrates used for CYP1A, 2C9, CYP2C19, CYP2D6, and CYP3A4 were phenacetin (50 µM), tolbutamide (500 µM), (S)-mephenytoin (20 µM), bufuralol (50 µM), and midazolam (30 µM), respectively. The CYP time-dependent inhibitors used were furafyllin, tienilic acid, ticlopidine, paroxetin and troleandomycin for CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A, respectively, at two concentrations. The samples were analysed by LC–MS/MS.
Chemical synthesis and analytical data
Extended Data
Extended Data Figure 1. Three screening-hit series yield new compound scaffolds against known targets.

a–d, BRD0026 exhibits the same mode of action as NITD609 and showed moderate in vitro potency against asexual (EC50 = 0.346 µ M) and late-sexual (EC50 = 1.98 µM) blood stages of the parasites and exhibited reduced potency against P. falciparum NITD609R (EC50 = 1.77 µM), a transgenic strain carrying a point mutation in P. falciparum ATPase4 (ref. 9). P. falciparum ATPase4 is the presumed molecular target of NITD609 (ref. 9). a, b, Three of the eight possible stereoisomers (R,S,R; S,S,S; and R,S,S) of BRD0026 have activity. c, Initial characterization of BRD0026 showed good solubility in PBS and low cytotoxicity. d, Treatment with BRD0026 resulted in a rapid increase in the parasite cytosolic Na+ concentration, while artesunate- or mefloquine-treated parasites maintained a constant cytosolic Na+ concentration. This result suggests that parasites treated with BRD0026 are not able to counter the influx of Na+ by actively extruding the cation, similar to the proposed mechanism for NITD609 (data are mean ± s.d.; two biological and two technical replicates). e–h, BRD7539 targets and inhibits P. falciparum DHODH. BRD7539 showed excellent in vitro potency against liver-stages (EC50 = 0.015 µM) and asexual blood-stages (EC50 = 0.010 µM) of the parasite, conferring markedly reduced potency against PfscDHODH19. This strain heterologously expresses the cytosolic S. cerevisiae DHODH, which does not require ubiquinone as an electron acceptor. Thus, this transgenic strain is resistant to inhibitors of mitochondrial electron transport chain functions19. BRD7539 was tested against three different P. falciparum strains with mutations in mitochondrial genes targeted by other antimalarial agents: (i) TM90C6B strain, containing a point mutation in the quinol oxidase domain of P. falciparum cytochrome b (Qo site) and resistant to atovaquone15; (ii) a P. falciparum CYTbG33V mutant strain, selected against IDI5994 and containing a point mutation in the quinone reductase site of P. falciparum cytochrome b (Qi site)15; and (iii) a P. falciparum DHODHE182D mutant strain, selected against Genz-666136 and containing a point mutation in the P. falciparum DHODH gene73. BRD7539 exhibited an approximately 59-fold shift in potency against the P. falciparum DHODHE182D strain, whereas potency was unaffected in the TM90C6B and P. falciparum CYTbG33V strains. BRD7539 also inhibits recombinant P. falciparum DHODH in an in vitro biochemical assay (IC50 = 0.033 µ M) but not the human orthologue. Altogether, these results demonstrate that BRD7539 targets P. falciparum DHODH. e, f, Only two (S,S,S and R,S,S) of eight possible stereoisomers of BRD7539 showed activity. g. In vitro growth inhibition assays showed no change in activity in P. falciparum CYTbG33V and TM90C6B strains but exhibited a tenfold change in potency in P. falciparum DHODHE182D strain, indicating that BRD7539 targets P. falciparum DHODH but not P. falciparum cytochrome bc1h, BRD7539 inhibited recombinant P. falciparum DHODH in vitro with an IC50 of 33 nM; no inhibition of the human orthologues was observed (data are mean ± s.d. for two biological and two technical replicates). i–m, BRD73842 targets and inhibits P. falciparum PI4K. BRD73842 showed excellent in vitro activity against asexual (EC50 = 0.069 µM), late-sexual blood-stage (EC50 = 0.643 µM) and liver-stage (EC50 = 0.459 µM) parasites. i, j, The structure of BRD73842 indicates the required stereochemistry for activity (R stereoisomer). k, Initial characterization of BRD73842 showed good solubility and limited cytotoxicity. To gain insight into the mechanism of action of BRD73842, two resistant P. falciparum lines were evolved against BRD73842 from four independent cultures (a total of over 4 × 109 inocula, see Extended Data Fig. 2a). After more than 3 months of drug pressure, the EC50 values increased approximately 10- to 20-fold. Two clones were obtained from each culture. Sequence analyses revealed that all clones contain non-synonymous SNVs in PF3D7_0509800, the locus that encodes P. falciparum PI4K (Supplementary Table 3). l, To confirm that PI4K is the molecular target of BRD73842, the compound was assayed against purified recombinant P. vivax PI4K protein. BRD73842 selectively inhibits the kinase activity of P. vivax PI4K (IC50 = 21 nM), but not human PI4K. P. falciparum PI4K has been identified as the molecular target of two recently described antimalarial compounds, KAI407 (ref. 20) and MMV048 (ref. 21).(data are mean ± s.d.; two biological and two technical replicates). m, The biphasic dose–response curve is a signature of P. falciparum PI4K inhibitors (data are mean ± s.d.; three biological and three technical replicates). The EC50 values reported in this study are derived from the first transition of the dose–response curves (indicated by arrow).
