

Abstract

We report the first enantioselective Rh-catalyzed Markovnikov hydroboration of unactivated terminal alkenes. Using a novel sp2–sp3 hybridized diboron reagent and water as a proton source, a broad range of alkenes undergo hydroboration to provide secondary boronic esters with high regio- and enantiocontrol.
The H. C. Brown asymmetric hydroboration of alkenes, reported in 1961, holds an important position in the history of asymmetric synthesis as the first example of a chemical transformation in which high enantioselectivity was conferred by a small molecule.1 Previously, high selectivity was the sole preserve of macromolecular structures, like enzymes. Although historically significant, and practical at the time, this method has been largely superseded by methods involving asymmetric metal catalysis, principally rhodium- and copper-catalyzed processes.2 Certain classes of alkenes, e.g., styrenes,3,4 electronically activated alkenes5−8 or alkenes bearing directing groups,9 give high regio- and enantiocontrol, but archetypal aliphatic terminal alkenes have not succumbed to asymmetric hydroboration through either metal-catalyzed or noncatalyzed processes. Although aliphatic terminal alkenes usually give the linear alkylboronic ester,10 recently disclosed copper-catalyzed hydroboration processes, employing bulky phosphine or NHC ligands, give the branched Markovnikov product.11,12 The process, however, has yet to be rendered asymmetric. A general catalytic asymmetric method for the generation of secondary alkylboronic esters from the abundant feedstock of aliphatic terminal alkenes13 remains an unmet challenge, which is now addressed in this paper.
We sought a process for adding a metal–boron bond across an alkene, placing a bulky metal at the less hindered terminal carbon atom. We were attracted to Nishiyama’s diboration reaction,14,15 which is postulated to proceed via a rhodium(III)–boryl species that undergoes insertion into the alkene, installing a secondary carbon–boron bond and generating a terminal rhodium(III)–alkyl species. The introduction of the second boron moiety then occurs through σ-bond metathesis (Scheme 1A). We surmised that if we could prevent the introduction of the second boron moiety, and protodemetalate instead, we could access the desired Markovnikov hydroboration products. However, introduction of a proton source was not sufficient to favor a protodemetalation pathway: addition of isopropyl alcohol to the standard Nishiyama diboration conditions with 4-phenyl-1-butene (2a) as substrate did not lead to the desired hydroboration product and diboration product 5 was formed exclusively (86%, 98:2 er, Table 1, Entry 1). We reasoned that if one of the boron centers of the diboron reagent was coordinatively saturated, the terminating σ-bond metathesis would be inhibited, allowing for the desired protodemetalation (Scheme 1B). For this, we envisioned using a mixed sp2–sp3 hybridized diboron species, in which one boron atom is bound to an amino diol ligand.16 The use of these “preactivated” diboron reagents would also enable the direct transfer of the sp2 boron to the rhodium(III) center, obviating the need for external base.
Scheme 1. Diverting Diboration into Hydroboration.
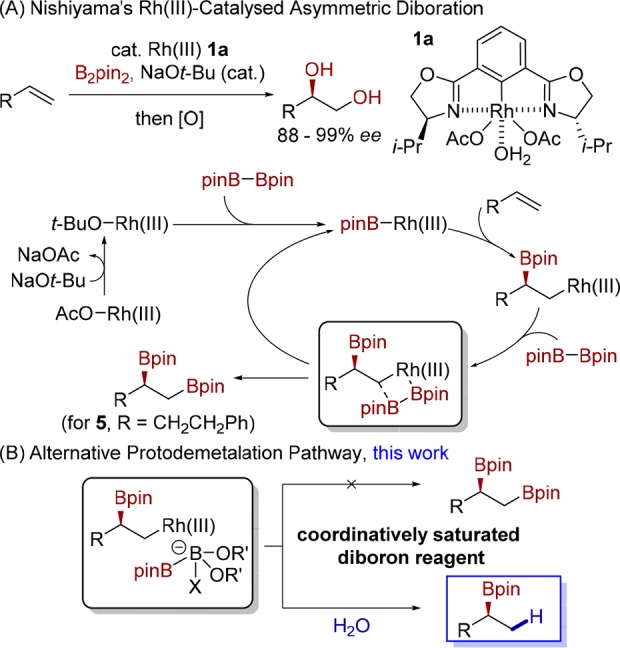
Table 1. Optimization of the Hydroboration Reactiona.

