

Abstract
Protein flexibility poses a major challenge in binding site identification. Several computational pocket detection methods that utilize small-molecule probes in molecular dynamics (MD) simulations have been developed to address this issue. Although they have proven hugely successful at reproducing experimental structural data, their ability to predict new binding sites that are yet to be identified and characterized has not been demonstrated. Here, we report the use of benzenes as probe molecules in ligand-mapping MD (LMMD) simulations to predict the existence of two novel binding sites on the surface of the oncoprotein MDM2. One of them was serendipitously confirmed by biophysical assays and X-ray crystallography to be important for the binding of a new family of hydrocarbon stapled peptides that were specifically designed to target the other putative site. These results highlight the predictive power of LMMD and suggest that predictions derived from LMMD simulations can serve as a reliable basis for the identification of novel ligand binding sites in structure-based drug design.
The identification and characterization of binding pockets is an important component of structure-based drug design. It is also often the first step in evaluating the druggability of a protein target.1,2 In recent decades, various computational algorithms and methods that rely on the use of static protein structures have been developed for rapid identification of binding pockets for ligand design.1 They are, however, severely limited by their dependence on available protein structures. Proteins are intrinsically flexible and frequently undergo conformational changes on ligand binding.3−6 A major concern is that cryptic binding pockets that are absent in the input structures and present themselves only in the presence of a suitable interacting ligand will be missed. This is often the case for hydrophobic pockets, which tend to remain occluded in polar solvents and open up only in the presence of less polar ligands.7
To address this issue, there have been recent efforts to develop molecular dynamics (MD)-based methods that incorporate small molecules into the protein’s solvent box for pocket detection.8−12 In these simulations, the probes interact dynamically with the protein surface, allowing for ligand-induced conformational changes. The use of hydrophobic probes is of particular interest because it reduces the solvent polarity, thus facilitating the opening and enlargement of hydrophobic pockets that may otherwise remain undetected in pure water simulations of the protein.7 Ligand-mapping MD (LMMD)13,14 is one of two probe-based MD simulation methods that employ hydrophobic probes for pocket detection. In contrast to the related site identification by ligand competitive saturation (SILCS) method,9 LMMD does not require the addition of artificial interligand repulsive energy terms because of the use of relatively low concentrations of hydrophobic probes to avoid ligand aggregation. LMMD simulations have been shown to be especially useful at revealing cryptic binding sites14 and were previously used to guide the design of a ligand to target a cryptic pocket.13 Recently, LMMD has also been established as a reliable method for the identification of hydrophobic peptide binding sites.15 To date, probe-based MD simulations have mostly been limited to the reproduction of known structural data. Unlike the non MD-based pocket detection methods,16 there have been no previous reports of the successful prediction of a previously unknown binding site by these simulations, although a recent study suggests that SILCS has the potential to propose alternative binding sites.17 A demonstration of the predictive power of probe-based MD simulations will provide confidence for and encourage their application in structure-based drug design projects. Here, we focus on the promising anticancer therapeutic target MDM2 as a prototypical example for the detection of novel ligand binding sites by LMMD.
The E3 ubiquitin ligase MDM2 is a potent inhibitor of the tumor suppressor protein p53,18 which plays an essential role in coordinating cellular responses, including cell cycle arrest, apoptosis, and senescence, to a variety of stress signals.19 MDM2 binds to the transactivation domain of p53 to block p53-mediated transactivation20 and targets it for ubiquitin-mediated proteolysis.21 It is overexpressed in many cancers and is thought to be one of the primary causes of p53 network inactivation in p53 wild-type (WT) tumors.22 Antagonists of the MDM2–p53 interaction can reactivate the p53 response, leading to cell cycle arrest and apoptosis in tumor cells.23,24 Several small-molecule inhibitors of the MDM2–p53 interaction have been developed, and some of them have reached clinical trials.25,26 These molecules mimic the three key binding residues (Phe19, Trp23, and Leu26) in the p53 transactivation domain, which binds as an amphipathic α-helix to a deep hydrophobic cleft in the N-terminal domain of MDM2.27
Besides small molecules, peptides derived from the transactivation domain of p53 have also been used to inhibit the MDM2–p53 interaction. Unlike small molecules, however, linear peptides are susceptible to proteolytic cleavage, lack a well-defined conformation prior to target engagement, and are poorly cell-permeable.28 These shortcomings can potentially be overcome by hydrocarbon stapling, in which two unnatural residues bearing olefin side chains of varying lengths are introduced into the α-helix of the peptide, followed by a ruthenium-catalyzed ring-closing metathesis reaction to form a covalent staple across one or two α-helical turns.29 Hydrocarbon stapling generally leads to enhanced helicity, protease resistance, and cell permeability.30,31 Three families of MDM2-binding hydrocarbon stapled peptides have been identified to date: one derived from the WT p53 sequence32 and two others from phage display studies.33,34 Two of these peptides have been cocrystallized with MDM2, revealing that the hydrocarbon staple also interacts with the protein surface, thus augmenting the hydrophobic interactions at the binding interface.35,36
In this Letter, we describe the use of LMMD to identify two new binding sites close to the consensus p53-binding cleft in MDM2. Subsequent biophysical and crystallographic studies showed that one of these sites is important for the binding of a novel set of hydrocarbon stapled peptides.