Extended Data Figure 2. Resistance selection of BRD38427 and BRD1095.

a, Over 3 months of intermittent and increasing resistance selection pressure of BRD73842 starting at 150 nM (EC99.9) or 0.5 µM (10× EC50) yielded two cultures showing a 13- to 16-fold EC50 shift. Two clonal lines from each culture were developed and subjected to whole-genome sequencing. b, Over 3 months of intermittent pressure of BRD1095 at 60 nM (EC99.9) or 150 nM (10 ×EC50) yielded three cultures showing a 3- to 67-fold EC50 shift. Two clonal lines from each culture were developed and subjected to whole-genome sequencing.
Extended Data Figure 3. In vivo blood-stage efficacy study of BRD7929.

a, BRD7929 shows single-dose in vivo efficacy in a P. berghei model of malaria. CD-1 mice were inoculated intravenously with approximately 2 × 107 P. berghei (ANKA GFP-luc) blood-stage parasites intravenously 24 h before treatment and BRD7929 was administered as a single 50, 25, or 12.5 mg kg−1 dose orally at 0 h (n = 4 for each group, this study was conducted once). Infections were monitored using IVIS. A single 100 mg kg−1 dose of artesunate results in rapid suppression of parasites, but owing to its short half-life, the parasites re-emerge very quickly. A single 25 mg kg−1 dose of BRD7929 resulted in 100% cure of the infected animals. One in four animals treated with a single oral dose of 12.5 mg kg−1 showed recrudescence at 6 days after treatment, but all other animals administered with 12.5 mg kg−1 were also completely parasite-free for 30 days. To ensure that no viable parasites remained, approximately 100 µl of combined blood samples from the four animals treated with 25 mg kg−1 of BRD7929 was intravenously injected into two naive mice and parasitaemia was monitored for an additional 30 days. No parasites were detected, suggesting that BRD7929 achieved a sterile cure for P. berghei with a single oral dose of as low as 25 mg kg−1. The same colour scale is used for the all images; not all time-point images are shown here. b, Bioluminescent intensity was quantified from each mouse and plotted against time. The dotted horizontal line represents the mean bioluminescence intensity level obtained from all the animals before the parasite inoculation. c, BRD7929 shows single-dose in vivo efficacy in a P. falciparum huRBC NSG mouse blood-stage model. huRBC NSG mice were inoculated intravenously with approximately 1 × 107 P. falciparum 3D7HLH/BRD blood-stage parasites 48 h before treatment and BRD7929 was administered as a single 50, 25, 12.5 or 6.12 mg kg−1 dose orally at 0 h (n = 2 for each group, this study was conducted once). Infections were monitored using the IVIS. No recrudescence was observed at doses as low as a single 12.5 mg kg−1 of BRD7929 in the infected animals. To ensure that no viable parasites remained, approximately 350 µl of combined blood samples from the two animals treated with 12.5 mg kg−1 of BRD7929 was cultured in vitro and monitored for an additional 30 days. No parasites were detected, suggesting that BRD7929 achieved a sterile cure for P. falciparum 3D7HLH/BRD with a single oral dose as low as 12.5 mg kg−1 (see Fig. 4a). The same colour scale is used for the all images; not all time point images are shown. Images of mice treated with vehicle on days 11 and 20 are not shown, because the bioluminescent signal was too high to show in the same colour scale as other images.
Extended Data Figure 4. In vivo liver-stage efficacy study of BRD7929 in a mouse malaria model.