| entry | 4 | solvent | H source | yield (%)b | rrc | erd |
|---|---|---|---|---|---|---|
| 1e | 4c | THF | i-PrOH | 0 | N/A | N/A |
| 2 | 4a | THF | i-PrOH | 1 | 77:23 | N/A |
| 3 | 4b | THF | i-PrOH | 7 | 97:3 | 90:10 |
| 4 | 4b | THF | MeOH | 23 | 95:5 | 89:11 |
| 5 | 4b | THF | TFE | 1 | 94:6 | N/A |
| 6 | 4b | THF | t-BuOH | 2 | 95:5 | N/A |
| 7 | 4b | THF | H2O | 15 | 95:5 | 89:11 |
| 8 | 4b | THF | BzOH | 1 | 98:2 | N/A |
| 9 | 4b | DME | H2O | 33 | 97:3 | 89:11 |
| 10 | 4b | heptane | H2O | 48 (44) | 98:2 | 89:11 |
| 11f | 4b | heptane | H2O | 79 (76) | 98:2 | 90:10 |
| 12f | 4a | heptane | H2O | 3 | 80:20 | N/A |
| 13f,g | 4c | heptane | H2O | (38) | 95:5 | 90:10 |
| 14f | 4d | heptane | none | 63 | 25:75 | 77:23 |
Reactions conducted with 0.38 mmol 2a.
Yields determined by GC analysis by using biphenyl as an internal standard; yields of isolated product in parentheses.
The branched/linear ratio (rr) was determined by GC analysis of the crude reaction mixture.
Determined by chiral SFC analysis following oxidation of 3a.
5 mol % NaOt-Bu was used as an additive; reaction conducted at 60 °C; diboration product 5 was isolated in 86% yield, 98:2 er.
Reaction conditions: 5 mol % catalyst 1a, 1.5 equiv boron source 4, 6 equiv proton source, 1 M concentration, 40 °C, 16 h.
Diboration product 5 was isolated in 6% yield, 81:19 er.
We thus treated alkene 2a with Santos’s diboron reagent 4a(16b) in the presence of Nishiyama’s [(S,S)-Rh(Phebox-i-Pr)(OAc)2H2O] catalyst 1a and isopropyl alcohol (Table 1, Entry 2) and were pleased to observe trace amounts of hydroboration product 3a, regioselectivity favoring the desired internal isomer (77:23) and no evidence of the diboration product. The use of a novel N-methyl-capped derivative of the Santos reagent, diboron reagent 4b (see SI for synthesis and characterization), provided the hydroboration product in 7% yield, and crucially, with high regioselectivity (rr, 97:3) and high enantioselectivity (er, 90:10; Table 1, Entry 3). Having validated our hypothesis, we set about optimizing the reaction (see SI for full details). Competing isomerization and reduction processes were limiting the yield of hydroboration product 3a, as determined by 1H NMR spectroscopy and GC–MS (see SI). A screen of proton sources revealed that methanol and water both provided an increase in yield compared with isopropyl alcohol, while maintaining good levels of regio- and enantiocontrol (Table 1, Entries 3–8). Water led to less isomerization and was thus chosen for further optimization studies. A solvent screen established heptane as the optimum solvent, leading to product 3a being isolated in 44% yield, 98:2 rr and 89:11 er (Table 1, Entry 10). Changing the ligand failed to provide improved enantioselectivity. The conditions were further optimized through a design of experiment (DoE) study using diboron reagent 4b, water as the proton source, and heptane as the solvent. Under the optimized conditions, 3a was isolated in a 76% yield, 98:2 rr and 90:10 er (Entry 11). Exchange of diboron reagent 4b with either B2pin2 or 4a under these conditions led to diminished yields (Entries 12 and 13). When B2pin2 was used, diboration was also observed (5, 6%, 81:19 er) confirming that the mixed sp2–sp3 hybridized diboron reagent is essential for shutting down the diboration pathway. This experiment also suggests that the preactivation of diboron reagent 4b, which is absent in B2pin2, promotes the hydroboration reaction. Use of pinacolborane in the absence of a proton source provided the hydroboration product in 63% yield, but with inverted regioselectivity (25:75, Entry 14).