The crystal structure of human MDM2 in complex with the p53 transactivation domain peptide (PDB code 1YCR)27 was used as the initial structure for the LMMD simulations. As cryptic binding sites tend to be hydrophobic in nature,7 benzene was chosen as the mapping ligand to better explore such sites. An initial set of 10 independent 5 ns LMMD simulations with different starting benzene distributions was performed on apo MDM2 after removing p53 from the complex structure. A relatively low benzene concentration (0.2 M) was used to prevent phase separation and ligand aggregation on the protein surface, which may cause denaturation.37 A second set of LMMD simulations was performed on p53-bound MDM2 to determine whether the binding sites that are discrete from the p53-binding cleft could be reproduced in the presence of the peptide.
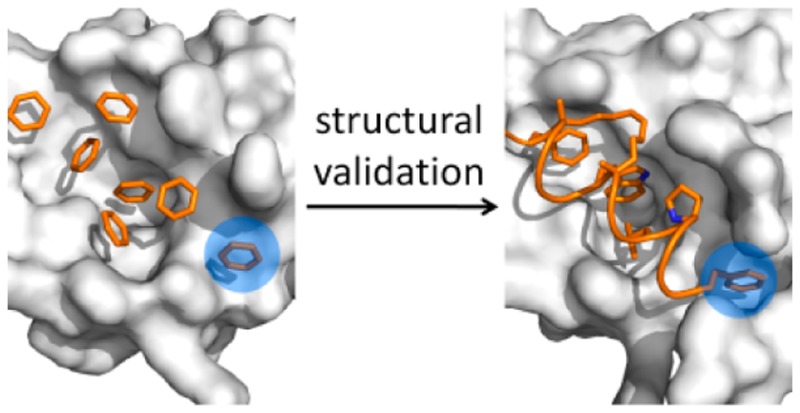
Analysis of the benzene occupancy maps generated from the apo and holo LMMD simulations revealed two putative discrete binding sites close to the p53-binding cleft, which have never been exploited by any known inhibitors of MDM2 and may be utilized to improve the affinity and selectivity of existing ligands. The first binding site lies between Tyr100 and Tyr104 and was mapped by the benzene probes in both apo and holo LMMD simulations. It was found to correspond to a crystal packing contact for a second nutlin molecule in the crystal structure of its complex with MDM2 (Figure 1A).38 This secondary nutlin interaction site is defined by seven residues: Val28, Met50, Tyr100, Ile103, Tyr104, Leu107, and Val109 (Figure 1B). These residues potentially constitute a cryptic binding pocket that is occluded by Tyr104 in the majority of MDM2 structures, including the one used for the LMMD simulations. There is experimental evidence to suggest that the region around this cryptic site may be relevant for nutlin binding at the p53-binding cleft;39 however, its ability to bind ligands has not been verified.
Figure 1.

Putative binding sites (blue circles) identified on the MDM2 surface (white). (A) Benzene occupancy maps (black mesh) overlaid on the structure of MDM2 complexed with nutlin-2 (PDB code 1RV1), showing detection of the second nutlin interaction site. The neighboring protomer is represented as white cartoon. (B) Residues forming the second nutlin interaction site. (C) Benzene occupancy maps overlaid on the structure of MDM2 complexed with p53 peptide (PDB code 1YCR), showing detection of the proximal P27 binding site. (D) Residues forming the proximal P27 binding site.
The second putative binding site was identified in the apo LMMD simulations within a region contiguous to where Pro27 of p53 binds (Figure 1C); however, it was not detected in the corresponding holo simulations because it was occluded by the C-terminal region of the p53 peptide. This “proximal P27 site” is essentially a relatively shallow hydrophobic basin constituted by Thr26, Met50, Lys51, Leu54, Tyr100, and Tyr104 (Figure 1D).
