

Abstract
Objective:
To assess the prevalence and clinical features of individuals affected by spinocerebellar ataxia 36 (SCA36) at a large tertiary referral center in the United States.
Methods:
A total of 577 patients with undiagnosed sporadic or familial cerebellar ataxia comprehensively evaluated at a tertiary referral ataxia center were molecularly evaluated for SCA36. Repeat primed PCR and fragment analysis were used to screen for the presence of a repeat expansion in the NOP56 gene.
Results:
Fragment analysis of triplet repeat primed PCR products identified a GGCCTG hexanucleotide repeat expansion in intron 1 of NOP56 in 4 index cases. These 4 SCA36-positive families comprised 2 distinct ethnic groups: white (European) (2) and Asian (Japanese [1] and Vietnamese [1]). Individuals affected by SCA36 exhibited typical clinical features with gait ataxia and age at onset ranging between 35 and 50 years. Patients also suffered from ataxic or spastic limbs, altered reflexes, abnormal ocular movement, and cognitive impairment.
Conclusions:
In a US population, SCA36 was observed to be a rare disorder, accounting for 0.7% (4/577 index cases) of disease in a large undiagnosed ataxia cohort.
Genetic cerebellar ataxia is a clinically heterogeneous disease that progressively destroys the cerebellum and consequently impairs balance and coordination in the affected individual.1 The diagnosis of cerebellar ataxia can be challenging because there are more than 500 genes associated with either primary or secondary ataxia, and many affected families remain undiagnosed for decades. The introduction of unbiased genomic diagnostic testing methods, such as clinical exome sequencing, into the diagnostic evaluation has greatly improved time to diagnosis,2,3 but is limited in its ability to detect certain forms of genetic mutation such as repeat expansion disorders.4,5 The spinocerebellar ataxias (SCAs) are a diverse group of neurodegenerative disorders characterized by an autosomal dominant pattern of inheritance and cerebellar symptoms, currently consisting of at least 43 distinct clinical entities, of which 11 are repeat expansion disorders.6 One of these conditions, spinocerebellar ataxia 36 (SCA36), is caused by a GGCCTG hexanucleotide repeat expansion in the first intron of the pre-mRNA processing gene, nucleolar protein 56 (NOP56).7 Individuals with this disorder show progressive, late-onset, ataxic symptoms affecting limb, trunk, and/or gait stability.7 To date, SCA36 prevalence has been reported in regions of Japan,7,8 Spain,9 France,8 Germany,8 and most recently, in China,10 but its prevalence in the United States is undetermined. Through combined methods of exome sequencing, linkage analysis, and fluorescent repeat primed PCR (RP-PCR), we molecularly identified SCA36 in 4 index cases, representing less than 1% of the undiagnosed population at a tertiary referral ataxia center.
METHODS
Patient recruitment and diagnostic evaluation.
All patients were initially seen at our tertiary referral ataxia center for disorders of gait and balance. All patients underwent a comprehensive clinical evaluation for acquired causes of ataxia prior to consideration of genetic causes.1,11 For enrollment in this study, patients were required to have negative testing for the most common genetic ataxias worldwide,6,12 specifically SCA1, SCA2, SCA3, SCA6, and SCA7, and Friedreich ataxia if clinically appropriate. Age at onset or mode of inheritance was not used as exclusion criteria to prevent bias against phenotypic variability3 or inaccurate assessment of familial association in relatives who could not be directly assessed clinically.
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained to collect DNA for genetic analysis. All patients were provided genetic counseling both before and after completion of the study. All study methods were approved by the Institutional Review Board of the University of California, Los Angeles.
Exome sequencing and array genotyping.
