

Abstract
Objective:
To describe the phenotypes, treatment response, and outcome of IgLON5 autoimmunity.
Methods:
Archived serum and CSF specimens from 367 patients known to harbor unclassified antibodies which stained neural synapses diffusely (mimicking amphiphysin-IgG) were reevaluated by indirect immunofluorescence assay (IFA) using a composite of mouse tissues and recombinant IgLON5-transfected cell-based assay (CBA, Euroimmun).
Results:
Available specimens (serum, 25; CSF, 9) from 26/367 patients (7%) had identical IFA appearance and robust IgLON5 CBA positivity. Clinical information was available for 20/26 patients; 13 were women. Median disease-onset age was 62 years (range, 46–75 years). Most patients had insidious onset and progression of neurological symptoms affecting movement and sleep predominantly. Sleep disorders were sleep-disordered breathing (11) and parasomnias (3). Brainstem disorders were gait instability (14), dysphagia (10), abnormal eye movements (7), respiratory dysfunction (6), ataxia (5), craniocervical dystonia (3), and dysarthria (3). Findings compatible with hyperexcitability included myoclonus (3), cramps (3), fasciculations (2), and exaggerated startle (2). Neuropsychiatric disorders included cognitive dysfunction (6), psychiatric symptoms (5), and seizures (1). Dysautonomia, in 9, affected bladder function (7), gastrointestinal motility (3), thermoregulation (3), and orthostatic tolerance (1). Just 2 patients had coexisting autoimmune disease. Brain MRI findings were nonspecific and CSF was noninflammatory in all tested. Seven of 9 immunotherapy-treated patients improved: 6 of those 7 were stable at last follow-up. Three untreated patients died. Each IgLON5-IgG subclass (1–4) was readily detectable in ≥80% of specimens using CBA.
Conclusions:
IgLON5-IgG is diagnostic of a potentially treatable neurological disorder, where autoimmune clues are otherwise lacking.
Since 2004, several autoimmune CNS disorders have been described, each characterized serologically by an antibody targeting the extracellular domain of a neural autoantigen.1 Pertinent examples include aquaporin-4, NMDA receptor (NR1 subunit), and glycine receptor.2–4 Subacute onset and rapid symptom progression, and responses to immune therapies are typical for these disorders, although not universal.5 Autoimmunity targeting the extracellular domain of an immunoglobulin-like cell adhesion molecule, IgLON5, is a reported exception to this general rule.6–8
IgLON5 is an adhesion molecule widely expressed in the CNS, but whose precise function is poorly understood. IgLON5 autoimmunity has been characterized as a progressive CNS disorder of insidious onset with prominent sleep and movement abnormalities. Progression to death due to respiratory failure is common.6 Reported neuropathologic findings of “tauopathy,” which unifies a number of disorders, such as progressive supranuclear palsy (PSP), and corticobasal degeneration, have also reinforced the concept of IgLON5 autoimmunity having a poor prognosis.9 Characteristic neuropathologic findings include a lack of inflammatory infiltrates, neuronal loss, gliosis, and neuronal accumulation of hyperphosphorylated tau protein (both 3-repeat and 4-repeat [3R + 4R] isoforms) found predominantly in the hypothalamus and the tegmental brainstem nuclei.9
In this article, we report our Mayo Clinic experience of IgLON5 autoimmunity in 20 patients, including neurological improvements, in response to immune therapies.
METHODS
Standard protocol approvals, registrations, and patient consents.
The Mayo Clinic Institutional Review Board approved this study (no. 08-006647).
Serological testing.
Specimens (serum and CSF) were evaluated by indirect immunofluorescence assay (IFA) on a composite substrate of mouse hippocampus, cerebral cortex, cerebellum, basal ganglia, thalamus, kidney, and stomach. Four-micrometer frozen cryosections were fixed, blocked, and incubated with patient serum or CSF for 40 minutes and then with appropriate secondary antibodies. IgLON5 specificity was confirmed by cell-based assay (CBA) on human embryonic kidney 293 cells that were transfected with the IgLON5 complementary DNA, fixed with 1% formalin and stored at 4°C (Euroimmun AG, Lubeck, Germany). The sera (1:10 dilution) or CSF (neat) was incubated with the transfected cells. The cells were then washed and exposed to fluorescein isothiocyanate–conjugated goat antihuman IgG (Southern Biotech, Birmingham, AL). For IgG subclass testing, antihuman secondary antibodies specific for IgG1 (Thermo Fisher, Waltham, MA), IgG2 (Southern Biotech), IgG3 (Invitrogen, Carlsbad, CA and The Binding Site, Birmingham, AL), and IgG4 (Southern Biotech) were used.
