

Abstract
The US Food and Drug Administration (FDA) issued a guidance document in 2010 on pharmacokinetic (PK) studies in renal impairment (RI) on the basis of observations that substances such as uremic toxins might result in altered drug metabolism and excretion. No specific recommendations for oncology drugs were included. We surveyed the publicly available FDA review documents of 29 small molecule oncology drugs approved between 2010 and the first quarter of 2015. The objectives were as follows: (i) summarize the impact of RI on PK at the time of the initial new drug application; (ii) identify limitations of the guidance; and (iii) outline an integrated approach to study the impact of RI on these drugs. Our survey indicates that the current FDA guidance does not appear to provide clear strategic or decision pathways for RI studies in terms of small molecule oncology drugs. The FDA review documents indicate an individualized approach to the review because of the complex pharmacologic nature of these drugs and patient populations. Overall, the strategy for carrying out a RI study during clinical development or as a postmarketing study requires integration with the totality of data, including mass balance, absolute bioavailability, drug–drug interaction, hepatic dysfunction, population PK, exposure–response analysis, the therapeutic window for best guidance, and determination of the optimal doses for special oncology populations.
Keywords: exposure–response, Food and Drug Administration, oncology, pharmacokinetics, renal impairment, small molecules
Introduction
In 1998, the US Food and Drug Administration (FDA) issued its first guidance on renal impairment studies – ‘Guidance for industry: pharmacokinetics in patients with impaired renal function – study design, data analysis and impact on dosing and labeling’, which was subsequently revised in 2010 1,2. Thereafter, in 2014, the European Medicines Agency (EMA) also issued a revised draft guideline on the evaluation of the pharmacokinetics (PK) of medicinal products in patients with decreased renal function 3. Consistently, both regulatory agencies require a dedicated renal impairment (RI) study in patients with varying degrees of RI for small molecule (SM) drugs intended for chronic use, irrespective of their elimination pathways (i.e. even for drugs that are eliminated by nonrenal pathways). For drugs mainly eliminated by nonrenal pathways, a reduced RI study in end-stage renal disease (ESRD) patients or in patients with severe RI can be carried out (a reduced PK study). If the results from the reduced study are positive, a full study with varying degrees of RI may be required (Fig. 1).
Fig. 1.
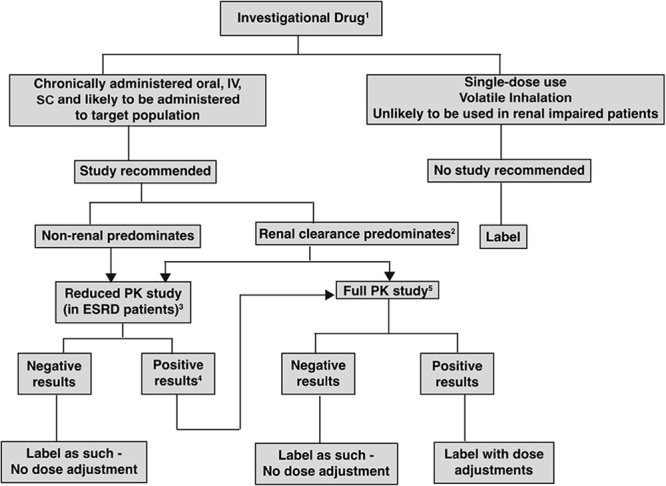
Decision tree for determining when a renal impairment study should be carried out according to the 2010 Food and Drug Administration Draft Guidance on RI. 1Metabolites (active/toxic) follow the same decision tree. 2The sponsor has the option of conducting a reduced study in end-stage renal disease (ESRD) patients or a full study. 3To be conducted in ESRD patients not yet on dialysis. 4The results are ‘positive’ when the pharmacokinetic (PK) changes are clinically significant on the basis of exposure–response of the drug. 5See section IV.B of the Food and Drug Administration draft guidance for the full PK study design or additional studies can be carried out including a population PK evaluation. IV, intravenously; SC, subcutaneously. From Appendix 1 of Guidance for industry: pharmacokinetics in patients with impaired renal function – study design, data analysis and impact on dosing and labeling by US FDA 2.
The rationale for the guidance stems from the knowledge that RI because of chronic kidney disease (CKD) is known to alter PK of drugs that are eliminated either by renal or by nonrenal pathways 4–7. Although the impact of RI on the PK and the potential adjustment of doses for drugs eliminated by renal pathways is well established, the awareness that drugs eliminated by nonrenal pathways can have their rate and extent of oral absorption, distribution, metabolism, and elimination altered by RI is a newer and a less established concept. Such indirect or secondary effects have been attributed to elevated uremic toxins and inflammation. These in turn result in altered expression and/or activities of plasma proteins, drug-metabolizing enzymes, and drug transporters. The proposed mechanisms for alterations in clinical PK [absorption, distribution, metabolism, excretion (ADME)], as a consequence of accumulated uremic toxins, are outlined in Table 1. PK in the RI patient population can further be complicated by drug removal by dialysis, formation and elimination of active or toxic metabolites, and drug–drug interactions (DDI). In addition to impacting PK, RI leads to physiological changes that may alter the response and/or tolerance to drugs. For consistency, hereafter, we will use RI to denote the clinical signs and symptoms of CKD.
Table 1.