Whole-exome sequencing was performed for 3 members of 2 families (families A and B, figure 1A) at the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine as part of the Baylor-Hopkins Center for Mendelian Genomics initiative. With 0.5 ng of DNA, an Illumina paired-end pre-capture library was generated as described in the BCM-HGSC protocol (hgsc.bcm.edu/content/protocols-sequencing-library-construction). Four of these libraries were pooled and then hybridized in solution to the HGSC CORE design13 (52 Mb; NimbleGen, Madison, WI) in accordance with the NimbleGen SeqCap EZ Exome Library SR User's Guide. Each capture pool, containing 4 samples, was sequenced in one lane using the Illumina HiSeq 2000 platform in paired-end mode, with sequencing-by-synthesis reactions extended for 101 cycles from each end and an additional 7 cycles for the index read. On average, each exome yielded 9.6 Gb of data with 93% of the targeted exome bases covered to a depth of 20-fold or greater. Variant calls were generated according to the GATK best practices (Genome Analysis Toolkit 3.4) using the hg19 reference genome and annotated using the SVS Annotation Suite (Golden Helix). Only variants causing nonsynonymous protein-coding changes or splice-site variants were included in the analysis. Annotated variants were filtered according to a minor allele frequency (MAF) using the Exome Aggregation Consortium (ExAC) database14 (r.0.2) (exac.broadinstitute.org) at an MAF of 0.1% for genes containing a single heterozygous variant, and 1% for genes containing multiple heterozygous variants or homozygous variants. Variants remaining after filtering were compared to a list of 2,256 genes identified by the keyword “ataxia” in the Online Mendelian Inheritance in Man database15 (omim.org) and assessed for their clinical significance as previously described.3
Figure 1. Pedigrees and Southern blot analysis of the SCA36 families identified in this study.

(A) Affected individuals (dark fill) and index cases (arrow) are indicated. +Genotyped individuals; *Individuals who had exome sequencing performed; gray fill indicates at-risk individuals, currently asymptomatic. All genotyped individuals in families A and B were included in the linkage analysis. (B) Southern blot analysis of patient DNA from probands of 3 of the identified SCA36 families, a negative control (−), and a positive control (+). The 3 Kb wild-type allele (arrow) and the larger expanded allele (*) are shown. SCA36 = spinocerebellar ataxia 36.
In parallel to the exome workflow, Illumina Infinium Human Exome v1-2 array data were generated for linkage analysis from 6 members of family A and 2 members of family B (figure 1A). Array data quality assessment included orthogonal confirmation of sample identity and purity using the Error Rate In Sequencing (ERIS) pipeline developed at the BCM:HGSC. Using an “e-GenoTyping” approach, ERIS screens all sequence reads for exact matches to probe sequences defined by the variant and position of interest. A successfully sequenced sample must meet quality control metrics of ERIS single nucleotide polymorphism (SNP) array concordance (>90%) and ERIS average contamination rate (<5%). These arrays capture more than 240,000 SNPs, which were carried forward into quality filtering, pedigree validation, and linkage analyses. All insertions/deletions and nonautosomal polymorphisms and all variants without an rs identifier were removed. In addition, SNPs were removed for high missingness (>5%) or mapping to identical locations (duplicates) resulting in 227,103 variants for analysis. Pedigrees were validated using the prePRIMUS QC pipeline to estimate pairwise kinship and were reconstructed in PRIMUS.16,17
Linkage analysis.
To identify overlapping haplotypes shared identically by descent (IBD) by all genotyped cases and not shared by genotyped controls in families A and B (figure 1A), multifamily parametric linkage analysis was conducted using a fully penetrant dominant model with no phenocopies (f0,1,2 = 0,1,1) using ALLEGRO.18 Logarithm of the odds (LOD) scores generated from linkage in these families are unable to meet genome-wide significance thresholds because of power (max LOD given the information across the pedigrees is 1.2); the purpose of these analyses was to identify candidate regions where IBD sharing was consistent with the model of inheritance for disease in both families for further analysis rather than to identify regions of genome-wide significant linkage. Rare (MAF <0.1% in ExAC), nonsynonymous and splice-site variants from exome sequencing falling within linkage regions (peaks reaching maximum LOD, ∼58.6 megabases genome wide) were identified and evaluated as potential disease candidates (figure e-1 at Neurology.org/ng).
SCA36 repeat expansion testing.