Patients.
We evaluated 367 archived specimens (serum, 334; CSF, 33) from patients referred to the Mayo Clinic Neuroimmunology Laboratory for evaluation of neurological autoimmunity (1997–2016), for whom tissue-based immunofluorescence assay (IFA) screening had demonstrated a pattern of diffuse neural synaptic (neuropil) staining resembling (but not meeting criteria for) amphiphysin-IgG (which is similar to that reported for IgLON5-IgG).6 Specimens were retested by IFA and by CBA for IgLON5-IgG.
Sera and CSF from 96 patients diagnosed at Mayo Clinic with the following disorders that might mimic IgLON5 autoimmunity were also evaluated by IFA and CBA for IgLON5-IgG: PSP, 50; multiple system atrophy (MSA), 40; progressive age-related tauopathy, 3; and glutamic acid decarboxylase 65 (GAD65) kDa isoform neurological autoimmunity mimicking PSP, 3.
Clinical evaluation.
Of the 26 patients determined to be IgLON5-IgG positive by IFA and CBA, clinical information was available in 20. Fifteen had a Mayo Clinic medical record that was reviewed extensively; comprehensive neurological evaluations were available for all (including history, physical examination findings, laboratory results, imaging data, and electrophysiologic testing). Patients from elsewhere (5) had data limited primarily to the neurological phenotype available and were not included in our analysis of sleep data. Among Mayo Clinic patients, polysomnography was available for review in 3 patients permitting detailed review of video-PSG findings, and portable overnight oximetry was available for 3 patients (including 1 patient who did not have polysomnography). Sleep diagnoses or reports of sleep symptoms were available only from medical records for an additional 10 patients, made prior to Mayo Clinic evaluation.
RESULTS
Serological results.
All 26 patients had the IgLON5-IgG characteristic pattern of staining by indirect IFA, and positivity was confirmed for all specimens by IgLON5 CBA. In the laboratory, 19 patients were identified retrospectively, 7 prospectively after initiation of the study (March 2016–January 2017), approximately 1 per month. Two of those 7 were evaluated in person by AM. The synaptic pattern of immunoreactivity was more intense in the cerebellum (granular layer more than molecular layer), midbrain, and thalamus than in the hippocampus and cerebral cortex (figure 1). The renal glomeruli and the smooth muscle of the stomach (but not myenteric nervous tissue) were also reactive with patient IgG in all cases. Unlike smooth muscle antibody, preabsorption of serum with bovine liver powder did not abrogate the smooth muscle staining by IgLON5-IgG. The median IgLON5-IgG antibody value in serum was 1:3,840 (range 1:480–1:15,360; normal value ≤120). All 4 IgG subclasses were detected using CBA in most patients tested (20/24): IgG1, 23/24; IgG2, 20/24; IgG3, 23/24; and IgG4, 22/24. Fluorescence was more intense for IgG1 and IgG4 than IgG2 and or IgG3 (figure 2). Just a single patient had 1 IgLON5-IgG subclass detected (IgG4, patient 15). Two patients had coexisting antibodies detected in serum (leucine-rich glioma-inactivated 1 antibody, 1 and GAD65 antibody, 1). None of the 96 control patients (degenerative disease or degenerative disease mimics) were IgLON5 antibody positive by IFA or CBA.
Figure 1. IgLON5 antibody tissue-based indirect immunofluorescence assay.