The proximity of the second nutlin interaction site to the p53-binding cleft raises the possibility of accessing it by extension of the p53 peptide. Suitable MDM2 structures, in which the identified cryptic pocket was present, were selected from the apo and holo LMMD simulations. The p53 peptide was modeled onto the apo MDM2 structure by extraction from the 1YCR crystal structure. Either a phenylalanine or a tyrosine residue was then appended to the peptide’s C-terminus, such that there was optimal overlap of the phenyl ring with the benzene molecule bound at the second nutlin interaction site (Figure 2A). The peptide in the holo MDM2 structure was similarly extended. We then replaced the WT p53 sequence in these extended peptides with that of a previously reported potent MDM2-binding and p53-activating stapled peptide, sMTide-02.33 The i, i + 7 staple used in sMTide-02, however, could not be transferred to these peptides because it would induce an additional helical turn that prevents the appended residue from accessing the second nutlin interaction site. Instead, a shorter i, i + 4 staple formed by two (R)-2-(4′-pentenyl)alanine residues (R5) was used to maintain the extended structure of the C-terminal residues. The R,R configuration of the hydrocarbon staple was preferred to the more common S,S configuration because it allows the staple to point toward and form intimate hydrophobic contacts with the MDM2 surface. These novel stapled peptides were termed YS-1 (TSFR5EYWR5LLPENF) and YS-2 (TSFR5EYWR5LLPENY), respectively.
Figure 2.
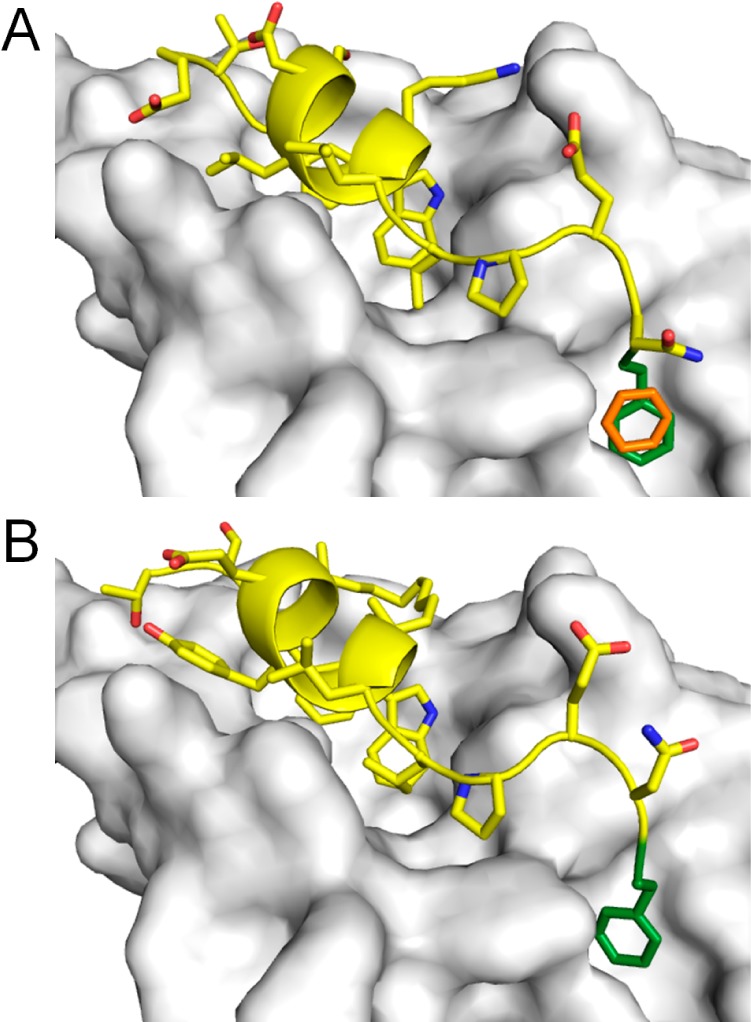
Design of extended stapled peptides. (A) A Phe residue (green) was appended to the C-terminus of the WT p53 peptide (PDB code 1YCR) such that it overlapped with the benzene molecule (orange) bound in the second nutlin interaction site. (B) Conformation of YS-1 after 50 ns of MD simulation.
MD simulations of the complexes of MDM2 with WT p53, sMTide-02, YS-1, and YS-2 were performed for 50 ns each to evaluate their stability. The structures of YS-1 and YS-2 did not deviate significantly from their initial minimized structures, with root-mean-square deviation (RMSD) values remaining below 1.6 Å (Figure S1). The Phe (Figure 2B) and Tyr extensions (not shown) remained bound at their targeted binding sites at the end of the simulations, indicating the stability of the predicted binding mode. Binding free energies of the various MDM2–peptide complexes were then estimated using the molecular mechanics/generalized Born surface area (MM/GBSA) method.40 The sMTide-02 peptide was predicted to have a higher binding affinity for MDM2 compared to WT p53 peptide (Table S5), in agreement with previous experimental results.33 Notably, the calculations suggest that both YS-1 and YS-2 are significantly more potent binders of MDM2 than sMTide-02 (Table S5). To validate the computational predictions, the stapled peptides were subsequently characterized in biophysical and X-ray crystallographic experiments.