DNA was isolated from the peripheral blood and purified using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany). Fluorescent RP-PCR was implemented,9 and PCR products were separated then analyzed on an ABI Prism 3730 XL Analyzer (Retrogen, San Diego, CA). Peak Scanner 2 software was used to assess for the presence of the SCA36 hexanucleotide repeat expansion in NOP56. This method specifically detects expanded alleles and does not assess normal NOP56 alleles.
Southern blot.
For confirmation of repeat expansions, Southern blot analysis was conducted on DNA samples of 3 affected individuals (figure 1B). Genomic DNA (10 μg) was digested overnight with BsmI (New England Biolabs, Ipswich, MA). Digested DNA was resolved on a 0.8% agarose gel, depurinated with 0.25 M HCl, denatured with 1.5 M NaCl, 0.5 M NaOH neutralized with 1.5 M NaCl, 0.5 M Tris-HCl (pH 7.0), and transferred with 20× saline-sodium citrate buffer onto a Hybond-N+ nylon membrane (GE Health care, Piscataway, NJ). DNA was immobilized on the membrane by UV cross-linking. A 464-bp probe for hybridization was synthesized from genomic DNA by PCR using primers10 flanking exons 2 and 3 of NOP56. The PCR-amplified probe DNA was used as template to make a 32P-labeled probe using random hexamer primers (#58875; Invitrogen, Carlsbad, CA), Klenow polymerase (#M0210S, NEB) and dNTP's containing [32P]-dCTP. Hybridization with the labeled probe was performed at 65°C overnight. The membrane was developed by X-ray film exposure.
RESULTS
Identification of 2 SCA36 families by exome sequencing and linkage analysis.
We independently performed exome sequencing on 3 individuals from 2 undiagnosed families (families A and B, figure 1A) with adult-onset dominant cerebellar ataxia given the high diagnostic yield of this testing in patients previously screened for the most common genetic forms of ataxia.2,3,19,20 However, we were unable to identify any potentially clinically relevant sequence variation associated with ataxia using standard methods.3 To address the possibility of a novel gene or a form of genetic variation not detected by exome sequencing (e.g., noncoding, copy number variants, or repeat expansions), we