Clinical pharmacokinetic (ADME) effects and proposed mechanisms of increased drug concentrations as a consequence of accumulated uremic toxins
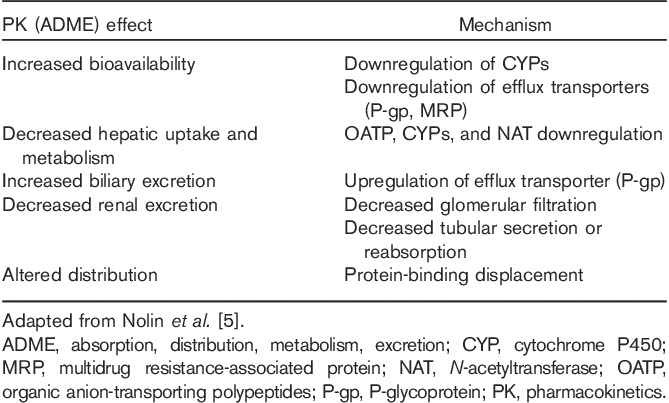
The proposed mechanisms for RI altering the PK of nonrenally cleared drugs have clinical relevance. Nolin et al. 5 reviewed drugs with altered PK in patients with RI. These drugs are substrates for a variety of metabolizing enzymes and transporters. The representative examples and their nonrenal pathway outlined in parentheses included bupropion (CYP2B6), cerivastatin [CYP2C8, CYP3A4, P-glycoprotein (P-gp)], organic anion-transporting polypeptide, multidrug resistance-associated protein, breast cancer resistance protein), and repaglinide (CYP2C8, CYP3A4, organic anion-transporting polypeptide, P-gp) 5. Cumulative evidence shows that RI may alter a variety of nonrenal ADME pathways in a clinically relevant manner.
The impact of the 1998 FDA guidance document on drug development was evaluated previously. Zhang et al. 8 surveyed new drug application (NDA) submissions for 94 SM new molecular entities approved between 2003 and 2007. The authors found a 17% increase in formal evaluations of the effect of RI on the PK of new molecular entities, from 44% before to 61% after publication of the 1998 guidance, which used a full study design 8. Thirty-seven of the surveyed NDAs were for nonrenally eliminated drugs (i.e. defined as <30% of the dose excreted unchanged in the urine), of which 23 (62%) had a dedicated RI study carried out. Approximately 55% of these studies showed at least 1.5-fold increase in area under the concentration–time curve from time 0 to the last time point with quantifiable concentration (AUCt). This resulted in labeling recommendations for dose adjustments in RI for six (46%) drugs. The guidance was subsequently updated in 2010 2.
The 2010 FDA draft guidance provides general practice information on assessing PK in patients with RI, but does not contain special considerations for oncology drug development. The impact of RI can be significant for SM oncology drugs owing to the following factors: (i) these drugs are often administered at or close to the maximum tolerable dose (MTD), and so any elevation of the PK exposure because of RI might be a safety concern 9; (ii) the prevalence of RI can be higher among cancer patients than general disease populations 10,11; (iii) cancer patients on certain chemotherapies are more likely to have deteriorated renal function as a result of direct nephrotoxic effects and/or develop comorbid and confounding conditions, increasing the chances of being intolerant to their established doses 12; and (iv) the impact of a DDI on a victim drug may be exacerbated during concomitant administration in patients with RI. As a result, important aspects of clinical pharmacology in developing SM oncology drugs include understanding the impact of RI on PK and safety, identifying the patients at risk, and adjusting the dose.
In light of the update and issuance of the guidance document in 2010 2, we reviewed the 29 SM oncology drugs approved by the FDA from 2010 to the first quarter of 2015 to assess the impact of RI on drug development decisions and the FDA review outcomes.
The overall objectives of our survey were as follows: (i) summarize available data to assess the impact of RI on PK at the initial NDA; (ii) discuss limitations of the FDA draft guidance; (iii) identify an integrated approach to assess the impact of RI on the PK of nonrenally eliminated SM oncology drugs, on the basis of the totality (or combinations) of data, including human mass balance, absolute oral bioavailability (BA), population PK, hepatic impairment, drug interaction, active metabolites, exposure–efficacy and/or exposure–safety (E–S) analyses, and therapeutic windows in the determination of the need for postmarketing RI PK studies. These assessments not only allow for a clear understanding of the FDA’s clinical pharmacology assessments/reviews, recommendations for labeling dose adjustments, and decisions on requesting further RI assessment [such as a postmarket requirement (PMR) study] but also outline an alternative path to the dedicated RI study to optimize the dose of SM drugs for cancer patients with varying degrees of RI.
Methods
The SM oncology drugs initially approved by the FDA between 2010 and March 2015 were identified using the oncology approvals section of the FDA website 13. Relevant information on each drug was extracted from the original United States Product Information (USPI) and the Summary Basis for Approval (SBA) documents provided by the FDA (Clinical Pharmacology and Biopharmaceutics Review section). The PMR or postmarketing commitment (PMC) of a dedicated RI study was confirmed with the FDA database for PMRs/PMCs. When different results in the USPI and the SBA document of a drug were included, the USPI result was selected for this survey.
The available information extracted and summarized included key PK parameters, metabolism and transport, active metabolites, DDI, human mass balance, available PK data from patients with impaired renal or hepatic function, and population PK analysis results included in the NDAs, as well as additional clinical pharmacology assessments as PMRs. Supplemental information from the published literature was also included as appropriate, but was not systematically applied to each drug.