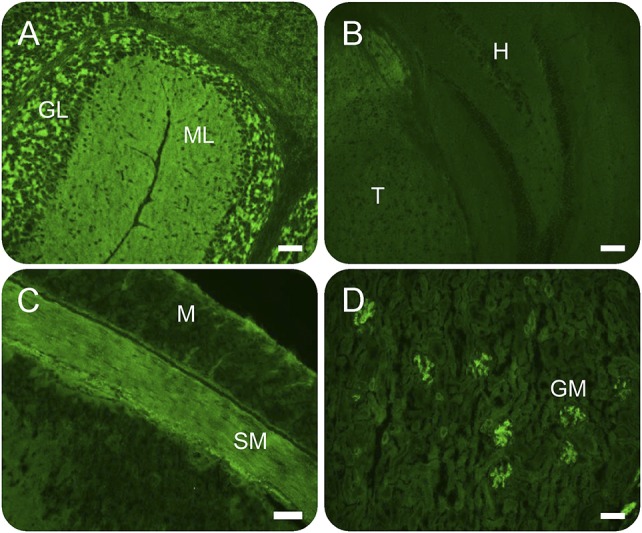
Immunoreactivity of patient serum IgLON5-IgG is more prominent in the cerebellum (A) than in the hippocampus (B). IgLON5-IgG immunoreactivity is present also in intestinal smooth muscle (C) and renal glomeruli (D). GL = granular layer of cerebellum; GM = renal glomeruli; H = hippocampus; M = intestinal mucosa; ML = molecular layer of cerebellum; SM = intestinal smooth muscle layer; T = thalamus. Scale bar: 0.5 mm.
Figure 2. IgLON5 antibody cell-based assay and IgG subtypes.

IgLON5-IgG subclasses were characterized in patient serum. Goat antihuman IgGs (reactive with all IgG [A] or 1 IgG subclass [C–F]) were used as secondary antibodies. IgG reactivity was more prominent for IgG1 (C) and IgG4 (F) than for IgG2 (D) or IgG3 (E). Patient IgG was nonreactive with mock-transfected cells (B). Scale bar: 0.5 mm.
Summary of demographic and clinical findings.
Thirteen of the 26 patients were women (50%); all were residing in the United States. All 14 patients where information regarding race was available were white. The median age of symptom onset was 62 years (range, 46–75 years). Clinical information was available for 20 patients. For those 20 patients, serum was available for testing in 19 and CSF in 8; all specimens were positive for IgLON5-IgG by both IFA and CBA. Symptoms evolved in an insidious and progressive manner in 15 patients (over several months to years) and subacutely in 5 patients (less than 1 month). The median duration of the disease from the onset of symptoms to last follow-up was 30 months (range, 2–156 months).
Clinical details are summarized in table 1. The predominant neurological manifestations of IgLON5 autoimmunity were brainstem disorders (dysphagia, dysarthria, eye movement disorders, and gait instability), sleep disorders (including sleep-disordered breathing and parasomnias), neuropsychiatric disorders (cognitive impairment and psychiatric symptoms), dysautonomia, and hyperexcitability (myoclonus, cramps, and exaggerated startle).
Table 1.
Demographic and clinical data for 20 IgLON5 autoimmune patients

Clinical course.
Sleep disorders.
Sleep disorders or sleep symptoms were reported in 12 of 15 patients where that history was available (75%, table 1), 5 of whom were known to have had sleep symptoms first. Of note, all 3 patients evaluated prospectively after the IgLON5 antibody serologic finding presented with sleep disorders. Obstructive sleep apnea (OSA) was diagnosed in 11 patients (although only 2 had this diagnosis confirmed by polysomnography in the course of their evaluations at Mayo Clinic, neither of these patients had audible stridor throughout sleep nor during wakefulness, and there was no report of recorded sleep or waking stridor nor history consistent with stridor provided by patients or their families in the remaining patients). Five patients had sleep apnea prior to the onset of neurological symptoms without temporal association to other neurological symptoms. A further 6 patients had the diagnosis of sleep apnea performed at the time of initial neurological evaluation or subsequently. Of 4 patients with sleep apnea discovered prospectively with known apnea severity, 3 were severe (Apnea-Hypopnea Index [AHI] 60, 47, and 38/hr), and 1 was moderate (AHI 15.4/hr), and all patients with severe OSA were treated successfully with nasal continuous positive airway pressure therapy, whereas the patient with moderate OSA was advised to obtain sleep medicine follow-up following a diagnostic study but was lost to further follow-up.
Three patients had parasomnias diagnosed following video-PSG review at our institution (by EKS). One was reported to have dream enactment behavior but had no recorded REM during polysomnogram. The second was reported to have dream enactment and had REM sleep without atonia. The third had an NREM parasomnia disorder of arousal (confusional arousals from N2 sleep during polysomnography).
One patient had stereotyped sleep-related complex motor behaviors diagnostic of nocturnal frontal lobe epilepsy (documented electrographic seizures corresponding to the clinical phenomena [shouting, followed by right arm jerking, and then stiffening] in the epilepsy monitoring unit).