Binding affinities of the stapled peptides for MDM2 were determined in competitive fluorescence polarization (FP) assays (Table 1). The designed stapled peptides exhibited approximately up to 5-fold improvement in binding potency relative to sMTide-02, in qualitative agreement with the computational predictions. Incorporation of a tyrosine instead of phenylalanine at the C-terminus of the stapled peptides made little difference to their binding affinities.
Table 1. Dissociation Constants (Kd) of Stapled Peptides As Determined by Competitive Fluorescence Polarization Titrations.
| peptide | sequence | Kd (nM) |
|---|---|---|
| sMTide-02 | Ac-17TSFR8EYWALLS527-NH2 | 34.4 ± 2.0 |
| YS-1 | Ac-17TSFR5EYWR5LLPENF30-NH2 | 9.9 ± 1.5 |
| YS-2 | Ac-17TSFR5EYWR5LLPENY30-NH2 | 7.4 ± 1.5 |
| YS-3 | Ac-17TSFR5EYWR5LLPEN29–NH2 | 36.1 ± 2.9 |
| YS-4 | Ac-17TSFR5EYWR5LLPENA30-NH2 | 27.7 ± 1.3 |
| YS-5 | Ac-17TSFR5EYWR5LLSENF30-NH2 | 11.6 ± 0.9 |
| YS-6 | Ac-17TSFR5EYWR5LLSENY30-NH2 | 8.8 ± 0.6 |
Crystal structures of the N-terminal domain of MDM2 in complex with YS-1 and YS-2 were solved to elucidate their binding modes. The two peptides bind to MDM2 very similarly, with only minor variations for some solvent-exposed residues (Figure 3A and Figure S3). Both stapled peptides adopt an α-helical structure with Phe19, Trp23, and Leu26 bound in their consensus pockets. The i, i + 4 hydrocarbon staple packs snugly against the MDM2 glycine shelf in a manner broadly similar to that seen for longer i, i + 7 staples of other MDM2-binding stapled peptides.35,36
Figure 3.
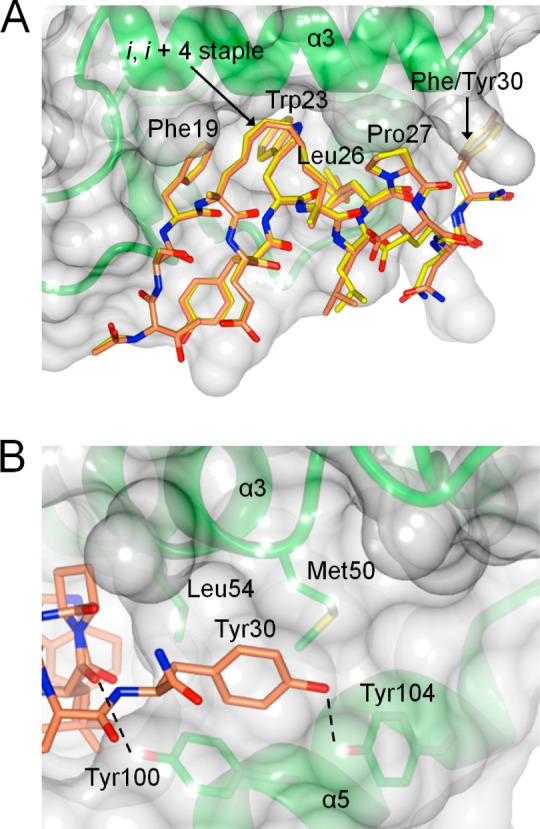
Crystal structures of MDM2 (green with translucent surface) bound to YS-1 and YS-2 (PDB codes 4UE1 and 4UD7, respectively). (A) MDM2 bound to YS-1 (yellow) with YS-2 (orange) superimposed. (B) Interactions of Tyr30 within the proximal P27 site, with hydrogen bonds represented as dashed lines.
Surprisingly, the last three residues of YS-1 and YS-2 do not adopt the linear conformation observed at the C-terminus of WT p53 when bound to MDM2; instead they extend the helical fold to create a kinked helix encompassing residues 19–30 (Figure 4A, residue numbers follow that in WT p53 sequence). While Pro27 acts as a helix breaker in the p53 peptide because of steric constraints and its inability to maintain the backbone hydrogen bonding network, it is unexpectedly incorporated into the helices of YS-1 and YS-2, allowing it to form close hydrophobic contacts with the MDM2 surface. It is likely that this is driven by the presence of the hydrocarbon staple, which has the ability to stabilize α-helices beyond the stapled α-helical turn.41 This results in the C-termini of the peptides packing into the cleft between helices α3 and α5 (Figure 3B), with Phe/Tyr30 bound at the proximal P27 site instead of the second nutlin binding site (Figure S5), contrary to the computational models. The presence of a proline in an α-helix is strongly linked to helix kinking,42 similar to what is observed here. A recent survey of the Protein Data Bank (PDB)43 reveals that this phenomenon is commonly found in long α-helices, occurring in 30% of membrane protein helices and 20% of length-matched (≥20 residues) globular protein helices.44 Tyr100 and Tyr104, which line the proximal P27 site, are both rotated toward the peptide, relative to the WT p53-MDM2 complex structure (Figure 3B). Tyr100 forms an edge-to-face interaction with Phe/Tyr30 and a hydrogen bond with the backbone carbonyl of Leu26 from the peptides, while rotation of Tyr104 allows it to form a hydrogen bond with the hydroxyl of Tyr30 and slightly occlude the putative second nutlin binding site.