a, BRD7929 shows single-dose causal prophylaxis in a P. berghei liver-stage model. CD-1 mice were inoculated intravenously with approximately 1 × 105 freshly dissected P. berghei ANKA luc-GFP sporozoites freshly dissected from A. stephensi salivary glands and immediately treated with a single oral dose of BRD7929 (25, 5, 1 or 0.2 mg kg−1). Infections were monitored using IVIS; mice were monitored until day 30 to ensure complete cure. No recrudescence was observed at doses as low as a single 5 mg kg−1 of BRD7929 in the infected animals (n = 4 for each group, study conducted once). The same colour scale is used for the all images. Not all time point images are shown. b, Bioluminescent intensity was quantified from each mouse and plotted against time. c, BRD7929 shows single-dose causal prophylaxis in a P. berghei liver-stage model up to 3 days before infection and two days after infection. CD-1 mice were infected with P. berghei and infections were monitored as described in a. Single oral doses of BRD7929 (10 mg kg−1) were administered at days 5, 3, and 1 before infection (days −5, −3 and − 1), on day 0, and on days 1 and 2 after infection (n = 4 for each group, this study was conducted once). All dosing regimens except for the day − 5 dose offered complete protection from infection for 32 days, indicating that BRD7929 has potent causal prophylaxis activity. The same colour scale is used for all images. Not all time-point images are shown.
Extended Data Figure 5. In vivo liver-stage efficacy study of BRD7929 in a humanized mouse model.

a, BRD7929 shows single-dose in vivo efficacy in a P. falciparum huHep FRG-knockout mouse liver-stage model. huHep FRG knockout mice were inoculated intravenously with approximately 1 × 105 P. falciparum (NF54HT-GFP-luc) sporozoites and BRD7929 was administered as a single 10 mg kg−1 oral dose 1 day after inoculation (n = 2 for each group, this study was conducted once). Infections were monitored using IVIS. The same colour scale is used for all images. No increase in bioluminescence intensity level was observed from the mice treated with BRD7929 (see Fig. 4b). b, Blood samples were also collected from each mouse 7 days after inoculation (the first day of the blood stage) and analysed for the presence of the blood-stage transcripts PF3D7_0812600 (P. falciparum UCE) using qRT–PCR32 (two biological replicates for each group and three technical replicates for each biological sample). Each dot represents a technical replicate of a sample and each horizontal line represents a mean of technical replicates from each mouse. The presence of the blood-stage parasite specific transcripts was detected from the control (vehicle) mice, while no amplification of the marker was detected after 40 amplification cycles (Ct value = 40) from the mice treated with BRD7929. Primer efficiency and sensitivity of the primer pairs for P. falciparum UCE have a detection limit ranging between 10 and 100 transcript copies33. Approximately 110 µl of combined blood samples from the two treated animals was also cultured in vitro and monitored for an additional 30 days but viable parasites were not detected.
Extended Data Figure 6. In vivo transmission-stage efficacy study of BRD7929.

a, Oral doses of BRD7929 2 days before feeding mosquitoes upon infected mice resulted in complete blocking of transmission at 5 mg kg−1, and reduced transmission activity at 1.25 mg kg−1 and 0.31 mg kg−1 (n = 2 for each group, this study was conducted once). b, Mosquitoes fed on vehicle-treated mice showed heavy infection 1 week after feeding, while mosquitoes fed on treated mice showed no or very few oocysts in the midguts. Representative images are shown; scale bars, 100 µm. c, To confirm that BRD7929 eliminates mature gametocytes in the host circulation rather than killing gametes, zygotes or ookinetes in the mosquito midgut, CD-1 mice infected with P. berghei (parasitaemia between 11 to 19%) were first treated with BRD7929 (oral, 25 mg kg−1). Infected mice were then exposed to female A. stephensi mosquitoes for blood feeding 1, 4 or 10 days after the treatment. Blood samples were also obtained before the blood feedings to measure the plasma concentration of remaining BRD7929 (n = 2 for each group, this study was conducted once). No oocysts were found in midguts dissected from mosquitoes from all time points, whereas 896.5, 170.5 and 8.6 ng ml−1 of the compound remained in the circulation 1, 4 and 10 days after respectively treatment, respectively, suggesting that BRD7929 eliminated mature gametocytes in the mice. d–f. In vivo transmission-stage efficacy study of BRD7929 (humanized mouse model). huRBC NSG mice were infected with the blood-stage P. falciparum 3D7HLH/BRD for 2 weeks to allow the gametocytes to mature fully and were treated with a single oral dose of BRD7929 (12.5 mg kg−1). n = 2 for each group, this study was conducted once. Blood samples collected from vehicle- and BRD7929-treated mice were tested for the presence of gametocyte-specific transcripts using mature gametocyte marker (PF3D7_1438800; d) and immature gametocyte marker (PF3D7_1477700; e). PF3D7_1120200 (P. falciparum UCE), a constitutively expressed gene, was used as a positive control marker for parasite detection (f). Data are mean ± s.d.; three technical replicates for each biological sample.
Extended Data Figure 7. Safety and resistance propensity profiling of the bicyclic azetidine series.