Neurological disorders.
Fourteen of 20 (70%) patients had gait instability accompanied by ataxia, 5; chorea, 2; parkinsonism, 5 (akinetic, rigid phenotype in all); or limb and axial stiffness, 1. Falls were frequent in 5 patients, 3 of whom were diagnosed with a neurodegenerative disorder by movement disorders subspecialists at our institution (PSP, 1; MSA, 1; or synucleinopathy not otherwise specified, 1). The patient diagnosed with PSP presented with early falls and was found to have axial more than appendicular rigidity and symmetric bradykinesia but did not have vertical gaze palsy. None of the other patients had isolated downgaze palsy or both up and downgaze paralyses typical of the PSP phenotype (Richardson syndrome).
Other brainstem symptoms comprised dysphagia, 10; respiratory dysfunction, 6 (3 of whom developed laryngospasm or respiratory failure requiring tracheostomy and 1 also had stridor); pulmonary aspiration, 3; diplopia, 5; and dysarthria, 3. Hyperkinetic movement disorders, present in 7, were myoclonus, 3; craniocervical dystonia, 3; tremor, 2; myorhythmia, 1; chorea, 1; and choreoathetosis, 1. Patient 1 had an akinetic rigid syndrome with supranuclear gaze palsy and orolingual myorhythmia. Whipple disease was considered but excluded. Patient 20 (the video at Neurology.org/nn) was initially thought to have a functional disorder because of behavioral abnormalities in addition to a movement disorder but was later recognized to have a frontal-subcortical syndrome with chorea.
Neuropsychiatric disorders affecting 8 patients (40%) included memory loss, 5; delirium, 4; hallucinations, 3; depression, 2; and executive dysfunction, 1. One patient had frontal lobe seizures.
Findings consistent with central or peripheral nervous system hyperexcitability were noted in 7 patients (35%). Patient 14 had hyperekplexia (paroxysmal whole-body spasms in response to startle) and was diagnosed with a stiff-person syndrome spectrum disorder. Seven patients had upper motor neuron signs: hyperreflexia, 8, extensor plantar responses, 3; and spasticity, 1.
Nine patients (45%) had autonomic dysfunction affecting bladder, 7; gastrointestinal motility (diarrhea or constipation), 3; sweating (anhidrosis), 3; and orthostatic hypotension, 1. Four patients had evidence of peripheral neuropathy. One had mononeuritis multiplex and sural nerve biopsy-proven small vessel vasculitis, and the other 3 had EMG evidence supporting peripheral neuropathy (axonal sensorimotor, 2 and motor demyelinating, 1).
Other diagnoses.
One patient had adenocarcinoma of the breast detected prospectively. An additional 2 patients had a history of cancer (prostate adenocarcinoma, 1 [detected 5 years prior to the onset of neurological symptoms] and remote history of non-Hodgkin lymphoma, 1). Two patients reported a history of autoimmune disease (one each of Hashimoto thyroiditis and recurrent iritis).
CSF studies.
CSF testing was largely unremarkable. Five patients had a mild elevation of protein concentration (median value, 57 mg/dL: range, 49–106 mg/dL [normal, 0–35 mg/dL]). None had elevations in white blood cells (more than 5/µL), IgG indices (>0.85), IgG synthesis rates (>12 mg/24 hours), or CSF-exclusive oligoclonal band numbers (≥4).
Imaging and electrophysiology.
Brain MRIs were also largely unremarkable: normal in 10 patients and demonstrated one or more nonspecific abnormalities in 10: leukoariotic changes, 6; mild-to-moderate cerebral atrophy, 6; and cerebellar atrophy, 2 (both had ataxia). No parenchymal spinal cord abnormalities were noted in any of 13 patients tested. EEG was abnormal in 1 patient with frontal lobe seizures. EMG revealed abnormal findings in 3 patients: axonal sensorimotor peripheral neuropathy, 1; cramps and fasciculations, 1; and mononeuritis multiplex, 1. EMG study using surface electrodes in the Movement Disorders Electrophysiology Laboratory study demonstrated cortical myoclonus in 1 patient and exaggerated exteroceptive response in another (patient 14 with stiff-person syndrome).
Treatment and outcomes.