The reaction was tolerant to a wide range of functional groups, including alkyl chlorides (3c), unprotected alcohols (3d), ketones (3h), amides (3i and 3k) and esters (3e–f, 3j, 3m, 3p and 3s) (Scheme 2). We observed enhanced levels of enantioselectivity (up to 95:5 er) with substrates possessing a carbonyl moiety at the position δ to the alkene (3f–3j). Desymmetrization of diene 2p proceeded smoothly furnishing the β-benzoyloxy boronic ester 3p in 80:20 dr, where the major diastereomer was formed in excellent enantioselectivity (99:1 er). Vinyl arenes were also suitable substrates for the hydroboration reaction (3q–3t), including the sterically encumbered 2-vinylnaphthalene (2r), which gave the desired product 3r in good yield (76%) and with excellent regio- and enantiocontrol (96:4 rr and 98:2 er). 1,1-Disubstituted and conjugated dienes failed to undergo the hydroboration reaction under the optimized conditions. When enantioenriched homoallylic boronic ester 2u was subjected to the reaction conditions by using both enantiomers of catalyst 1a, matched/mismatched reactivity was observed (Scheme 3A). Employing catalyst (S,S)-1a provided anti-1,3-bis(boronic ester) 3u in moderate dr (84:16). Using catalyst (R,R)-1a, however, provided the corresponding syn diastereoisomer 3v in excellent dr (93:7). Showcasing the exceptional functional-group tolerance of this reaction and its compatibility with basic amines, hydroxyl groups, and heterocycles, quinine underwent hydroboration under slightly modified conditions to provide, after oxidation, the secondary alcohol 3w in 38% yield and 93:7 dr (Scheme 3B). Using catalyst (R,R)-1a revealed mismatched behavior providing alcohol 3x in 39% yield and 30:70 dr.
Scheme 2. Scope of the Hydroboration Reaction.

Reactions were conducted with 0.38 mmol of 2. Quoted yields are those of isolated product and are based on an average of values obtained from two experiments. Regioselectivity (rr) was determined by GC–MS analysis of the crude reaction mixtures, unless otherwise stated. Enantioselectivity (er) was determined by either chiral HPLC, SFC or GC analysis following oxidation (and in some cases further derivatization–see SI) of the isolated products (3), unless otherwise stated.
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by chiral SFC or HPLC analysis of the boronic ester (3).
Alcohol 3i was obtained following an oxidative work up using H2O2/NaOH.
Determined by LCMS analysis of the crude reaction mixture.
Scheme 3. Hydroboration of an Enantioenriched Substrate.

Determined by GCMS analysis of the crude reaction mixture.
Determined by 13C NMR analysis of isolated material.