Figure 4.

Comparison of YS-2 (orange) to (A) WT p53 (yellow, PDB code 1YCR), (B) M06 (magenta, PDB code 4UMN) and SAH-p53-8 (cyan, PDB code 3V3B).
The binding of the N-termini of YS-1 and YS-2 (residues 17–23) is very similar to that of two other MDM2-binding stapled peptides, SAH-p53-8 and M06 (Figure 4B). However, the different stapling strategies resulted in residues 24–28 being bound in a slightly different manner. The introduction of the second stapling point at residue 24 of YS-1 and YS-2 causes Leu25 and Leu26 to be displaced further along the helical axis. This results in a more expanded helical structure with a 1.7 Å displacement at the Cα position of Leu26 (compared to SAH-p53-8), which forces Leu26 into a position and conformation that is midway between that of SAH-p53-8/M06 and WT p53. Residues 27–29 then adopt a tighter helical conformation than in SAH-p53-8, bringing Asn29 of SAH-p53-8 and Gln29 in YS-1 and YS-2 closer together.
MD simulations show that this unexpected binding mode of the stapled peptides is stable (Figure S6). Although MM/GBSA analysis suggests that adoption of the crystallographic binding mode results in a loss of binding free energy compared to the predicted binding mode, YS-1 and YS-2 were predicted to be tighter MDM2 binders than sMTide-02 (Table S5), in agreement with the FP assay results. The unanticipated binding modes of the stapled peptides observed in the crystal structures highlight the challenges in characterizing multiple conformational states with their distinct thermodynamic profiles.45 Fine balances between enthalpic and entropic contributions often make it difficult to predict the behavior of the protein and its ligand upon complex formation.46
The effect of Phe/Tyr30 on the interaction with MDM2 was further investigated by synthesizing two control stapled peptides, one lacking the C-terminal aromatic residue (YS-3) and the other having alanine as the C-terminal residue (YS-4), and then evaluating their binding affinities in competitive FP binding assays. The dissociation constants (Kd) of YS-3 and YS-4 were 5-fold and 4-fold higher than that of YS-2, respectively. The slightly lower Kd of YS-4 compared to YS-3 could be due to the weak hydrophobic interaction of the methyl side chain of Ala30 with the proximal P27 site. Together with the crystal structures of MDM2 bound to YS-1 and YS-2, these results suggest that the proximal P27 site is a functional binding site that plays an important role in ligand binding to MDM2.
The secondary structures of unbound YS-1 and YS-2 were characterized by circular dichroism spectroscopy. Both peptides show very low overall α-helicity as compared to sMTide-02, based on their molar circular dichroism value at 222 nm (Figure S7). To increase the α-helicity of the designed peptides, we replaced Pro27 in YS-1 and YS-2 with a serine residue, generating the respective analogs YS-5 and YS-6. As predicted, YS-5 and YS-6 exhibited a marked increase in α-helicity relative to YS-1 and YS-2 (Figure S7). However, enhancing peptide α-helicity did not improve the binding affinity (Table 1). This could be due to the loss of favorable hydrophobic interactions of Pro27 with the MDM2 surface when it is replaced by serine, which negates the reduced entropic cost of increasing α-helicity.
In this proof-of-concept study, we have used a probe-based MD method called LMMD to detect novel binding sites on the surface of the anticancer protein target MDM2. Two adjacent putative binding sites on the N-terminal domain of MDM2 close to the p53 binding pocket were identified. Through biophysical binding assays and X-ray crystallography, we serendipitously confirmed the proximal P27 site as a functional ligand binding site by using hydrocarbon stapled peptides that were designed to target the downstream putative site. We measured a 5-fold improvement in binding affinity for stapled peptides that interact with the proximal P27 binding site (YS-2 versus YS-3, Table 1). Because of its proximity to the p53-binding cleft, it may be exploited to enhance the binding potencies of current MDM2 ligands. Further optimization of inhibitor structure, such as the use of alternative staple architectures47 and replacement of Pro27 with helix-stabilizing aliphatic residues that retain hydrophobic interactions with the MDM2 surface, may also result in binding affinity improvements greater than those observed here.