Results
Overview of survey results
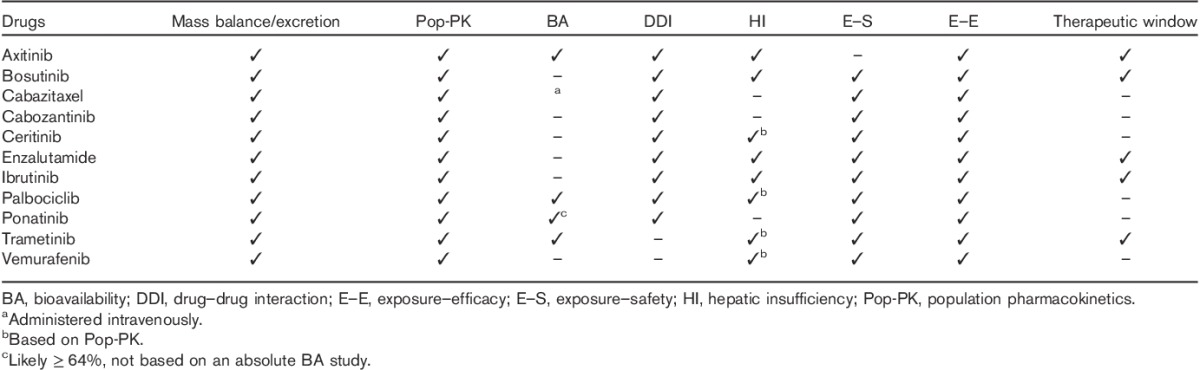
A total of 29 SM oncology drugs were approved by the FDA between 2010 and the first quarter of 2015 (Table 2). FDA guidelines suggest that PK studies may be important not only when the drug or the principal active metabolite undergoes considerable renal elimination (i.e. if the fraction of the dose excreted in the urine is at least 30%) but also if a drug is primarily metabolized or secreted in bile. This is because RI can inhibit some pathways of hepatic and gut drug metabolism, and alter drug transport expression. The contribution of renal excretion toward overall drug elimination was assessed in 14C human mass balance studies for 27 oncology drugs, with the exception of belinostat, for which the percentage of renal elimination was determined by other assessments, and for carfilzomib, where no human study was carried out and excretion data were reported from a rat ADME study (Table 2).
Table 2.
Small molecule oncology drugs approved by the Food and Drug Administration since 2010
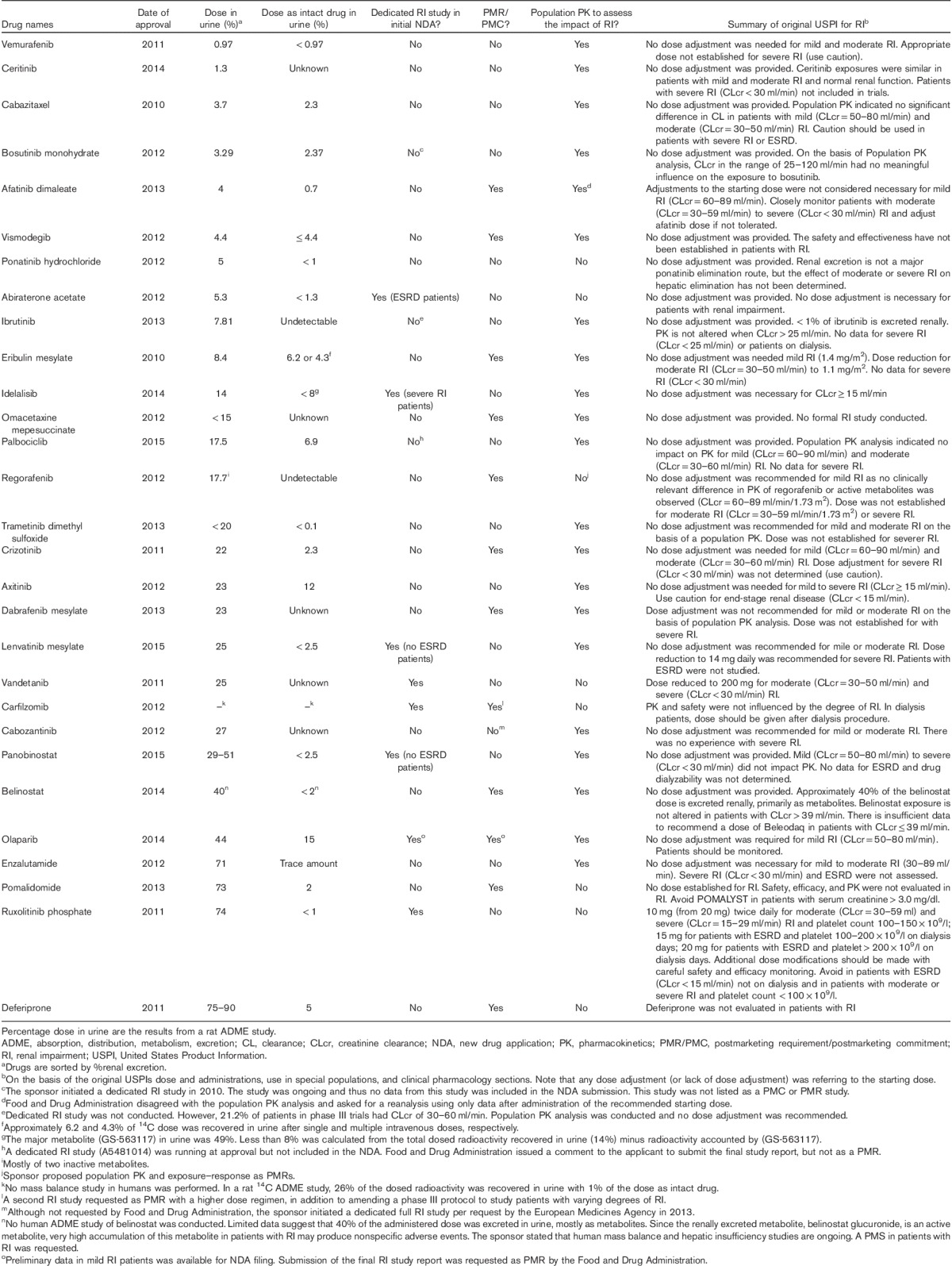
For all the drugs tested, excretion of intact drug in urine was less than 30%. Of these drugs, axitinib and olaparib had the highest urinary excretion of intact drug (12 and 15%, respectively). All other tested drugs had urinary excretion values of less than 10% (Table 2). The total 14C dose recovered in urine ranged from 0.97% (vemurafenib) to 75–90% (deferiprone), with 21 drugs having less than 30% of the 14C-labeled dose excreted in the urine, and seven drugs had at least 30% of total 14C excretion in the urine, which included panobinostat, belinostat, olaparib, enzalutamide, pomalidomide, ruxolitinib phosphate, and deferiprone (Table 2).