a, Results of in vitro cytotoxicity, phototoxicity and CYP inhibition assays. * Phototoxicity was assessed using the NIH 3T3 neutral red assay at Cyprotec; †CYP1A, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A; ‡CYP1A, CYP2C9, CYP2D6, CYP3A. b, Histopathology analysis of mice treated with a high dose (100 mg kg−1) of BRD7929. CD-1 mice were orally treated with 100 mg kg−1 BRD7929 and organs were collected 10 days after treatment. No significant tissue damage was detected. Representative images are shown here. Scale bars, 200 µm. c, d, Measurement of the minimal inoculum for resistance of BRD7929. Cultures containing various numbers of inoculum (1 × 105–1 × 109) were exposed to a constant level of drug pressure (EC90). Parasites developed resistance to atovaquone at the lowest inoculum of 1 × 107 but not to BRD7929.
Extended Data Table 1.
In vitro potency of BRD3444, BRD7929 and BRD3316 against multiple parasite stages
| Species (strain) | Stage | EC50 (µM)
|
||
|---|---|---|---|---|
| BRD3444 | BRD7929 | BRD3316 | ||
| P. falciparum (Dd2) | Blood | 0.009 | 0.005 | 0.019 |
| P. falciparum (3D7HLH/BRD) | Blood | 0.009 | ||
| P. falciparum (3D7) | Gametocyte (IV-V) | 0.663 | 0.160 | |
| P. falciparum (NF54) | Gametocyte(ID/D)* | 0.270/< 10 | ||
| P. falciparum (NF54) | Gametocyte (E / L)† | 0.282/1.44 | ||
| P. falciparum (NF54) | Gamete formation (M / F)‡ | ~1.00/0.804 | ||
| P. falciparum (NF54) | Liver | 1.31 | 0.340 | |
| P. berghei (ANKA) | Liver | 0.140 | 0.162 | |
| P. cynomolgi (M) | Liver (SF/LF)¶ | 3.34/2.86 | 0.933/1.04 | |
Data indicate the results of a standard membrane-feeding assay71. Indirect (ID) exposure refers to parasites treated with varying drug concentrations for 24 h before mosquito feeding, while direct (D) refers to parasites treated with a single drug concentration (10 µM) immediately before blood feeding.
Activity against early- (E, stages I–III) and late- (L, stages IV–V) stage gametocytes was assessed according the protocol described previously55.
Activity against male (M) and female (F) stage-V gametocytes was assessed in a dual gamete formation assay as described previously71. This assay (a standard membrane-feeding assay) is designed to determine the ability of compounds to either kill the mature P. falciparum male and female gametocytes directly or damage them in such a way that they cannot undergo onward development and form gametes in the mosquito midgut.
Activity against P. cynomolgi in primary rhesus hepatocytes was performed as described previously72. This assay measures inhibition of both the small form (SF, hypnozoite-like) and large form (LF, schizont) of intrahepatic Plasmodium.
Extended Data Table 2.
Structure–activity relationship study of the bicyclic azetidine series
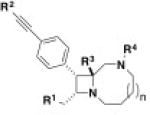
| |||||||
|---|---|---|---|---|---|---|---|
|
PfDd2 EC50(µM) |
PfcPheRS IC50(µM) |
R1 | R2 | R3 | R4 | n | |
| BRD8805 | 0.003 | 0.033 | -NMe2 | -Ph | -H |
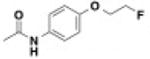
|
1 |
| BRD7929 | 0.009 | 0.023 | - | - | - |
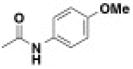
|
1 |
| BRD1095 | 0.010 | 0.046 | -NH2 | - | - | - | 1 |
| BRD3444 | 0.011 | 0.033 | -OH | - | - | - | 1 |
| BRD3316 | 0.022 | 0.029 | -O(CH2)2CO2H | - | - | - | 1 |
| BRD4716 | 0.024 | 0.086 | -NM/Pr | - | - | - | 1 |
| BRD2132 | 0.048 | 0.179 | -NMe(CH2)2F | - | - | - | 1 |
| BRD0185 | 0.087 | 0.097 | -OH | - | - | - | 2 |
| BRD8493 | 0.116 | 0.162 |
|
- | - | - | 1 |
| BRD6479 | 0.158 | 0.233 |
|
- | - | - | 1 |
| BRC4873 | 0.261 | 0.221 | -OH | -2-CNPh | - | - | 1 |
| BRD9599 | 0.850 | 0.366 | - | -Ph | - | - | 0 |
| BRD2936 | 1.87 | 29.4 | - | - | -CH2OH | - | 1 |
| 3RD5349 | 8.32 | 30.9 | - | - | -H |
|
1 |
| BRD5774 | 12.2 | 23.4 | - |
|
- |
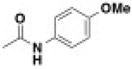
|
1 |
| BHD8260 | 19.5 | 34.6 | - | -Ph | - |
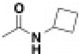
|
1 |
The structures of 16 bicyclic azetidine analogues with varying potency against asexual blood-stage parasites (Dd2), along with their corresponding inhibition of the P. falciparum PheRS activity in a biochemical assay. Aminoacylation inhibition activities were characterized using purified recombinant PheRS in which a range of inhibitor concentrations was used to determine IC50 values. The biochemically derived IC50 values correlate extremely well (r2 = 0.89) with the EC50 determined usingthe blood-stage parasite growth-inhibition assay (see Fig. 3d).