separately performed genotyping using a cytogenomic SNP array and subsequent linkage analysis in both families. Among these data, we identified 2 regions on chromosome 20 that reached maximum theoretical LOD scores across families A and B containing a known clinical ataxia gene, NOP56, causing disease through repeat expansion (figure e-1). Exome analysis had indicated that families A and B did not possess any rare protein-coding variants in any genes potentially associated with their phenotype in any of the identified linked regions (figure e-1). The absence of any clinically relevant rare variants in ataxia genes within the sequenced affected family members (figure 1A) prompted us to evaluate these 2 families for the repeat expansion in NOP56 associated with SCA36.7
Identification of an expanded GGCCTG repeat in NOP56 in families A and B.
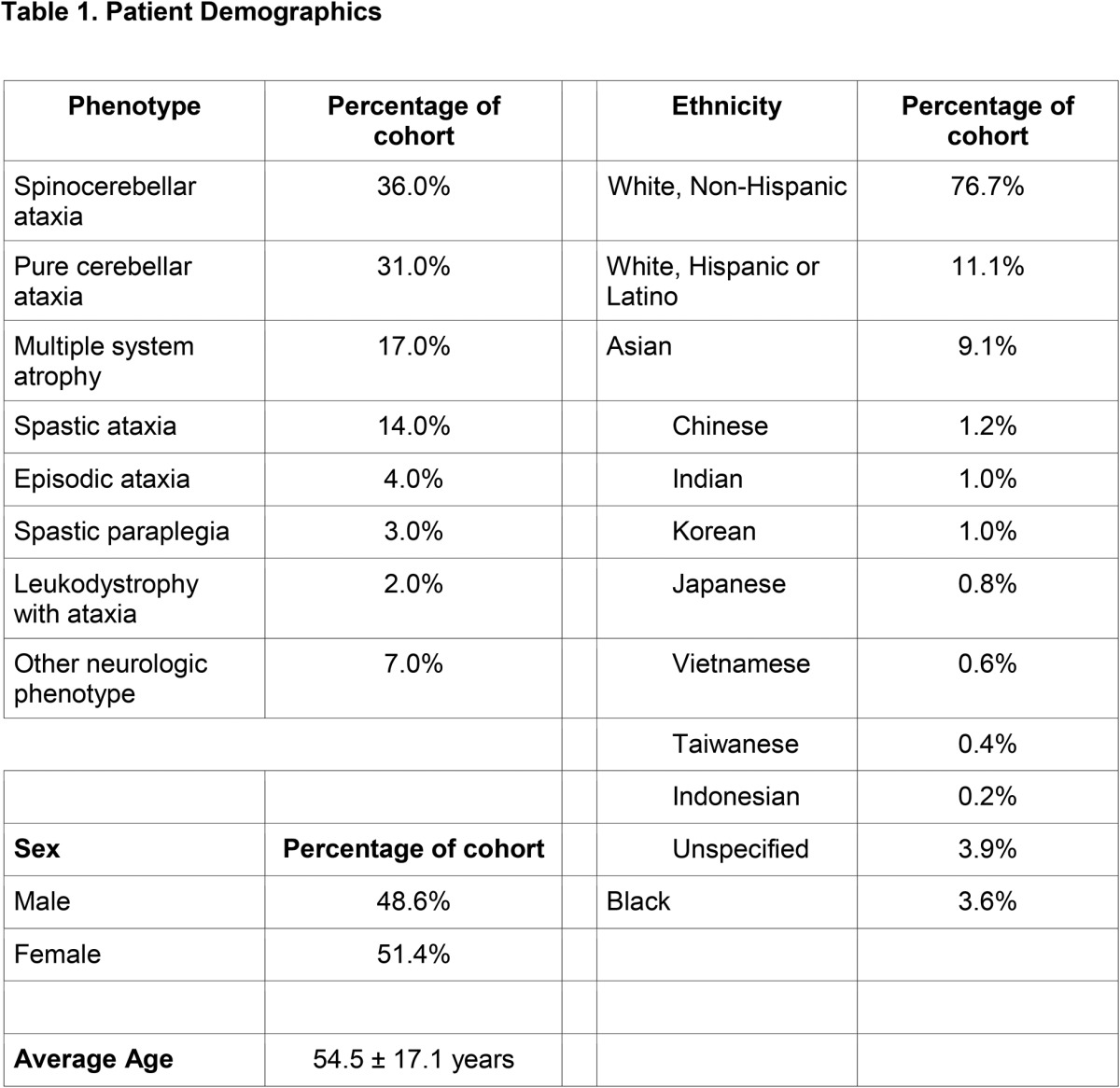
RP-PCR analysis was used to assess for expansion in the NOP56 gene. This method specifically detects the presence of an expansion through the generation of multiple products of varying size from different amplification sites, leading to a stuttering effect (figure e-2). RP-PCR identified the pathogenic hexanucleotide repeat expansion in intron 1 of NOP56 in both families A and B (figure e-2), confirming the diagnosis of SCA36. Because RP-PCR lacks the ability to quantitate expansion size, Southern blot analysis was used as a second confirmatory technique, detecting NOP56 repeat expansions in the probands from families A and B (figure 1B). Given that SCA36 is considered a rare repeat expansion disorder, but has been observed to have varying prevalence in different ethnic populations,7–10 the finding of 2 consecutive SCA36 families prompted us to evaluate our entire clinical cohort for the disease to determine the prevalence in a US ataxic population. We analyzed 577 index cases using fluorescent RP-PCR. The demographics of this patient population including age, sex, ethnicity, and phenotype are shown in table 1. Only 2 additional positive cases were identified, thus bringing the total to 4/577 cases (0.7%).
Table 1.
Patient demographics
Clinical characteristics of SCA36 in US families.