Investigations were then undertaken to shed light on the mechanism. We established 13C kinetic isotope effects (KIEs) using the Singleton 13C natural abundance NMR technique (Scheme 4A; see SI).17 Thus, triisopropylbenzoate-protected homoallylic alcohol 2e, which gives minimal side products, was subjected to the standard reaction conditions on a 2 mmol scale over two runs. The reaction was stopped at 60% and 52% conversion and the starting material was reisolated from the reaction mixture and subjected to 13C NMR analysis. Negligible 12C/13C KIEs were observed at the methylene carbon atoms. Significant 12C/13C KIEs were, however, observed for both olefinic carbon atoms, suggesting that migratory insertion of the alkene into the rhodium–boron bond is, or occurs before, the first irreversible step of the catalytic cycle. We then conducted 1H/2H KIE experiments (Scheme 4B; see SI). The rates for the reactions conducted using both H2O and D2O were determined. No primary KIE was observed, suggesting that protodemetalation is not rate determining. We then conducted further experiments to obtain more information about the nature of the protodemetalation process. We ruled out that the rhodium–carbon bond was being reduced by some hydridic species by conducting the reaction with two different isotopomers of isopropyl alcohol-d1 (Scheme 4C). No deuterium incorporation was observed when isopropyl alcohol-2-d1 (6, a deuteride source) was employed. Deuterio-3a, however, was generated with 77% deuterium incorporation when isopropanol-OD 7 was used. Two possible mechanisms are consistent with these studies. Either the migratory insertion of the alkene into the rhodium–boron bond is the first irreversible step of the catalytic cycle, followed by rapid protodemetalation, or alternatively, reversible migratory insertion occurs before a rate-determining binding of a water molecule to the rhodium center, followed by rapid intramolecular protodemetalation. To differentiate between these two pathways, the reaction was conducted under the standard reaction conditions using a 1:1 mixture of H2O and D2O (Scheme 4D). The product was obtained with 83% hydrogen incorporation, ruling out a nonreversible binding event of the water molecule prior to protodemetalation. We thus propose the mechanism outlined in Scheme 4E. The rhodium(III) catalyst undergoes transmetalation with the diboron reagent, which is activated by internal nitrogen coordination. Following alkene coordination, migratory insertion of the alkene into the rhodium–boron bond generates a primary rhodium–alkyl species with the boron moiety installed at the secondary position. This is the first irreversible step of the catalytic cycle. Subsequent protodemetalation involving a water molecule then provides the secondary alkyl boronic ester product and regenerates the active rhodium(III) catalyst. We propose that the Markovnikov selectivity derives from regioselective migratory insertion and is controlled by a combination of steric and electronic factors. Isomerization and reduction side-products likely arise from the presence of small quantities of rhodium–hydride species (perhaps formed from competing β-hydride elimination of the β-boron rhodium–alkyl intermediate). The absolute configuration of the hydroboration products was the same as that observed in Nishiyama’s diboration reaction. The slightly lower levels of enantioselectivity observed in the hydroboration reaction compared to the diboration reaction most likely results from the different ligand attached to the rhodium center (hydroxide or acetate versus tert-butoxide in Nishiyama’s system).
Scheme 4. Mechanistic Studies.