Extended Data Table 3.
In vitro and in vivo pharmacokinetic properties of the bicyclic azetidine series
| BRD3444* | BRD1095* | BRD7929* | BRD7929† | BRD3316* | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pf, Dd2 EC50 (nM) | 9 | 10 | 9 | 23 | |||||||
| PBS solubility (µM) | < 1 | 25 | 15 | 91 | |||||||
| Mouse Plasma protein binding (%) | 99.9 | 99.3 | 99.9 | ||||||||
| Mouse Clint (µL/min/mg) | 248 | < 20 | 21 | 38 | |||||||
| Human Clint (µL/min/mg) | 142 | < 20 | 31 | 34 | |||||||
| HepG2 CC50 (µM) | > 50 | 15.6 | 9 | > 50 | |||||||
| hERG IC50 (µM) | 5.2 | 5.1 | 2.1 | > 10 | |||||||
| Route (mg/kg) | IV (3) | PO (10) | IV (3) | PO (10) | IV (2.5) | IV (2.5)‡ | PO (10) | PO (3) | PO (9) | IV (3.2) | PO (13) |
|
|
|
|
|
|
|||||||
| Cmax (µM) | 0.6 | 0.6 | 0.54 | 0.21 | 0.6 | 6.8 | |||||
| Tmax (hr) | 0.5 | 4 | 8 | 12 | 12 | 1 | |||||
| T1/2 (hr) | 3.7 | 3.2 | 28.8 | N.C | 32 | 2.3 | 2.4 | ||||
| AUC0-t (µM*hr) | 1.2¶ | 4¶ | 7¶ | 11.7¶ | 3.5¶ | 9# | 11¶ | 6.4¶ | 19.7¶ | 13..2¶ | 33.5¶ |
| AUC0-inf (µM*hr) | 1.4 | 4 | 14.9 | 11.2 | 7.2 | 22.6 | 13.2 | 33.5 | |||
| MRT0-inf (hr) | 2.8 | 39.2 | 40.5 | 45 | 35.4 | 37.8 | 3.3 | 3.9 | |||
| Vss (L/kg) | 12 | 16 | 24 | 19 | 1.4 | ||||||
| F (%) | 86 | 50 | 79.5§ | 63 | |||||||
| CL (mL/min/kg) | 72 | 6.7 | 9.9 | 7.1 | 7.1 | ||||||
BRD3444 and BRD1095 were formulated in 70% PEG400 and 30% aqueous glucose (5% in H2O) for intravenous and oral dosing and pharmacokinetics were determined in CD-1 mice as described in Methods. Pharmacokinetic studies of BRD3444 and BRD1095 were performed by ChemPartner Co., Ltd and were estimated by a non-compartmental model using WinNonlin 6.2. BRD7929 and BRD3316 were formulated in 10% ethanol, 4% Tween, 86% saline for both intravenous and oral dosing. Pharmacokinetics in P. falciparum 3D7HLH/BRD-infected NSG mice was determined on dried blood spot samples from infected NSG mice using standard methods. Pharmacokinetics parameters for BRD7929 and BRD3316 were estimated by a non-compartmental model using proprietary Eisai software. Clint, intrinsic clearance; CL, clearance; MRT, mean resistance time; N.C, not calculated owing to insufficient data; Vss, steady-state volume of distribution.
Pharmacokinetic in CD-1 mice.
Pharmacokinetic in P. falciparum 3D7HLH/BRD-infected NSG mice.
Intravenously determined in a separate assay over 72 h to determine half-life.
t = 24 h.
t = 72 h.
Per cent value based on initial intravenous study at 24 h.