SCA36 was observed in 4 index cases from a cohort of 577 patients (families A–D; figure 1A). An additional 2 positive cases were subsequently identified (1 each from families A and B, respectively). The ancestral origins of these 4 families were either white, non-Hispanic, European (2 families), or Asian (1 Vietnamese and 1 Japanese family). The clinical features of these 6 individuals (5 men and 1 woman, mean age at onset: 44.5 ± 5.8 years) are summarized in table 2. All affected individuals exhibited an ataxia gait, consistent with cerebellar dysfunction, on examination. Other symptoms included limb ataxia or spasticity and altered reflexes. Saccadic ocular pursuit was present in 5 of 6 individuals, predominantly saccadic pursuit or overshoot, whereas hearing impairment was reported in only 1 of 6 individuals. Two of 6 patients presented with mild cerebellar atrophy detected using neuroimaging.
Table 2.
Clinical findings of SCA36 patients reported in this study

DISCUSSION
We evaluated 577 index cases seen at a tertiary referral ataxia center in the United States and identified the NOP56 hexanucleotide repeat expansion in 4 families. When compared with other reports, the prevalence of SCA36 in the United States (n = 577, 0.7%) appears to be less frequent than in most other countries evaluated to date, including Spain (n = 160, 6.3%),9 France (n = 270, 4.4%),8 Western Japan (n = 251, 3.6%),7 and Eastern Japan (n = 231, 2.2%)8 and is more similar to that of China (n = 512, 0.6%)10 and Germany (n = 175, 0.0%)8 (table 3). It should be noted that our cohort was of general undiagnosed patients with ataxia, unselected by mode of inheritance or other features, in whom the most frequent genetic etiologies had been previously excluded. As a tertiary referral center, we cannot estimate the number of patients with more common genetic etiologies identified in the community, and therefore not referred, so the overall prevalence in all undiagnosed ataxia cases is likely lower. Consistent with previous reports, SCA36-positive individuals in our cohort descended from European and East Asian backgrounds. Despite the majority of our cohort descending from European or Asian ancestry (table 1), we observe a much lower prevalence than countries such as Spain, France, or Japan.7–9 This may stem partially from the inclusion criteria defining our cohort, but likely also reflects regional variation across Europe and Asia, as noted in the studies from China and Germany,8,10 for example. One of our families (family B) identifies their family origins as emanating from Vietnam, which would be the first reported case of SCA36 from that country.
Table 3.
Comparison of clinical characteristics of US SCA36 patients with other SCA36 populations globally

Clinical examination of those 6 affected SCA36 patients in our cohort confirms many of the previously reported SCA36 symptoms, including gait and limb ataxia, dysarthria, increased lower limb reflexes, polyneuropathy, saccadic pursuit, and cognitive impairment (table 3). Facial or tongue fasciculations are typically seen in more than 50% of reported SCA36-positive cases with the exception of France (12%) and were present in 2 of 6 (33%) SCA36-positive individuals in this study. Hearing impairment is present in more than 40% of previously reported SCA36-positive individuals and was also reported in 2 of 6 individuals from family B (figure 1A and table 2), although detailed audiologic studies were not conducted on all the affected patients in this study. In summary, SCA36 represents a small percentage of the undiagnosed cerebellar ataxia population in the United States. Given that SCA36 is a repeat expansion disorder that would not be detected by the high-yield genomic diagnostic tests performed early in a patient's evaluation,3 repeat expansion testing should be considered in individuals with appropriate clinical phenotypes and either a sporadic or dominant mode of inheritance.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all the patients and their families who contributed to this study. B.L.F.thanks Xizhe Wang and Stephanie Pang for technical assistance.
GLOSSARY
- ERIS
Error Rate In Sequencing (ERIS)
- IBD
identically by descent
- LOD
logarithm of the odds
- MAF
minor allele frequency
- RP-PCR
repeat primed PCR
- SCA
spinocerebellar ataxia
- SCA36
spinocerebellar ataxia 36
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
B.L.F., J.E.B., M.J.S., and J.R.L. contributed to the conception and design of the research project, and J.M.V., T.D., and L.E.P. were responsible for its execution. S.P., B.L.F., and D.H.G. supervised the clinical data, samples, and patient care. B.Q. and Z.Y. contributed to the RP-PCR protocol design and control sample preparation. E.B., D.M., R.G., J.R.L., and B.L.F. contributed to next-generation sequencing design and analysis. L.E.P. and J.E.B. conducted all bioinformatics analyses. D.A. and R.B. performed the Southern blot analysis as shown in figure 1B. J.M.V., T.D., and B.L.F. wrote the manuscript, and all authors were responsible for its review and critique.