In summary, we report the first asymmetric hydroboration of unactivated terminal alkenes. Secondary alkyl boronic esters are formed in good yields and high levels of enantioselectivity. Very high levels of regioselectivity are obtained without the need for directing groups or electronic biasing of the alkene substrates. Efforts to probe further the mechanism of this novel hydroboration method are currently underway.
Acknowledgments
We thank EPSRC (EP/I038071/1), H2020 ERC (670668) and Bristol University for financial support. J.R.S. and M.H. thank the Bristol Chemical Synthesis Centre for Doctoral Training, funded by the EPSRC (EP/G036764/1), AstraZeneca and the University of Bristol for a PhD studentship. We thank Professor Barry Carpenter for useful discussions and Dr. Hazel A. Sparkes for assistance with X-ray crystallography.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05149.
The authors declare no competing financial interest.
Supplementary Material
References
- a Brown H. C.; Zweifel G. J. Am. Chem. Soc. 1961, 83, 486. 10.1021/ja01463a055. [DOI] [Google Scholar]; b Brown H. C.; Singaram B. Acc. Chem. Res. 1988, 21, 287. 10.1021/ar00152a001. [DOI] [Google Scholar]; c Thomas S. P.; Aggarwal V. K. Angew. Chem., Int. Ed. 2009, 48, 1896. 10.1002/anie.200805604. [DOI] [PubMed] [Google Scholar]
- a Crudden C. M.; Edwards D. Eur. J. Org. Chem. 2003, 2003, 4695. 10.1002/ejoc.200300433. [DOI] [Google Scholar]; b Carroll A.-M.; O’Sullivan T. P.; Guiry P. J. Adv. Synth. Catal. 2005, 347, 609. 10.1002/adsc.200404232. [DOI] [Google Scholar]; c Schiffner J. A.; Müther K.; Oestreich M. Angew. Chem., Int. Ed. 2010, 49, 1194. 10.1002/anie.200906521. [DOI] [PubMed] [Google Scholar]
- For selected examples of rhodium-catalyzed asymmetric hydroboration of styrene derivatives:; a Hayashi T.; Matsumoto Y.; Ito Y. J. Am. Chem. Soc. 1989, 111, 3426. 10.1021/ja00191a049. [DOI] [Google Scholar]; b Brown J. M.; Hulmes D. I.; Layzell T. P.. J. Chem. Soc., Chem. Commun., 1993, 1673. 10.1039/c39930001673. [DOI] [Google Scholar]
- For copper-catalyzed asymmetric hydroboration of styrene derivatives:; a Noh D.; Chea H.; Ju J.; Yun J. Angew. Chem., Int. Ed. 2009, 48, 6062. 10.1002/anie.200902015. [DOI] [PubMed] [Google Scholar]; b Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 3160. 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovnikov hydroboration of perfluoroalkylethylenes to give racemic product:; a Brown H. C.; Chen G.-M.; Jennings M. P.; Ramachandran P. V. Angew. Chem., Int. Ed. 1999, 38, 2052.. [DOI] [PubMed] [Google Scholar]; b Ramachandran P. V.; Jennings M. P.; Brown H. C. Org. Lett. 1999, 1, 1399. 10.1021/ol9909583. [DOI] [Google Scholar]
- Xi Y.; Hartwig J. F. J. Am. Chem. Soc. 2016, 138, 6703. 10.1021/jacs.6b02478. [DOI] [PubMed] [Google Scholar]
- For selected examples of copper-catalyzed asymmetric β-borylation of α,β-unsaturated carbonyl compounds:; a Lee J.-E.; Yun J. Angew. Chem., Int. Ed. 2008, 47, 145. 10.1002/anie.200703699. [DOI] [PubMed] [Google Scholar]; b Chen I.-H.; Yin L.; Itano W.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2009, 131, 11664. 10.1021/ja9045839. [DOI] [PubMed] [Google Scholar]
- For examples of copper-catalyzed hydroboration of dienes and strained alkenes:; a Sasaki Y.; Zhong C.; Sawamura M.; Ito H. J. Am. Chem. Soc. 2010, 132, 1226. 10.1021/ja909640b. [DOI] [PubMed] [Google Scholar]; b Parra A.; Amenós L.; Guisán-Ceinos M.; López A.; García Ruano J. L.; Tortosa M. J. Am. Chem. Soc. 2014, 136, 15833. 10.1021/ja510419z. [DOI] [PubMed] [Google Scholar]; c Guisán-Ceinos M.; Parra A.; Martín-Heras V.; Tortosa M. Angew. Chem., Int. Ed. 2016, 55, 6969. 10.1002/anie.201601976. [DOI] [PubMed] [Google Scholar]