Here, we also present the first ever crystal structures of an i, i + 4 (R,R) stapled peptide bound to its protein target. The structures of these complexes reveal the rare and unusual incorporation of a proline residue into the α-helix of the stapled peptides. This implies that the hydrocarbon staple is able to exert a strong helix stabilization effect that extends to residues beyond the staple, including those with low α-helix propensities such as proline. We also report four modified stapled peptides with MDM2 binding affinities superior to that of the parent peptide sMTide-02, indicating their potential as templates for the development of a new family of potent p53-activating stapled peptides. Studies are now underway to evaluate their biological activities and develop variants with enhanced potencies.
We have used LMMD simulations to identify a novel binding site on the N-terminal domain of MDM2 close to the p53-binding cleft. The work described here is a significant step forward for the emerging field of probe-based MD because to the best of our knowledge, this is the first study to utilize such simulations to successfully predict and subsequently validate with definitive biophysical and structural data a previously unknown binding site. The results obtained corroborate the use of probe-based MD techniques to identify novel binding sites and inform structure-based drug design.
Acknowledgments
We are grateful for funding from the A*STAR Joint Council Office. J.R. and C.E.J. are supported by grants from Astex Pharmaceuticals Ltd and Cancer Research UK (grant reference C240/A15751). We thank Diamond Light Source for access to beamlines I04 and I04-1 and Dr. Arnaud Basle for data collection.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.6b01525.
Computational and experimental details; additional MD simulation details; supplementary tables and figures (PDF)
Accession Codes
The crystal structures of MDM2 in complex with YS-1 and YS-2 were deposited in the PDB under the accession codes 4UE1 and 4UD7, respectively.
Author Contributions
‡ Y.S.T., J.R., and C.J.B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Henrich S.; Salo-Ahen O. M. H.; Huang B.; Rippmann F.; Cruciani G.; Wade R. C. Computational Approaches to Identifying and Characterizing Protein Binding Sites for Ligand Design. J. Mol. Recognit. 2010, 23, 209–219. 10.1002/jmr.984. [DOI] [PubMed] [Google Scholar]
- Fauman E. B.; Rai B. K.; Huang E. S. Structure-Based Druggability Assessment—Identifying Suitable Targets for Small Molecule Therapeutics. Curr. Opin. Chem. Biol. 2011, 15, 463–468. 10.1016/j.cbpa.2011.05.020. [DOI] [PubMed] [Google Scholar]
- Betts M. J.; Sternberg M. J. E. An Analysis of Conformational Changes on Protein-Protein Association: Implications for Predictive Docking. Protein Eng., Des. Sel. 1999, 12, 271–283. 10.1093/protein/12.4.271. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.; Ultsch M. H.; de Vos A. M.; Wells J. A. Convergent Solutions to Binding at a Protein-Protein Interface. Science 2000, 287, 1279–1283. 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- Teague S. J. Implications of Protein Flexibility for Drug Discovery. Nat. Rev. Drug Discovery 2003, 2, 527–541. 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- Withers I. M.; Mazanetz M. P.; Wang H.; Fischer P. M.; Laughton C. A. Active Site Pressurization: A New Tool for Structure-Guided Drug Design and Other Studies of Protein Flexibility. J. Chem. Inf. Model. 2008, 48, 1448–1454. 10.1021/ci7004725. [DOI] [PubMed] [Google Scholar]
- Eyrisch S.; Helms V. What Induces Pocket Openings on Protein Surface Patches Involved in Protein-Protein Interactions?. J. Comput.-Aided Mol. Des. 2009, 23, 73–86. 10.1007/s10822-008-9239-y. [DOI] [PubMed] [Google Scholar]
- Seco J.; Luque F. J.; Barril X. Binding Site Detection and Druggability Index from First Principles. J. Med. Chem. 2009, 52, 2363–2371. 10.1021/jm801385d. [DOI] [PubMed] [Google Scholar]