Supplementary Material
Acknowledgments
This work was supported in part by the Bill and Melinda Gates Foundation (grant OPP1032518 to S.L.S., grant OPP1054480 to E.A.W. and D.F.W., grant OPP1023607 to S.N.B.), the Global Health Innovative Technology Fund (grant G2014-107 to S.L.S.), Medicines for Malaria Venture and the Wellcome Trust (grant WT078285 to C.H.M.K.), a New Investigator and Open Operating Grant from Canadian Institute of Health Research (grant FRN 142393 to J.E.B.) and Medicines for Malaria Venture (grant 12-2400 to V.M.A.). S.L.S. is an Investigator at the Howard Hughes Medical Institute. Mi.M. was supported by a fellowship from the National Science Foundation (DGE1144152). The authors thank R. Elliott, K. Duncan, and O. Vandal as well as J. Burrows, J. Duffy, F. Escudié and colleagues for discussions and access to invaluable scientific and experimental resources; K. Emmith for assistance with data processing and management; I. Goldowitz for assistance with establishing a gametocyte assay; N. van der Werff for technical assistance with the P. cynomolgi assay; J. Kotz and B. Melillo for discussions and assistance with the manuscript; J. Pu, M. Leighty, B. Braibant, S. LeQuement and J. Beaudoin for assistance with compound synthesis; E. Garcia-Rivera for assistance with molecular modelling; A. Hakura for performing the Ames test; Broad Institute Comparative Medicine Platform and Facility for assistance with animal studies; and the Broad Institute Compound Management and analytical teams for assistance with compound access and characterization. We also acknowledge WuXi AppTec and ChemPartner Co., Ltd for in vitro and in vivo pharmacokinetics assays, and Cyprotek for the phototoxicity analysis. P. falciparum scDHODH transgenic strain was a gift from A. B. Vaidya, P. falciparum 3D7HLH strain from D. Fidock and P. falciparum NF54HT-GFP-luc from S. H. Kappe.
Footnotes
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions The author contributions are detailed in the Supplementary Information.
The whole-genome sequencing analysis data in this study have been deposited to the Short Read Archive; accession code SRP064988 and the x-ray crystal structure data for BRD7929 have been deposited to the Cambridge Structural Database: deposition number 1429949. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Wells TN. Discovering and developing new medicines for malaria control and elimination. Infect. Disord. Drug Targets. 2013;13:292–302. doi: 10.2174/1871526513666131129160156. [DOI] [PubMed] [Google Scholar]
- 2.Flannery EL, Chatterjee AK, Winzeler EA. Antimalarial drug discovery— approaches and progress towards new medicines. Nat. Rev. Microbiol. 2013;11:849–862. doi: 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ariey F, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campo B, Vandal O, Wesche DL, Burrows JN. Killing the hypnozoite— drug discovery approaches to prevent relapse in Plasmodium vivax. Pathog. Glob. Health. 2015;109:107–122. doi: 10.1179/2047773215Y.0000000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hameed PS, et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat. Commun. 2015;6:6715. doi: 10.1038/ncomms7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiménez-Díaz MB, et al. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rγnull mice engrafted with human erythrocytes. Antimicrob. Agents Chemother. 2009;53:4533–4536. doi: 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips MA, et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 2015;7:296ra111. doi: 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts L, Enserink, Malaria M. Did they really say … eradication? Science. 2007;318:1544–1545. doi: 10.1126/science.318.5856.1544. [DOI] [PubMed] [Google Scholar]
- 9.Rottmann M, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamo F-J, et al. Thousands of chemical starting points for antimalarial lead identifcation. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 11.Guiguemde WA, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meister S, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science. 2011;334:1372–1377. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Comer E, et al. Diversity-oriented synthesis-facilitated medicinal chemistry: toward the development of novel antimalarial agents. J. Med. Chem. 2014;57:8496–8502. doi: 10.1021/jm500994n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heidebrecht RW, Jr, et al. Diversity-oriented synthesis yields a novel lead for the treatment of malaria. ACS Med. Chem. Lett. 2012;3:112–117. doi: 10.1021/ml200244k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukens AK, et al. Diversity-oriented synthesis probe targets Plasmodium falciparum cytochrome b ubiquinone reduction site and synergizes with oxidation site inhibitors. J. Infect. Dis. 2015;211:1097–1103. doi: 10.1093/infdis/jiu565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dancík V, Seiler KP, Young DW, Schreiber SL, Clemons PA. Distinct biological network properties between the targets of natural products and disease genes. J. Am. Chem. Soc. 2010;132:9259–9261. doi: 10.1021/ja102798t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke MD, Schreiber SL. A planning strategy for diversity-oriented synthesis. Angew. Chem. Int. Edn Engl. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 18.Marcaurelle LA, et al. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: discovery of macrocyclic histone deacetylase inhibitors. J. Am. Chem. Soc. 2010;132:16962–16976. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specifc role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 20.McNamara CW, et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 2013;504:248–253. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghidelli-Disse S, et al. Identifcation of Plasmodium PI4 kinase as target of MMV390048 by chemoproteomics. Malar. J. 2014;13(suppl. 1):38. [Google Scholar]