STUDY FUNDING
This work was supported in part by a US National Human Genome Research Institute and National Heart Lung and Blood Institute grant (U54HG006542) to the Baylor-Hopkins Center for Mendelian Genomics, the National Institute for Neurologic Disorders and Stroke (grant R01NS082094 to Dr. Fogel), and the National Ataxia Foundation (Young Investigator Award to Dr. Fogel). The research described was supported by the NIH/National Center for Advancing Translational Science UCLA Clinical and Translational Science Institute grant UL1TR000124. Dr. Fogel acknowledges the support through donations to the University of California by the Rochester Ataxia Foundation. The authors also received support from the Spanish Institute of Health Carlos III (grant PI12/00742) and FEDER funds. Z.Y. was supported by a grant from Fundación Carolina. R.B. was supported by grants from the NIH, National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR059847), and the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK092590). D.A. was supported by the American Heart Association (15POST25090134).
DISCLOSURE
J.M. Valera, T. Diaz, and L.E. Petty report no disclosures. B. Quintáns has been a consultant for Genomic Consulting S.L. Z. Yáñez reports no disclosures. E. Boerwinkle has received research support from NIH. D. Muzny has received research support from NIH and the Simons Foundation. D. Akhmedov has received research support from the American Heart Association. R. Berdeaux has received research support from NIH. M.J. Sobrido has received speaker honoraria from Actelion Pharmaceuticals; has served on the editorial boards of Human Mutation and Applied & Translational Genomics; is the CEO of Genomic Consulting; has a private clinical neurology practice and performs genetic diagnosis of ataxias; and has received research support from Actelion Pharmaceuticals, Instituto de Salud Carlos III (Spain), and the Asociación Galega de Ataxias (AGA). R. Gibbs has received travel funding/speaker honoraria from Fusion Conference; has served on the editorial board of Genome Research; and has received research support from NIH. J.R. Lupski has stock ownership in 23andMe; is a paid consultant for Regeneron Pharmaceuticals; has stock options in Lasergen; is a member of the scientific advisory boards of Baylor Genetics Laboratories, Lasergen Inc., Regeneron Pharmaceuticals, and BioPontis Alliance for Rare Diseases; receives publishing royalties from Humana Press; is an employee of Baylor College of Medicine (the Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the chromosomal microarray analysis and clinical exome sequencing offered in the Baylor Genetics Laboratory [bmgl.com]); has received research support from NIH; and is a coinventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. D.H. Geschwind has served on the scientific advisory board for Ovid Therapeutics; has served on the editorial boards of Cell, Molecular Autism, Molecular Neuropsychiatry, Nature, Nature Genetics, Nature Neuroscience, Neurology, Neuron, New England Journal of Medicine, Public Library of Science (PLoS), Genetics, Science, and Translational Psychiatry; holds patents for Peripheral Gene Expression Biomarkers for Autism, Genetic Risk Factor for Neurodegenerative Disease, Compositions and Methods for Diagnosing and Treating Brain Cancer and Identifying Neural Stem Cells, Genetic Variants Underlying Human Cognition: Novel Diagnostic and Therapeutic Targets, Peripheral Gene Expression Biomarkers for Autism, Brain Gene Expression Changes Associated with Autism Spectrum Disorders, Full Biomarkers in Friedreich's Ataxia (provisional patent application), Signaling Networks Causing Neurodevelopmental Disorders In Human Neurons, Genes Dysregulated in Autism: Potential Biomarkers and Therapeutic Pathways, Peripheral Gene Expression Biomarkers for Autism, A Genetic Target for Treatment of Individuals with Neurocognitive Spectrum Disorders, Neuronal Regeneration, Frataxin Knock-Down Mouse, Jakmip1 Knockout Mouse, and Cyfip1 Transgenic Mouse; receives publishing royalties from Oxford University Press; has been a consultant for Ovid Therapeutics Ltd; has received research support from Takeda Pharmaceutical Company, NIH, the Simons