Population PK analysis was also used to assess the impact of RI on PK. These analyses were usually carried out when limited (or no) data were available from a dedicated RI study conducted before the NDA filing (Table 2). Although the PK data from patients with severe RI were either lacking or limited in most cases, population PK analyses have been a key approach to assess the impact of RI on PK and complement the findings from the mass balance studies. The use of the population PK approach has allowed dose selection for patients with RI and to inform the regulatory decision on the need for a postmarketing RI study. When data were considered informative, population PK analysis either confirmed the lack of RI impact on PK (e.g. vemurafenib, ceritinib, idelalisib, and trametinib) or informed dose adjustment for studied degrees of RI (e.g. eribulin). When data were not informative, further RI assessment was usually required (e.g. afatinib and belinostat).
On the basis of the FDA’s SBA documents, population PK for RI assessment was not performed for 8 drugs in the initial NDAs. This was either because a conclusion was drawn on the basis of a dedicated RI study (i.e. abiraterone, vandetanib, carfilzomib, and ruxolitinib) or limited PK data were available from patients with RI (i.e. ponatinib, regorafenib, and pomalidomide), or when a PMR RI study was well expected on the basis of urinary excretion data in the mass balance study (e.g. deferiprone).
Table 3 categorizes RI studies carried out for the oncology drugs into urinary recovery, timing of the NDA, and review outcome. For the 29 drugs surveyed, a total of 20 RI studies were carried out, nine of them before the NDA submission, and 11 as PMRs during the review of the NDA. An RI study was carried out during the development of carfilzomib, but it was considered unsatisfactory because of the low dose used and the FDA issued a PMR for a second RI study. For both bosutinib and olaparib, the RI study was being carried out during the NDA review. No data or only preliminary data in mild RI patients were available for bosutinib and olaparib, respectively, to inform the initial USPI.
Table 3.
Renal impairment studies performed for 29 small molecule oncology drugs approved by the Food and Drug Administration from 2010 through the first quarter of 2015

Five of the six drugs with 30% or more renal dose recovery had a dedicated RI study, except for enzalutamide, whereas nine out of 21 drugs with less than 30% renal recovery had no dedicated RI, study carried out, or required as a PMR. For the nine drugs with a dedicated RI study before their NDA filing, the percentage of dose recovery in urine ranged from 3.29 to 74%, and with five (56%) drugs having less than 30% renal dose recovery. Among these drugs, vandetanib and ruxolitinib had specific dosing adjustments for RI in their initial USPIs on the basis of results from dedicated RI studies. Vandetanib and ruxolitinib had 25 and 74% total 14C renal dose recovery, respectively. No dose adjustment was considered necessary (or was provided) for abiraterone, bosutinib, idelalisib, lenvatinib, carfilzomib, panobinostat, and olaparib for tested degrees of RI (Table 2).
For the nine drugs with a 14C human mass balance data and a dedicated RI study issued as a PMR/PMC, the percentage dose recovery in urine ranged from 4 to 75–90% and seven (64%) drugs had less than 30% renal dose recovery. For the 10 drugs without a dedicated RI study carried out or requested, nine (90%) drugs had less than 30% renal dose recovery, except for enzalutamide with 71% radioactive dose excreted in urine (Tables 2 and 3).
To better understand regulatory expectations, and how to efficiently assess the impact of RI on SM oncology drug PK, we focused on three drugs with a dedicated RI study issued as a PMR, despite low renal excretion (14C dose recovery in urine<10%). These drugs include afatinib, eribulin, and vismodegib (Table 3). In addition, we summarized the characteristics of 10 drugs that were approved without a dedicated RI study either before the NDA or as a PMR/PMC (Table 3). These drugs included axitinib, cabazitaxel, cabozantinib, palbociclib, ceritinib, ibrutinib, ponatinib, trametinib, vemurafenib, and enzalutamide (Table 3). For bosutinib and olaparib, dedicated RI studies were ongoing at the time of NDA submission. The reason for carrying out a RI study for bosutinib was not entirely clear in our assessment of the review summaries. The FDA SBA document suggests that this might have been related to the lack of informative population PK and/or mass balance data. We included bosutinib in this section because the initial USPI was based on the available data in the NDA (no data from a dedicated RI study) and reflected the regulatory interpretation of the available RI data at the initial approval.
For each drug, its general PK properties and DDIs were first summarized as the background for discussion. The mass balance study results were then discussed to understand the contributions of renal and hepatic (and/or intestinal) elimination. However, the urinary dose recovery of a drug following an oral administration is by design an underestimation of the relative renal contribution and overestimation of fecal elimination if the oral BA is incomplete. Thus, absolute oral BA, when available, must be coupled with mass balance data to properly assess the impact of RI. However, population PK analysis does not have this limitation. When PK data were available from patients with varying degrees of RI, population PK analysis enables assessment of any direct or secondary effect of RI on PK. Additional relevant data, when available, were also included for discussion. For example, DDI or hepatic dysfunction data may allow for the estimation of uremic toxin effects on PK.