- 22.Sharma A, Sharma A. Plasmodium falciparum mitochondria import tRNAs along with an active phenylalanyl-tRNA synthetase. Biochem. J. 2015;465:459–469. doi: 10.1042/BJ20140998. [DOI] [PubMed] [Google Scholar]
- 23.Pham JS, et al. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int. J. Parasitol. 2014;4:1–13. doi: 10.1016/j.ijpddr.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herman JD, et al. The cytoplasmic prolyl-tRNA synthetase of the malaria parasite is a dual-stage target of febrifugine and its analogs. Sci. Transl. Med. 2015;7:288ra77. doi: 10.1126/scitranslmed.aaa3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussain T, Yogavel M, Sharma A. Inhibition of protein synthesis and malaria parasite development by drug targeting of methionyl-tRNA synthetases. Antimicrob. Agents Chemother. 2015;59:1856–1867. doi: 10.1128/AAC.02220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Novoa EM, et al. Analogs of natural aminoacyl-tRNA synthetase inhibitors clear malaria in vivo. Proc. Natl Acad. Sci. USA. 2014;111:E5508–E5517. doi: 10.1073/pnas.1405994111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoepfner D, et al. Selective and specifc inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe. 2012;11:654–663. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Istvan ES, et al. Validation of isoleucine utilization targets in Plasmodium falciparum. Proc. Natl Acad. Sci. USA. 2011;108:1627–1632. doi: 10.1073/pnas.1011560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhatt TK, et al. A genomic glimpse of aminoacyl-tRNA synthetases in malaria parasite Plasmodium falciparum. BMC Genomics. 2009;10:644. doi: 10.1186/1471-2164-10-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaughan AM, et al. A transgenic Plasmodium falciparum NF54 strain that expresses GFP-luciferase throughout the parasite life cycle. Mol. Biochem. Parasitol. 2012;186:143–147. doi: 10.1016/j.molbiopara.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 31.Vaughan AM, et al. Plasmodium falciparum genetic crosses in a humanized mouse model. Nat. Methods. 2015;12:631–633. doi: 10.1038/nmeth.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang H-H, et al. Persistence of Plasmodium falciparum parasitemia after artemisinin combination therapy: evidence from a randomized trial in Uganda. Sci. Rep. 2016;6:26330. doi: 10.1038/srep26330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joice R, et al. Plasmodium falciparum transmission stages accumulate in the human bone marrow. Sci. Transl. Med. 2014;6:244re5. doi: 10.1126/scitranslmed.3008882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding XC, Ubben D, Wells TN. A framework for assessing the risk of resistance for anti-malarials in development. Malar. J. 2012;11:292. doi: 10.1186/1475-2875-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dandapani S, Marcaurelle LA. Grand challenge commentary: Accessing new chemical space for ‘undruggable’ targets. Nat. Chem. Biol. 2010;6:861–863. doi: 10.1038/nchembio.479. [DOI] [PubMed] [Google Scholar]
- 36.Lovering F, Bikker J, Humblet C. Escape from fatland: increasing saturatio as an approach to improving clinical success. J. Med. Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 37.Lowe JT, et al. Synthesis and profling of a diverse collection of azetidine-based scafolds for the development of CNS-focused lead-like libraries. J. Org. Chem. 2012;77:7187–7211. doi: 10.1021/jo300974j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baragaña B, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522:315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Younis Y, et al. 3,5-Diaryl-2-aminopyridines as a novel class of orally active antimalarials demonstrating single dose cure in mice and clinical candidate potential. J. Med. Chem. 2012;55:3479–3487. doi: 10.1021/jm3001373. [DOI] [PubMed] [Google Scholar]
- 40.Burrows JN, van Huijsduijnen RH, Möhrle JJ, Oeuvray C, Wells TNC. Designing the next generation of medicines for malaria control and eradication. Malar. J. 2013;12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng PY, Tang Y, Knosp WM, Stadler HS, Shaw JT. Synthesis of diverse lactam carboxamides leading to the discovery of a new transcription-factor inhibitor. Angew. Chem. Int. Edn Engl. 2007;46:5352–5355. doi: 10.1002/anie.200700762. [DOI] [PubMed] [Google Scholar]
- 42.Yu C, et al. High-throughput identifcation of genotype-specifc cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016;34:419–423. doi: 10.1038/nbt.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaccard P. Lois de distribution forale dans la zone alpine. Bull. Soc. Vaud. Sci. Nat. 1902;38:69–130. [Google Scholar]
- 44.Rogers D, Hahn M. Extended-connectivity fngerprints. J. Chem. Inf. Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 45.Pipeline Pilot v. 8.5.0.200. Accelrys Software Inc.; 2011. http://cscenter.pbsci.ucsc.edu:9944/ [Google Scholar]