- Guvench O.; MacKerell A. D. Jr. Computational Fragment-Based Binding Site Identification by Ligand Competitive Saturation. PLoS Comput. Biol. 2009, 5, e1000435. 10.1371/journal.pcbi.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A.; Nevins N.; Lakdawala A. S.; Bahar I. Druggability Assessment of Allosteric Proteins by Dynamics Simulations in the Presence of Probe Molecules. J. Chem. Theory Comput. 2012, 8, 2435–2447. 10.1021/ct300117j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexa K. W.; Carlson H. A. Improving Protocols for Protein Mapping through Proper Comparison to Crystallography Data. J. Chem. Inf. Model. 2013, 53, 391–402. 10.1021/ci300430v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O. Computational Functional Group Mapping for Drug Discovery. Drug Discovery Today 2016, 10.1016/j.drudis.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y. S.; Sledz P.; Lang S.; Stubbs C. J.; Spring D. R.; Abell C.; Best R. B. Using Ligand-Mapping Simulations to Design a Ligand Selectively Targeting a Cryptic Surface Pocket of Polo-Like Kinase 1. Angew. Chem., Int. Ed. 2012, 51, 10078–10081. 10.1002/anie.201205676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y. S.; Spring D. R.; Abell C.; Verma C. The Use of Chlorobenzene as a Probe Molecule in Molecular Dynamics Simulations. J. Chem. Inf. Model. 2014, 54, 1821–1827. 10.1021/ci500215x. [DOI] [PubMed] [Google Scholar]
- Tan Y. S.; Spring D. R.; Abell C.; Verma C. S. The Application of Ligand-Mapping Molecular Dynamics Simulations to the Rational Design of Peptidic Modulators of Protein–Protein Interactions. J. Chem. Theory Comput. 2015, 11, 3199–3210. 10.1021/ct5010577. [DOI] [PubMed] [Google Scholar]
- Wassman C. D.; Baronio R.; Demir O.; Wallentine B. D.; Chen C.-K.; Hall L. V.; Salehi F.; Lin D.-W.; Chung B. P.; Hatfield G. W.; Chamberlin A. R.; Luecke H.; Lathrop R. H.; Kaiser P.; Amaro R. E. Computational Identification of a Transiently Open L1/S3 Pocket for Reactivation of Mutant p53. Nat. Commun. 2013, 4, 1407. 10.1038/ncomms2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzl G. A.; Huang W.; Yu W.; Giardina B. J.; Zhou Y.; MacKerell A. D.; Wilks A.; Xue F. Iminoguanidines as Allosteric Inhibitors of the Iron-Regulated Heme Oxygenase (Hemo) of Pseudomonas aeruginosa. J. Med. Chem. 2016, 59, 6929–6942. 10.1021/acs.jmedchem.6b00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat M. H. G.; Jones S. N.; Vousden K. H. Regulation of p53 Stability by MDM2. Nature 1997, 387, 299–303. 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Vogelstein B.; Lane D.; Levine A. J. Surfing the p53 Network. Nature 2000, 408, 307–310. 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Momand J.; Zambetti G. P.; Olson D. C.; George D.; Levine A. J. The mdm-2 Oncogene Product Forms a Complex with the p53 Protein and Inhibits p53-Mediated Transactivation. Cell 1992, 69, 1237–1245. 10.1016/0092-8674(92)90644-R. [DOI] [PubMed] [Google Scholar]
- Haupt Y.; Maya R.; Kazaz A.; Oren M. MDM2 Promotes the Rapid Degradation of p53. Nature 1997, 387, 296–299. 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Rayburn E.; Zhang R. W.; He J.; Wang H. MDM2 and Human Malignancies: Expression, Clinical Pathology, Prognostic Markers, and Implications for Chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. 10.2174/1568009053332636. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Lain S.; Verma C. S.; Fersht A. R.; Lane D. P. Awakening Guardian Angels: Drugging the p53 Pathway. Nat. Rev. Cancer 2009, 9, 862–873. 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Ventura A.; Kirsch D. G.; McLaughlin M. E.; Tuveson D. A.; Grimm J.; Lintault L.; Newman J.; Reczek E. E.; Weissleder R.; Jacks T. Restoration of p53 Function Leads to Tumour Regression in Vivo. Nature 2007, 445, 661–665. 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Hoe K. K.; Verma C. S.; Lane D. P. Drugging the p53 Pathway: Understanding the Route to Clinical Efficacy. Nat. Rev. Drug Discovery 2014, 13, 217–236. 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Aguilar A.; Bernard D.; Wang S. Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction (MDM2 Inhibitors) in Clinical Trials for Cancer Treatment. J. Med. Chem. 2015, 58, 1038–1052. 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie P. H.; Gorina S.; Marechal V.; Elenbaas B.; Moreau J.; Levine A. J.; Pavletich N. P. Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- Vlieghe P.; Lisowski V.; Martinez J.; Khrestchatisky M. Synthetic Therapeutic Peptides: Science and Market. Drug Discovery Today 2010, 15, 40–56. 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Schafmeister C. E.; Po J.; Verdine G. L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. 10.1021/ja000563a. [DOI] [Google Scholar]