Foundation, Adelson Medical Research Foundation, The Tau Consortium, and Ovid Therapeutics Ltd; and receives license fee payments for Mouse model of Friedreich Ataxia. S. Perlman has received research support from the University of California, Los Angeles and the National Ataxia Foundation. J.E. Below has received research support from Sanofi Innovation Awards Program, Tulane National Primate Research Center, and NIH. B.L. Fogel has received speaker honoraria from the American Academy of Neurology; has received travel funding from the National Ataxia Foundation; has received speaker honoraria and travel funding from American Physician Institute for Advanced Professional Studies; and has received research support from NIH and the National Ataxia Foundation. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Shakkottai VG, Fogel BL. Autosomal dominant spinocerebellar ataxia. Neurol Clin 2013;31:987–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pyle A, Smertenko T, Bargiela D, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 2015;138:276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fogel BL, Lee H, Deignan JL, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol 2014;71:1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fogel BL, Lee H, Strom SP, et al. Clinical exome sequencing in neurogenetic and neuropsychiatric disorders. Ann NY Acad Sci 2016;1366:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fogel BL, Satya-Murti S, Cohen BH. Clinical exome sequencing in neurologic disease. Neurol Clin Pract 2016;6:164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–894. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi H, Abe K, Matsuura T, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 2011;89:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obayashi M, Stevanin G, Synofzik M, et al. Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J Neurol Neurosurg Psychiatry 2015;86:986–995. [DOI] [PubMed] [Google Scholar]
- 9.García-Murias M, Quintáns B, Arias M, et al. “Costa da Morte” ataxia is spinocerebellar ataxia 36: clinical and genetic characterization. Brain 2012;135:1423–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee YC, Tsai PC, Guo YC, et al. Spinocerebellar ataxia type 36 in the Han Chinese. Neurol Genet 2016;2:e68 doi: 10.1212/NXG.0000000000000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fogel B, Perlman S. Cerebellar disorders: balancing the approach to cerebellar ataxia. In: Tuite PJ, Gálvez-Jiménez N, editors. Uncommon Causes of Movement Disorders. New York: Cambridge University Press; 2011:198–216. [Google Scholar]
- 12.Fogel BL, Lee JY, Lane J, et al. Mutations in rare ataxia genes are uncommon causes of sporadic cerebellar ataxia. Mov Disord 2012;27:442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bainbridge MN, Wang M, Yuanqing W, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol 2011;12:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Exome Aggregation Consortium (ExAC). ExAC Browser. Cambridge, MA: 2014. Available at: http://biorxiv.org/content/early/2015/10/30/030338. Accessed December 2015. [Google Scholar]
- 15.McKusick-nathans Institute of Genetic Medicine; Johns Hopkins Medicine; National Human Genome Research Institute. Online Mendelian Inheritance in Man. Avaiable at: http://omim.org/. Accessed December 2015. [Google Scholar]
- 16.Staples J, Qiao D, Cho MH, et al. PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. Am J Hum Genet 2014;95:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staples J, Ekunwe L, Lange E, et al. PRIMUS: improving pedigree reconstruction using mitochondrial and Y haplotypes. Bioinformatics 2016;32:596–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gudbjartsson DF, Jonasson K, Frigge ML, et al. Allegro, a new computer program for multipoint linkage analysis. Nat Genet 2000;25:12–13. [DOI] [PubMed] [Google Scholar]
- 19.Ohba C, Osaka H, Iai M, et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 2013;14:225–232. [DOI] [PubMed] [Google Scholar]
- 20.Sawyer SL, Schwartzentruber J, Beaulieu CL, et al. ; FORGE Canada Consortium. Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Hum Mutat 2014;35:45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.