- 46.R Core Development Team. R: A Language and Environment for Statistical Computing v. 3.0. R Foundation for Statistical Computing; Vienna, Austria: 2013. http://www.R-project.org/ [Google Scholar]
- 47.Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008;26:120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- 48.March S, et al. A microscale human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax. Cell Host Microbe. 2013;14:104–115. doi: 10.1016/j.chom.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.March S, et al. Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens. Nat. Protocols. 2015;10:2027–2053. doi: 10.1038/nprot.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–343. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 51.Ponnudurai T, et al. Infectivity of cultured Plasmodium falciparum gametocytes to mosquitoes. Parasitology. 1989;98:165–173. doi: 10.1017/s0031182000062065. [DOI] [PubMed] [Google Scholar]
- 52.Guguen-Guillouzo C, et al. High yield preparation of isolated human adult hepatocytes by enzymatic perfusion of the liver. Cell Biol. Int. Rep. 1982;6:625–628. doi: 10.1016/0309-1651(82)90187-4. [DOI] [PubMed] [Google Scholar]
- 53.Dembele L, et al. Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PLoS One. 2011;6:e18162. doi: 10.1371/journal.pone.0018162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mazier D, et al. Complete development of hepatic stages of Plasmodium falciparum in vitro. Science. 1985;227:440–442. doi: 10.1126/science.3880923. [DOI] [PubMed] [Google Scholar]
- 55.Dufy S, Avery VM. Identifcation of inhibitors of Plasmodium falciparum gametocyte development. Malar. J. 2013;12:408. doi: 10.1186/1475-2875-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stone WJR, et al. A scalable assessment of Plasmodium falciparum transmission in the standard membrane-feeding assay, using transgenic parasites expressing green fuorescent protein-luciferase. J. Infect. Dis. 2014;210:1456–1463. doi: 10.1093/infdis/jiu271. [DOI] [PubMed] [Google Scholar]
- 57.Manary MJ, et al. Identifcation of pathogen genomic variants through an integrated pipeline. BMC Bioinformatics. 2014;15:1–14. doi: 10.1186/1471-2105-15-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ross LS, et al. In vitro resistance selections for Plasmodium falciparum dihydroorotate dehydrogenase inhibitors give mutants with multiple point mutations in the drug-binding site and altered growth. J. Biol. Chem. 2014;289:17980–17995. doi: 10.1074/jbc.M114.558353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansen M, et al. Inhibitor binding in a class 2 dihydroorotate dehydrogenase causes variations in the membrane-associated N-terminal domain. Protein Sci. 2004;13:1031–1042. doi: 10.1110/ps.03533004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burke JE, et al. Structures of PI4KIIIβ complexes show simultaneous recruitment of Rab11 and its efectors. Science. 2014;344:1035–1038. doi: 10.1126/science.1253397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burke JE, Williams RL. Dynamic steps in receptor tyrosine kinase mediated activation of class IA phosphoinositide 3-kinases (PI3K) captured by H/D exchange (HDX-MS) Adv. Biol. Regul. 2013;53:97–110. doi: 10.1016/j.jbior.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 63.Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biasini M, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jacobson MP, et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 66.Sherlin LD, et al. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA. 2001;7:1671–1678. [PMC free article] [PubMed] [Google Scholar]
- 67.Cestari I, Stuart K. A spectrophotometric assay for quantitative measurement of aminoacyl-tRNA synthetase activity. J. Biomol. Screen. 2013;18:490–497. doi: 10.1177/1087057112465980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Angulo-Barturen I, et al. A murine model of falciparum-malaria by in vivo selection of competent strains in non-myelodepleted mice engrafted with human erythrocytes. PLoS One. 2008;3:e2252. doi: 10.1371/journal.pone.0002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ekland EH, Schneider J, Fidock DA. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J. 2011;25:3583–3593. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Joice R, et al. Inferring developmental stage composition from gene expression in human malaria. PLOS Comput. Biol. 2013;9:e1003392. doi: 10.1371/journal.pcbi.1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ruecker A, et al. A male and female gametocyte functional viability assay to identify biologically relevant malaria transmission-blocking drugs. Antimicrob. Agents Chemother. 2014;58:7292–7302. doi: 10.1128/AAC.03666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zeeman A-M, et al. KAI407, a potent non-8-aminoquinoline compound that kills Plasmodium cynomolgi early dormant liver stage parasites in vitro. Antimicrob. Agents Chemother. 2014;58:1586–1595. doi: 10.1128/AAC.01927-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lukens AK, et al. Harnessing evolutionary ftness in Plasmodium falciparum for drug discovery and suppressing resistance. Proc. Natl Acad. Sci. USA. 2014;111:799–804. doi: 10.1073/pnas.1320886110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.