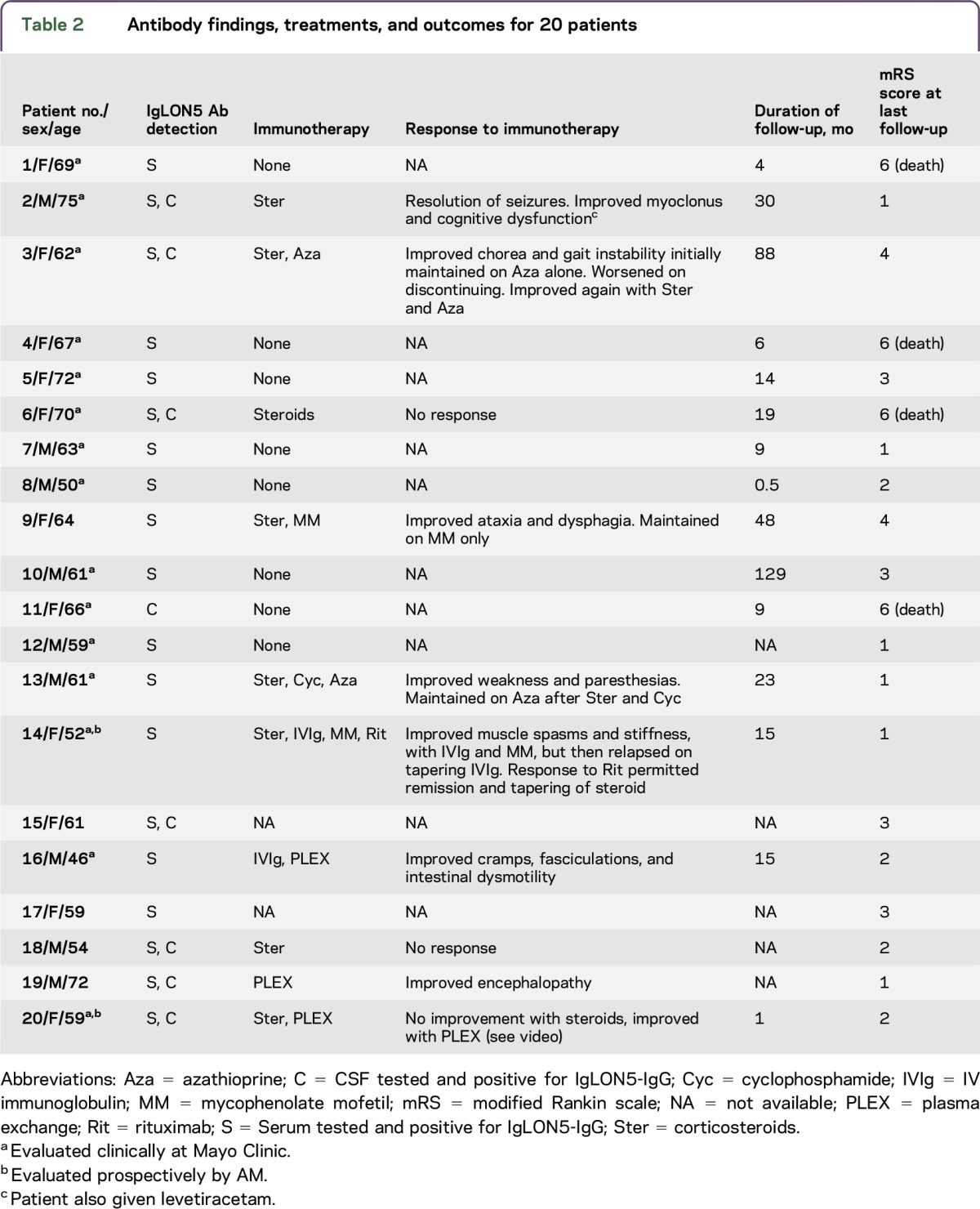
Ten patients received one or more immunotherapies and had follow-up available (table 2). Improvements were encountered in 5 of 7 patients who received steroids, all 3 who received plasma exchange, both who received IVIg, both who received azathioprine, 1 of 2 who received mycophenolate mofetil, and 1 who received rituximab. The improvements resulted in sustained near full recovery, 2; sustained partial improvements, 5; and initial improvement with subsequent worsening, 1. Two patients had no response to immunotherapy, one of whom eventually died (table 2). Four patients in total were reported to have died, 3 had not received immunotherapy. The median modified Rankin score at last follow-up was 2.5 (range, 1–6); median duration from evaluation to last follow-up was 15 months (range, 0.5–129 months).