- Verdine G. L.; Hilinski G. J. All-Hydrocarbon Stapled Peptides as Synthetic Cell-Accessible Mini-Proteins. Drug Discovery Today: Technol. 2012, 9, e41–e47. 10.1016/j.ddtec.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Nevola L.; Giralt E. Modulating Protein-Protein Interactions: The Potential of Peptides. Chem. Commun. 2015, 51, 3302–3315. 10.1039/C4CC08565E. [DOI] [PubMed] [Google Scholar]
- Bernal F.; Tyler A. F.; Korsmeyer S. J.; Walensky L. D.; Verdine G. L. Reactivation of the p53 Tumor Suppressor Pathway by a Stapled p53 Peptide. J. Am. Chem. Soc. 2007, 129, 2456–2457. 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. J.; Quah S. T.; Jong J.; Goh A. M.; Chiam P. C.; Khoo K. H.; Choong M. L.; Lee M. A.; Yurlova L.; Zolghadr K.; Joseph T. L.; Verma C. S.; Lane D. P. Stapled Peptides with Improved Potency and Specificity That Activate p53. ACS Chem. Biol. 2013, 8, 506–512. 10.1021/cb3005148. [DOI] [PubMed] [Google Scholar]
- Chang Y. S.; Graves B.; Guerlavais V.; Tovar C.; Packman K.; To K.-H.; Olson K. A.; Kesavan K.; Gangurde P.; Mukherjee A.; Baker T.; Darlak K.; Elkin C.; Filipovic Z.; Qureshi F. Z.; Cai H.; Berry P.; Feyfant E.; Shi X. E.; Horstick J.; Annis D. A.; Manning A. M.; Fotouhi N.; Nash H.; Vassilev L. T.; Sawyer T. K. Stapled α-Helical Peptide Drug Development: A Potent Dual Inhibitor of MDM2 and Mdmx for p53-Dependent Cancer Therapy. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E3445–E3454. 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S.; Kutchukian P. S.; Verdine G. L.; Huber R.; Holak T. A.; Lee K. W.; Popowicz G. M. Structure of the Stapled p53 Peptide Bound to MDM2. J. Am. Chem. Soc. 2012, 134, 103–106. 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]
- Chee S. M. Q.; Wongsantichon J.; Tng Q. S.; Robinson R.; Joseph T. L.; Verma C.; Lane D. P.; Brown C. J.; Ghadessy F. J. Structure of a Stapled Peptide Antagonist Bound to Nutlin-Resistant MDM2. PLoS One 2014, 9, e104914. 10.1371/journal.pone.0104914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster T. J.; Mackerell A. D. Jr.; Guvench O. Balancing Target Flexibility and Target Denaturation in Computational Fragment-Based Inhibitor Discovery. J. Comput. Chem. 2012, 33, 1880–91. 10.1002/jcc.23026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L. T.; Vu B. T.; Graves B.; Carvajal D.; Podlaski F.; Filipovic Z.; Kong N.; Kammlott U.; Lukacs C.; Klein C.; Fotouhi N.; Liu E. A. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Hernychova L.; Man P.; Verma C.; Nicholson J.; Sharma C.-A.; Ruckova E.; Teo J. Y.; Ball K.; Vojtesek B.; Hupp T. R. Identification of a Second Nutlin-3 Responsive Interaction Site in the N-Terminal Domain of MDM2 Using Hydrogen/Deuterium Exchange Mass Spectrometry. Proteomics 2013, 13, 2512–2525. 10.1002/pmic.201300029. [DOI] [PubMed] [Google Scholar]
- Srinivasan J.; Cheatham T. E.; Cieplak P.; Kollman P. A.; Case D. A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate–DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. 10.1021/ja981844+. [DOI] [Google Scholar]
- Phillips C.; Roberts L. R.; Schade M.; Bazin R.; Bent A.; Davies N. L.; Moore R.; Pannifer A. D.; Pickford A. R.; Prior S. H.; Read C. M.; Scott A.; Brown D. G.; Xu B.; Irving S. L. Design and Structure of Stapled Peptides Binding to Estrogen Receptors. J. Am. Chem. Soc. 2011, 133, 9696–9699. 10.1021/ja202946k. [DOI] [PubMed] [Google Scholar]
- Barlow D.; Thornton J. Helix Geometry in Proteins. J. Mol. Biol. 1988, 201, 601–619. 10.1016/0022-2836(88)90641-9. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilman H. R.; Shi J.; Deane C. M. Helix Kinks Are Equally Prevalent in Soluble and Membrane Proteins. Proteins: Struct., Funct., Genet. 2014, 82, 1960–1970. 10.1002/prot.24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenley A. T.; Muddana H. S.; Gilson M. K. Entropy–Enthalpy Transduction Caused by Conformational Shifts Can Obscure the Forces Driving Protein–Ligand Binding. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 20006–20011. 10.1073/pnas.1213180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastidar S. G.; Lane D. P.; Verma C. S. Multiple Peptide Conformations Give Rise to Similar Binding Affinities: Molecular Simulations of p53-MDM2. J. Am. Chem. Soc. 2008, 130, 13514–13515. 10.1021/ja804289g. [DOI] [PubMed] [Google Scholar]
- Tan Y. S.; Lane D. P.; Verma C. S. Stapled Peptide Design: Principles and Roles of Computation. Drug Discovery Today 2016, 10.1016/j.drudis.2016.06.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.