Individual drugs that had less than 10% renal excretion but a dedicated renal impairment study was issued as a postmarketing requirement
In the human mass balance studies of afatinib, eribulin, and vismodegib, 4, 8.4, and 4.4% of the 14C doses, respectively, were recovered in urine. For these three drugs, no dedicated RI study was included in the initial NDAs, but upon FDA review, dedicated RI studies were issued as PMRs. For vismodegib, additional data were provided to the FDA following the 2012 US approval, including hepatic impairment, DDI, and population PK data. This allowed the FDA to determine PMR as fulfilled. The characteristics of these three drugs are summarized below:
Afatinib
Afatinib dimaleate [Gilotrif, molecular weight (MW)=718; Boehringer Ingelheim Pharmaceuticals, Ridgefield, Connecticut, USA] is a kinase inhibitor indicated for the first-line treatment of patients with metastatic non-small-cell lung cancer with certain epidermal growth factor receptor mutations. The recommended dose is 40 mg orally once daily on an empty stomach. Afatinib is a Biopharmaceutics Classification System (BCS) I or III molecule with absolute BA. PK steady state was achieved following 8 days of dosing with 2.8-fold accumulation.
Afatinib has unique biotransformation among the SM oncology drugs reviewed. The main biotransformation products of afatinib are nonspecific and nonenzymatic conjugates, with cysteine on endogenous proteins by Michael addition. This might largely impact the disposition of afatinib and render afatinib less dialyzable. The human mass balance study showed that fecal and urinary recoveries accounted for 85 and 4%, respectively, of the orally administered dose.
Population PK analysis carried out by the FDA using pooled data including two patients with severe RI suggested that mild and moderate RI increased the trough concentration (Ctrough) of afatinib by 14 and 37%, respectively. The impact of RI on the PK of afatinib in the pivotal trial seems more significant with 27 and 85% increases in the Ctrough in patients with mild and moderate RI, respectively. These increases may be clinically relevant as E–S analyses indicated that higher concentrations of afatinib were associated with an increased risk of Common Terminology Criteria for Adverse Events: grade≥3 adverse events (AEs), grade≥2 diarrhea, or grade≥2 rash/acne. The absolute contribution of renal excretion and the effect of varying degrees of RI on afatinib elimination were unknown because of the lack of absolute oral BA data.
In summary, carrying out a RI study as a PMR for afatinib appeared to be supported by a total of three factors: (i) observed increases in trough concentrations in patients with moderate RI; (ii) the results of the E–S analysis; and (iii) the therapeutic window, where the concentrations resulting from the approved dose in RI patients in the pivotal trial would approach or exceed those of the MTD.
Eribulin
Eribulin mesylate (Halaven, MW=826; Eisai Co., Woodcliff Lake, New Jersey, USA) is a microtubule inhibitor indicated for the treatment of patients with metastatic breast cancer. The recommended dose is 1.4 mg/m2 intravenously on days 1 and 8 of a 21-day cycle. Eribulin showed linear PK with respect to dose with no accumulation with weekly administration and low plasma protein binding (<65%). The total body clearance (CL) and volume (V) were ∼1.81 l/h/m2 and 55.8 l, respectively. The renal CL was estimated to be 0.093–0.372 l/h/m2.
The metabolism of eribulin is limited, with the concentration of circulating metabolites representing less than 0.6% of that of eribulin. Clinical studies indicated that there is no CYP3A4 or P-gp related DDIs for eribulin. In the human mass balance study, 82 and 9% of the 14C dose were recovered in feces and urine, respectively. Unchanged drug was the major species, accounting for 88 and 91% of the recovered dose in feces and urine, respectively.
Before filing the NDA, no dedicated RI study was carried out. E–S analyses indicated an increase in neutropenia with increased concentrations. Intensive PK data from patients with varying degrees of RI indicated that eribulin AUC increased by two-fold in patients with moderate RI [creatinine clearance (CLcr) 30–50 ml/min]. Population PK analysis confirmed the correlation between CLcr and CL, supporting the observation that moderate RI can lead to an increase in eribulin exposure. There were no patients with severe RI enrolled in clinical studies with eribulin. A dedicated hepatic insufficiency (HI) study was carried out and showed a 1.8–2.5-fold increase in eribulin exposure, when administered to patients with mild and moderate HI, respectively. In the USPI, dose reduction was recommended for both RI and HI, and a dedicated reduced RI study was requested as a PMR.
Eribulin is the only drug in Table 2 where the plasma exposure was impacted by both renal and hepatic impairment. As eribulin is reported to have minimal renal excretion, low protein binding, insignificant metabolism, and no identified interaction with transporters, it is not clear how moderate RI could increase eribulin exposure by two-fold either by direct or by secondary mechanisms. Perhaps, the results from the PMR RI study will help to provide clarity for these disparate results.
In summary, carrying out a RI study for eribulin as a PMR appeared to be supported by the combination of four factors including the following: (i) results of the mass balance study that eribulin was minimally metabolized; (ii) the observations of the impact of RI on PK; (iii) the association of PK exposure with increased neutropenia; and (iv) an unfavorable therapeutic window, where the approved dose is the MTD.
Vismodegib
Vismodegib (Erivedge, MW=421; Genentech, Inc., South San Francisco, California, USA) is a hedgehog pathway inhibitor initially approved for the treatment of adults with metastatic basal cell carcinoma or with locally advanced basal cell carcinoma. The recommended daily dose is 150 mg orally.