Table 2.
Antibody findings, treatments, and outcomes for 20 patients
DISCUSSION
IgLON5 antibody is a rare finding in our laboratory (1 per month), which evaluates approximately 150,000 specimens per annum. The clinical course of patients encountered was usually insidious in onset and progression. Neurological disorders tended to be multifocal, although consistent with prior reports, findings of brainstem encephalopathy (affecting primarily movement, gait, and balance) and sleep were most common.6–8,10–13 Sleep disorders were frequent, but the clinical spectrum seemed more heterogenous than previous reports of patients presenting to sleep clinics. We encountered both mild and severe sleep disorders, possibly due to our screening process (for antibody rather than for clinical phenotype). It is possible that sleep disorders may have been overlooked in some patients (polysomnography was absent in all but 3) or worsened after the patients left Mayo Clinic (duration of follow-up was just 15 months). Notably, all 3 patients evaluated prospectively by the coauthors had sleep disorders.
Generic clues to an autoimmune diagnosis (subacute presentation, history of autoimmune disease, an inflammatory CSF, or inflammatory-appearing brain imaging) were infrequent, and thus, antibody testing of serum or CSF for IgLON5-IgG was critical. At this early stage, it would seem that serum and CSF have equal diagnostic sensitivity, although in general, when screening for all these rare autoimmune encephalitides in aggregate, it is recommended to test both serum and CSF.14
We encountered diverse movement disorders, including parkinsonism, ataxia, dystonia, chorea, and myoclonus.6,8,12 Patient 14, with stiff-person syndrome, did not have GAD65 antibody or glycine receptor antibody detected, but responded to immunotherapy nonetheless.15 Although not phenotypically classic, in some rare instances, IgLON5 autoimmunity may clinically resemble a tauopathy, such as PSP, or a synucleinopathy, such as MSA. The PSP-typical phenotypic vertical eye movement abnormalities were absent in the case initially diagnosed as such in our series. Furthermore, the pathology of IgLON5 autoimmunity more resembles that encountered in primary age-related tauopathy (3R + 4R isoforms) rather than PSP (4R tau isoforms only).16,17 Patient 1 had myorhythmia and supranuclear gaze palsy, resembling what has been reported for CNS Whipple disease.18
A paraneoplastic cause of IgLON5 autoimmunity appears to be rare, although patients were not systematically evaluated for the detected cancers (adenocarcinomas) during their evaluation at Mayo Clinic. At this early stage, screening broadly, one time, for age, sex, and other risk factor appropriate neoplasms among IgLON5-IgG–positive patients seems appropriate, given the track record for most autoimmune neurological disorders having some degree of oncological significance.1
We observed a range of responses to treatments used among IgLON5 autoimmune cases and a range of neurological prognoses. The improvements we report are in contrast to the original report of this disorder and the largest series to date (22 cases) where robust immunotherapy responses were rare and death occurred in 60%.6,13 Occasional individual cases of immunotherapy responsiveness have been reported.11,12 However, sudden death, despite initial responses to immune treatments, has also been reported.11
Similar to what has been reported previously, death was common,6 although in our patients, those who had not received immune therapies faired worst. Although the finding of tau deposition in the brain intuitively could lead one to conclude that IgLON5 autoimmunity is a secondary event in an otherwise neurodegenerative disorder, other data point to IgLON5-IgG having pathogenic effects. In vitro treatment of hippocampal neurons with IgG1 (but not IgG4) from affected patients has been shown to cause internalization of IgLON5 clusters from the neuronal cell surface.19 All IgLON5-IgG subclasses (1–4) were detectable in most of our patients, although IgG1 and IgG4 were most intense by indirect immunofluorescent CBA. Our findings essentially confirm that an initial report that demonstrated, quantitatively, IglON5-IgG is predominantly represented in IgG1 and IgG4 subclasses.19 The lack of inflammatory findings in CSF in our patients, and at autopsy in prior reports, may support IgG1-mediated IgLON5 internalization, without complement activation, as a key pathophysiologic mechanism.6,9
The discrepancies in outcome between our patients and those previously reported may be explained by differences in case ascertainment. IgLON5 autoimmunity was originally described among a cohort of patients identified by the clinical phenotype.6 By contrast, our patients were encountered on a serologic basis during a broad screen for neural antibodies using tissue-based IFA. Sleep symptoms were often present, but they were not always prominent, and respiratory failure occurred in a minority. Three patients had laryngospasm or respiratory failure requiring tracheostomy, 2 of whom subsequently died. Laryngospasm with sudden death is also reported among patients with antineuronal nuclear antibody type 2 (ANNA-2, Ri) autoimmunity. Other phenotypic similarities with ANNA-2 autoimmunity are dystonia (including jaw dystonia), eye movement disorders, and parkinsonism.20 We plan to prospectively analyze a larger sample of patients with IgLON5 autoimmunity syndrome in detail to more fully characterize the spectrum of sleep and respiratory disorders in this patient population.