- For selected examples of directed rhodium-catalyzed asymmetric hydroboration:; a Rubina M.; Rubin M.; Gevorgyan V. J. Am. Chem. Soc. 2003, 125, 7198. 10.1021/ja034210y. [DOI] [PubMed] [Google Scholar]; b Smith S. M.; Thacker N. C.; Takacs J. M. J. Am. Chem. Soc. 2008, 130, 3734. 10.1021/ja710492q. [DOI] [PubMed] [Google Scholar]; c Smith S. M.; Takacs J. M. J. Am. Chem. Soc. 2010, 132, 1740. 10.1021/ja908257x. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shoba V. M.; Thacker N. C.; Bochat A. J.; Takacs J. M. Angew. Chem., Int. Ed. 2016, 55, 1465. 10.1002/anie.201509137. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chakrabarty S.; Takacs J. M. J. Am. Chem. Soc. 2017, 139, 6066. 10.1021/jacs.7b02324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of transition-metal-catalyzed anti-Markovnikov hydroboration of terminal alkenes: Rh:; a Männig D.; Nöth H. Angew. Chem., Int. Ed. Engl. 1985, 24, 878. 10.1002/anie.198508781. [DOI] [Google Scholar]; b Beletskaya I.; Pelter A. Tetrahedron 1997, 53, 4957. 10.1016/S0040-4020(97)00001-X. [DOI] [Google Scholar]; Ir:; c Yamamoto Y.; Fujikawa R.; Umemoto T.; Miyaura N. Tetrahedron 2004, 60, 10695. 10.1016/j.tet.2004.09.014. [DOI] [Google Scholar]; Co:; d Obligacion J. V.; Chirik P. J. J. Am. Chem. Soc. 2013, 135, 19107. 10.1021/ja4108148. [DOI] [PubMed] [Google Scholar]; e Zhang L.; Zuo Z.; Leng X.; Huang Z. Angew. Chem., Int. Ed. 2014, 53, 2696. 10.1002/anie.201310096. [DOI] [PubMed] [Google Scholar]; Fe:; f Zhang L.; Peng D.; Leng X.; Huang Z. Angew. Chem., Int. Ed. 2013, 52, 3676. 10.1002/anie.201210347. [DOI] [PubMed] [Google Scholar]; g Obligacion J. V.; Chirik P. J. Org. Lett. 2013, 15, 2680. 10.1021/ol400990u. [DOI] [PubMed] [Google Scholar]; h Greenhalgh M. D.; Thomas S. P. Chem. Commun. 2013, 49, 11230. 10.1039/c3cc46727a. [DOI] [PubMed] [Google Scholar]; Cu:; i Kubota K.; Yamamoto E.; Ito H. J. Am. Chem. Soc. 2013, 135, 2635. 10.1021/ja3104582. [DOI] [PubMed] [Google Scholar]
- Ligand-controlled copper-catalysed hydroboration of terminal alkynes to provide α-vinylboronates:Jang H.; Zhugralin A. R.; Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 7859. 10.1021/ja2007643. [DOI] [PubMed] [Google Scholar]
- Copper-catalysed Markovnikov hydroboration of terminal alkenes:; a Sakae R.; Hirano K.; Miura M. J. Am. Chem. Soc. 2015, 137, 6460. 10.1021/jacs.5b02775. [DOI] [PubMed] [Google Scholar]; b Su W.; Gong T.-J.; Lu X.; Xu M.-Y.; Yu C.-G.; Xu Z.-Y.; Yu H.-Z.; Xiao B.; Fu Y. Angew. Chem., Int. Ed. 2015, 54, 12957. 10.1002/anie.201506713. [DOI] [PubMed] [Google Scholar]; c Iwamoto H.; Kubota K.; Ito H. Chem. Commun. 2016, 52, 5916. 10.1039/C6CC00782A. [DOI] [PubMed] [Google Scholar]; d Kerchner H. A.; Montgomery J. Org. Lett. 2016, 18, 5760. 10.1021/acs.orglett.6b03090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on the enantioselective functionalization of terminal alkenes:Coombs J. R.; Morken J. P. Angew. Chem., Int. Ed. 2016, 55, 2636. 10.1002/anie.201507151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toribatake K.; Nishiyama H. Angew. Chem., Int. Ed. 2013, 52, 11011. 10.1002/anie.201305181. [DOI] [PubMed] [Google Scholar]
- For alternative examples of asymmetric transition-metal-catalyzed alkene diboration reactions: Rh:; a Morgan J. B.; Miller S. P.; Morken J. P. J. Am. Chem. Soc. 2003, 125, 8702. 10.1021/ja035851w. [DOI] [PubMed] [Google Scholar]; b Trudeau S.; Morgan J. B.; Shrestha M.; Morken J. P. J. Org. Chem. 2005, 70, 9538. 10.1021/jo051651m. [DOI] [PubMed] [Google Scholar]; Pt:; c Kliman L. T.; Mlynarski S. N.; Morken J. P. J. Am. Chem. Soc. 2009, 131, 13210. 10.1021/ja9047762. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Coombs J. R.; Haeffner F.; Kliman L. T.; Morken J. P. J. Am. Chem. Soc. 2013, 135, 11222. 10.1021/ja4041016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Neeve E. C.; Geier S. J.; Mkhalid I. A. I.; Westcott S. A.; Marder T. B. Chem. Rev. 2016, 116, 9091. 10.1021/acs.chemrev.6b00193. [DOI] [PubMed] [Google Scholar]; b Gao M.; Thorpe S. B.; Santos W. L. Org. Lett. 2009, 11, 3478. 10.1021/ol901359n. [DOI] [PubMed] [Google Scholar]
- Singleton D. A.; Thomas A. A. J. Am. Chem. Soc. 1995, 117, 9357. 10.1021/ja00141a030. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.