Vismodegib shows nonlinear PK with respect to dose, with no increase in steady-state plasma concentration on increasing the daily dose from 150 to 270 or 540 mg. The unusual PK properties of vismodegib have been attributed to two distinct processes: (i) solubility-limited absorption related to the poor and pH-dependent solubility of vismodegib and (ii) high-affinity saturable plasma protein binding. In the single-dose and multiple-dose absolute BA study of vismodegib in healthy individuals, it was observed that the single-dose BA was ∼32% and decreased considerably with multiple dosing 14. Vismodegib is a BCS class II molecule.
CYP3A4 inhibitors and inducers did not significantly alter the systemic exposure of vismodegib. When vismodegib exposure was evaluated with food, it was observed that a high-fat meal increased single-dose vismodegib exposure relative to the fasted state by up to 38%, with no corresponding change in the steady-state concentration (Css). Vismodegib elimination involves multiple pathways, with the most predominant metabolites being formed by cytochrome P450 (CYP) enzymes CYP2C9 and CYP3A4.
In a human mass balance study, 86.6% of the administered 14C vismodegib dose was recovered over a 56-day collection period, on average with 82.2 and 4.4% doses recovered in feces and urine, respectively. On the basis of the available data at the time of the NDA submission, the relative contribution of renal function to the overall systemic exposure of vismodegib could not be determined. A dedicated RI study was requested as a PMR at the time of NDA approval.
Subsequent to the US approval of vismodegib, the sponsor completed a study in patients with hepatic impairment, a DDI study with vismodegib as a victim, and a population PK analysis. Despite the observed metabolism of vismodegib by CYP3A4 and CYP2C9, the results from the HI study showed that there was no effect of mild, moderate, or severe HI on vismodegib PK; thus, any indirect effect of elevated uremic toxins on CYP enzymes in RI patients is not likely to alter the PK of vismodegib. This is further supported by results from a DDI study with vismodegib as a victim of CYP3A4 and CYP2C9 inhibition. The DDI study showed that inhibition of CYP enzymes did not alter the plasma exposure of vismodegib, presumably because of the complicated plasma protein binding and absorption kinetics. This provides further evidence that uremic toxins (less potent enzyme inhibitors or suppressors) would be unlikely to impact the PK of vismodegib. In addition, and perhaps most importantly, the results of a population PK analysis showed that RI (CLcr=30–80 ml/min) did not influence the PK of vismodegib 15. Finally, E–S analysis in the NDA showed no relationship of observed PK exposure with AEs.
In summary, a totality of results from a mass balance study, a DDI study, a hepatic impairment study, a population PK analysis, and E–S analysis provided support that there was a low risk for RI to impact the PK and safety of vismodegib, and thus formed the basis for FDA to consider the PMR fulfilled and support the dose selection for patients with RI.
Individual drugs that did not have a dedicated renal impairment study for the new drug application or as a postmarketing requirement
In this section, the 11 identified SM oncology drugs approved by the FDA during our survey period are reviewed in instances where a dedicated RI study was neither carried out during development nor required as a PMR, with the exception of bosutinib for the reason provided previously.
Vemurafenib
Vemurafenib (Zelboraf, MW=490; Genentech, Inc., South San Francisco, California, USA) is a kinase inhibitor indicated for the treatment of patients with nonresectable or metastatic melanoma with the BRAFV600E mutation. The recommended dose is 960 mg orally twice a day without regard to food. The PK of vemurafenib is approximately dose linear with (presumably) low oral BA on the basis of its BCS classification as a poorly soluble and permeable molecule (BCS class IV). Vemurafenib binds extensively to plasma proteins (>99%) and shows 7.4-fold accumulation at steady state. The apparent distribution volume (V/F) and apparent clearance (CL/F) are reported to be 106 l and 31 l/day, respectively.
Vemurafenib is primarily metabolized by CYP3A4. Metabolites of vemurafenib represent less than 5% of the parent compound in circulation. Vemurafenib is an inhibitor of multiple CYP enzymes as well as a substrate and an inhibitor of the efflux transporter P-gp. Results from clinical DDI studies indicate that vemurafenib can increase the plasma exposures of drugs that are CYP1A2, CYP2D6, and/or CYP2C9 substrates.
Vemurafenib was minimally excreted in the urine (0.97%) following an oral dose. However, as the absolute oral BA of vemurafenib was not determined, a definitive conclusion on the relative importance of renal elimination could not be made. Notably, an absolute BA study of vemurafenib is currently ongoing (https://clinicaltrials.gov/ct2/show/NCT02441465), which will allow a better understanding of the contribution of renal elimination toward vemurafenib disposition. To assess the impact of RI on the PK of vemurafenib, a population PK analysis was carried out including data from patients with normal renal function (n=353), mild (n=94), moderate (n=11), and severe RI (n=1), respectively. The results from this analysis indicated that RI did not alter the PK of vemurafenib. A limitation of this population PK analysis is that it might not mechanistically address the question of whether the PK of vemurafenib could be impacted by any secondary changes in CYP3A4 and/or P-gp activity or expression (e.g. because of inhibition by uremic toxins and/or hepatic impairment). However, carrying out a dedicated RI study with a small sample size (e.g. 6/group) would also unlikely provide additional information to dose selection in this special population, at least for patients with mild and moderate RI.