IgLONs are members of the immunoglobulin superfamily of cell adhesion molecules abundantly expressed in neurons. The IgLON proteins contain 3 immunoglobulin domains followed by a glycosylphosphatidylinositol anchor protein.21 Metalloproteinase-dependent regulated shedding of IgLONs from the surface of mature cortical neurons has been reported to regulate neurite outgrowth and synaptic plasticity.21 Our data support IgLON5 being abundant, not just in the nervous system but also in the renal glomerulus, which has afferent and efferent innervation to regulate blood flow to the glomerular basement membrane and a diverse repertoire of neural antigens.22–25 Like ANNA-3 autoimmunity, where renal podocyte staining on tissue IFA is also recognized, renal manifestations are not known to accompany IgLON5 autoimmunity.26
In the appropriate clinical context (adults with unexplained sleep disorder, movement disorders, bulbar symptoms, CNS hyperexcitability), IgLON5 autoimmunity should be considered. Generic clues to an autoimmune diagnosis are lacking in most cases, although responses to immune therapies may be favorable.
ACKNOWLEDGMENT
The authors thank the Mayo Clinic Center for Individualized Medicine and the Department of Laboratory Medicine and Pathology, Mayo Clinic, for funding, and Vickie Mewhorter, John Schmeling, and Nancy Peters for excellent technical support.
GLOSSARY
- AHI
Apnea-Hypopnea Index
- ANNA
antineuronal nuclear antibody
- CBA
cell-based assay
- GAD65
glutamic acid decarboxylase 65
- MSA
multiple system atrophy
- OSA
obstructive sleep apnea
- PSP
progressive supranuclear palsy
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
J.A.H.: data collection, analysis and interpretation, and drafting of manuscript. L.K., K.F., and S.L.: supplied critical reagents and critical revision of manuscript. K.A.J., E.K.S.L., S.R.H., N.K., A.G., V.A.L., and S.J.P.: data collection and analysis and critical revision of manuscript. A.M.: study concept and design, data collection, analysis, and interpretation, critical revision of manuscript, and study supervision.
STUDY FUNDING
Mayo Clinic Center for Individualized Medicine and the Department of Laboratory Medicine and Pathology, Mayo Clinic.
DISCLOSURE
J.A. Honorat reports no disclosures. L. Komorowski is employed by Euroimmun AG. K.A. Josephs received research support from NIH/NIA, NIH, MINDS, NIH/NIDCD, and The Data Foundation. K. Fechner is employed by Euroimmun AG. E.K. St Louis served on the scientific advisory board for Inspire, Inc; is an editorial board member for and guest coeditor of Continuum; and received research support from Axovant, Merck, Sunovion Inc, Mayo Clinic CTSA, and National Center for Advancing Translational Science. S.R. Hinson reports no disclosures. S. Lederer is employed by Euroimmun Medizinische Labordiagnostika AG. N. Kumar and A. Gadoth report no disclosures. V.A. Lennon holds a patent for and receives royalties from RSR/Kronus for sale of aquaporin-4 autoantibody testing kits and for commercial aquaporin-4 autoantibody testing performed outside Mayo Clinic; received research support from the NIH; has a potential financial interest in the technology “Aquaporin-4 as an aid for cancer diagnosis”; and receives license fee payments for Non-Mayo sites performing “home brew” diagnostic testing for aquaporin-4 autoantibody. S.J. Pittock holds patents for that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker; consulted for Alexion and Medimmune; and received research support from Grifols, Medimmune, and Alexion, RO1 NS065829-01. A. McKeon has a patent pending for GFAP and MAP1B as markers of neurological autoimmunity and paraneoplastic disorders; consulted for Grifols, Medimmune, and Euroimmun; and received research support from Medimmune and Euroimmun. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.McKeon A, Pittock SJ. Paraneoplastic encephalomyelopathies: pathology and mechanisms. Acta Neuropathol 2011;122:381–400. [DOI] [PubMed] [Google Scholar]
- 2.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutchinson M, Waters P, McHugh J, et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology 2008;71:1291–1292. [DOI] [PubMed] [Google Scholar]
- 5.Tobin WO, Lennon VA, Komorowski L, et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014;83:1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabater L, Gaig C, Gelpi E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol 2014;13:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hogl B, Heidbreder A, Santamaria J, Graus F, Poewe W. IgLON5 autoimmunity and abnormal behaviours during sleep. Lancet 2015;385:1590. [DOI] [PubMed] [Google Scholar]
- 8.Simabukuro MM, Sabater L, Adoni T, et al. Sleep disorder, chorea, and dementia associated with IgLON5 antibodies. Neurol Neuroimmunol Neuroinflamm 2015;2:e136 doi: 10.1212/NXI.0000000000000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gelpi E, Hoftberger R, Graus F, et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol 2016;132:531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruggemann N, Wandinger KP, Gaig C, et al. Dystonia, lower limb stiffness, and upward gaze palsy in a patient with IgLON5 antibodies. Mov Disord 2016;31:762–764. [DOI] [PubMed] [Google Scholar]
- 11.Schroder JB, Melzer N, Ruck T, et al. Isolated dysphagia as initial sign of anti-IgLON5 syndrome. Neurol Neuroimmunol Neuroinflamm 2017;4:e302 doi: 10.1212/NXI.0000000000000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haitao R, Yingmai Y, Yan H, et al. Chorea and parkinsonism associated with autoantibodies to IgLON5 and responsive to immunotherapy. J Neuroimmunol 2016;300:9–10. [DOI] [PubMed] [Google Scholar]
- 13.Gaig C, Graus F, Compta Y, et al. Clinical manifestations of the anti-IgLON5 disease. Neurology 2017;88:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology 2011;76:1108–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKeon A, Martinez-Hernandez E, Lancaster E, et al. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol 2013;70:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 2010;23:394–400. [DOI] [PubMed] [Google Scholar]
- 17.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lynch T, Fahn S, Louis ED, Odel JG. Oculofacial-skeletal myorhythmia in Whipple's disease. Mov Disord 1997;12:625–626. [PubMed] [Google Scholar]
- 19.Sabater L, Planaguma J, Dalmau J, Graus F. Cellular investigations with human antibodies associated with the anti-IgLON5 syndrome. J Neuroinflammation 2016;13:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pittock SJ, Parisi JE, McKeon A, et al. Paraneoplastic jaw dystonia and laryngospasm with antineuronal nuclear autoantibody type 2 (anti-Ri). Arch Neurol 2010;67:1109–1115. [DOI] [PubMed] [Google Scholar]
- 21.Sanz R, Ferraro GB, Fournier AE. IgLON cell adhesion molecules are shed from the cell surface of cortical neurons to promote neuronal growth. J Biol Chem 2015;290:4330–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denton KM, Luff SE, Shweta A, Anderson WP. Differential neural control of glomerular ultrafiltration. Clin Exp Pharmacol Physiol 2004;31:380–386. [DOI] [PubMed] [Google Scholar]
- 23.Daniel C, Albrecht H, Ludke A, Hugo C. Nestin expression in repopulating mesangial cells promotes their proliferation. Lab Invest 2008;88:387–397. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Wu J, Wu H, et al. UCH-L1 expression of podocytes in diseased glomeruli and in vitro. J Pathol 2009;217:642–653. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Lehtonen S, Chen YC, Heikkila E, Panula P, Holthofer H. Neph3 associates with regulation of glomerular and neural development in zebrafish. Differentiation 2012;83:38–46. [DOI] [PubMed] [Google Scholar]
- 26.Chan KH, Vernino S, Lennon VA. ANNA-3 anti-neuronal nuclear antibody: marker of lung cancer-related autoimmunity. Ann Neurol 2001;50:301–311. [DOI] [PubMed] [Google Scholar]