Additional support for not carrying out a dedicated RI study as a PMR came from the population PK analysis to assess the potential impact of hepatic impairment on the PK. The data included patients with normal hepatic function (n=158), mild (n=58), moderate (n=27), and severe HI (n=3), respectively, from a phase III study, with baseline bilirubin as a marker of hepatic function. The analysis indicated that there was no significant change in the CL/F or V/F of vemurafenib in patients with HI. The HI data suggest that the PK of vemurafenib is unlikely to be impacted by RI even if RI were to lead to secondary impaired hepatic function (e.g. inhibition of hepatic enzymes by uremic toxins).
As vemurafenib is mainly metabolized by CYP3A4, the FDA requested a reduced HI study and DDI studies with vemurafenib as an inhibitor of CYP3A4 as PMRs. Data from these studies should provide a more definitive assessment of changes in vemurafenib PK because of altered hepatic enzyme activity irrespective of direct or indirect causes, such as accumulation of uremic toxins (Table 1).
E–S analysis showed no association of PK exposure and AEs. In the USPI of vemurafenib, no dose adjustment is recommended for mild and moderate RI and, as conservatively stated, an appropriate dose has not been established for patients with severe RI.
In summary, the totality of vemurafenib data appeared to provide adequate justification for not carrying out a dedicated PMR RI study, which included the following: (i) minimal urinary excretion in the mass balance study; (ii) a population PK analysis with predominantly mild and moderate RI patients, which showed no effect of RI on PK; (iii) PK in HI patients (predominantly mild and moderate grades), which indicated no significant PK changes and suggested no effect of uremic toxins on vemurafenib PK; and (iv) an E–S analysis indicating no association of PK exposure and AEs.
Ceritinib
Ceritinib (Zykadia, MW=558; Novartis, East Hanover, New Jersey, USA) is a kinase inhibitor indicated for the treatment of patients with anaplastic lymphoma kinase-positive metastatic non-small-cell lung cancer. The recommended dose is 750 mg orally once daily on an empty stomach. Ceritinib has poor solubility and permeability (BCS class IV) and (presumably) low absolute oral BA. The PK steady state was reached following ∼15 days of dosing with 6.2-fold accumulation. The plasma protein binding (97% bound) and the large apparent distribution volume (4230 l) limit the amount of drug available to renal elimination. Ceritinib is the main circulating drug component in plasma. It is a substrate-dependent and a time-dependent inhibitor of CYP3A4, as well as a substrate of P-gp. Clinically significant DDIs were observed between ceritinib and CYP3A4 perpetrators. Concomitant treatment with ketoconazole increased ceritinib AUC and Cmax by 2.9-fold and 22%, respectively.
Following an oral dose of 14C-ceritinib, 92.3 and 1.3% of the radiolabeled doses were recovered in feces and urine, respectively. The 1.3% urinary recovery is likely an underestimation of the actual contribution of renal elimination; however, even with assumed low absolute BA (e.g. 20%), renal excretion would account for only 6.5% of the absorbed dose. This seems consistent with the population PK analysis (including 97 and 22 patients with mild and moderate RI, respectively) that showed no apparent effect on PK for mild and moderate RI. Exposure–response (E–R) analysis indicated a relationship of PK exposure to grade 3–4 AEs and other events.
Even though the results of the E–R analysis indicated a relationship between PK exposure to high-grade AEs, not carrying out a RI study appeared to be supported by a combination of data including the following: (i) a low amount of the drug in urine in the mass balance study; (ii) the ketoconazole DDI study, where the effect of CYP3A and P-gp inhibition enables an estimate of potential effect of elevated uremic toxins on ceritinib PK, and (iii) a population PK analysis showing no effect in mild to moderate RI patients.
Although there was no RI study issued as a PMR, on the basis of the available data on ceritinib metabolism and DDI potential, several clinical pharmacology studies were issued as PMRs. These studies included a food effect study, CYP3A and CYP2C9 related DDI studies, and a HI study.
Cabazitaxel
Cabazitaxel (Jevtana, MW=836; Sanofi-Aventis, Bridgewater, New Jersey, USA) is a microtubule inhibitor administered in combination with prednisone for the treatment of hormone-refractory metastatic prostate cancer. It is indicated at 25 mg/m2 every 3 weeks as a 1 h (intravenous) infusion with oral prednisone 10 mg daily. Plasma concentrations of cabazitaxel are described by a three-compartment PK model with α, β, and γ half-lives of 4 min, 2 h, and 95 h, respectively. Cabazitaxel shows approximately dose-linear PK without apparent accumulation. On the basis of the population PK analysis, steady-state volume of distribution and plasma CL of cabazitaxel were 4864 l and 48.5 l/h, respectively.
Cabazitaxel is a substrate of P-gp and is metabolized extensively by CYP3A4/5 (80–90%). On the basis of in-vitro studies, cabazitaxel is unlikely to be a perpetrator of CYP enzymes or transporters at clinically relevant concentrations.
In a human mass balance study, ∼80% of the 14C dose administered was recovered within 2 weeks of dosing. Cabazitaxel was primarily recovered in feces as metabolites (76% of the administered dose) and minimally recovered in urine (3.7% of the administered dose). Unlike vemurafenib with unknown absolute oral BA, a low amount of urinary recovery was observed after an intravenous dose of cabazitaxel, indicating that the absolute contribution of renal excretion to elimination would be negligible as absolute BA is 100% with intravenous administration. The population PK analysis, which included data from one severe, 14 moderate, and 59 mild RI patients, confirmed these findings. It is reasonable to believe that RI is unlikely to alter the PK of cabazitaxel directly on the basis of the totality of the available information.
The initial USPI stated that caution should be exercised in patients with severe RI or ESRD. Our review of the regulatory documents did not uncover a definitive reason for this wording, but we assume that the wording may have been based on two factors consistent with the guidance document: (i) insufficient amount of data from patients with severe RI and (ii) possible metabolism changes secondary to RI that could impact the PK of cabazitaxel. Future results from a PMR HI study may further form the basis of any dose-adjustment decision for patients with primary HI or extrapolation of a compromised metabolism of cabazitaxel secondary to elevated uremic toxins with RI.
Bosutinib
Bosutinib monohydrate (Bosulif, MW=530; Pfizer, New York City, New Jersey, USA) inhibits the Bcr-Abl kinase and Src-family kinases, and is indicated for the treatment of adult patients with chronic myelogenous leukemia (CML). The recommended dose is 500 mg administered orally once daily. Bosutinib is a BCS class IV compound and should be administered with food. Bosutinib binds extensively to plasma proteins (>99% bound) and shows dose-proportional increases in AUC and Cmax. Bosutinib is primarily metabolized by CYP3A4, with the metabolites M2 and M6 (both inactive) accounting for 44% of the parent drug exposure in the plasma. Induction or inhibition of CYP3A4 significantly alters the exposure of bosutinib. The apparent volume of distribution (Vss/F) is 6080 l for bosutinib.
In the human mass balance study, 91 and 3% of the 14C doses were recovered in feces and urine, respectively. However, as the absolute oral BA is unknown, the contribution of renal elimination could not be determined from this result alone.
Bosutinib Cmax and AUC increase approximately two-fold in patients with Child–Pugh A, B, and C hepatic impairment compared with healthy volunteers. As a result, a reduced daily dose of 200 mg (60% dose reduction) is recommended for CML patients with HI. These data suggest the possibility that uremic toxins may alter the metabolism/excretion of bosutinib in RI patients.
However, population PK analysis indicated that although the CL of bosutinib is reduced by ∼30% in patients with moderate RI (CLcr<25 ml/min), this effect was not considered to be clinically significant. Regulatory review documents indicated that the data in the population PK analysis were considered to be limited and were highly variable. In addition, because of a dosing error, the 14C radioactivity administered was 10-fold lower than the protocol specified dose (0.01 μCi instead of 0.1 μCi), calling into question the reliability of the human mass balance study results.
The sponsor initiated a dedicated RI study in 2010 (before the NDA submission). However, the data were not available when the NDA was submitted. Notably, the initial USPI stated that on the basis of population PK analysis, CLcr ranging from 25 to 120 ml/min had no meaningful influence on the exposure to bosutinib. Consequently, no dose adjustment was recommended for patients with RI. E–R analyses showed no relationship for the primary efficacy endpoint of major cytogenetic remission/major cytogenetic response at 24 weeks and no clinical meaningful association with safety events. Bosutinib was approved at the MTD dose with upward dose titration on the basis of tolerability.
In summary, the totality of data appeared to provide justification for not carrying out a dedicated RI study with bosutinib. These data included the following: (i) a population PK analysis showing no clinically relevant changes with RI and (ii) an E–R analysis showing no exposure correlation to safety or efficacy. Although the approved bosutinib dose of 500 mg is the MTD, the E–S analysis provides support for the label language of allowing for an upward dose titration to 600 mg for patients not achieving an adequate cytogenetic response 9.
Ponatinib
Ponatinib hydrochloride (Iclusig, MW=569; ARIAD Pharmaceuticals, Cambridge, Massachusetts, USA) inhibits the activity of Bcr-Abl and mutant Bcr-Abl kinases as well as other tyrosine kinases. It is indicated for the treatment of adult patients with CML or Philadelphia chromosome-positive acute lymphoblastic leukemia or blast phase chronic myeloid leukemia. The recommended dose is 45 mg orally once daily. Ponatinib binds extensively to plasma proteins (>99.9% bound). Following the daily administration of 45 mg of ponatinib, the CL/F, V/F, and T1/2 were 35 l/h, 1223 l, and 24 h, respectively. Ponatinib is mainly metabolized by CYP3A4. When coadministered with ketoconazole, ponatinib AUC0-∞ and Cmax increased by 78 and 47%, respectively. A ponatinib metabolite, AP24567, with a four-fold less potency than the parent molecule and with plasma exposures less than 2% of ponatinib, was not expected to be clinically relevant.
Fecal and urinary excretion accounted for 87 and 5% of the 14C-ponatinib dose, respectively. Although the absolute oral BA was not determined, at least 64% of the ponatinib dose undergoes phase I and II metabolism, suggesting a moderate to high absolute oral BA. Taken together, the available PK data suggest that renal excretion likely accounts for less than or equal to 8% of the absorbed dose of ponatinib.
Unlike the drugs discussed above, no meaningful PK data or analysis were available for patients with RI or HI who were administered ponatinib. Therefore, the interpretation of the contribution of the kidney to ponatinib elimination was solely based on the human mass balance data. These limited data support the USPI precautionary language, which states, ‘Although renal excretion is not a major route of ponatinib elimination, the potential for moderate or severe RI to affect hepatic elimination has not been determined’. In addition, a dedicated HI study was requested as a PMR, but a dedicated RI study was not required.
In summary, it appeared that not carrying out a RI study as a PMR is supported by a combination of data including: (i) a mass balance study showing a low degree of urinary elimination; (ii) a DDI study with ketoconazole showing that potent CYP inhibitors have an effect, thus allowing for extrapolation to a uremic toxin effect; (iii) a high projected BA, thus suggesting that an increase in absorption from RI would likely not increase exposure to a clinically relevant degree; and (iv) although the approved dose is the MTD, and E–S analysis showed a dose and AE relationship, emerging AEs can be effectively managed by typical dose reduction and interruption per USPI.
Ibrutinib
Ibrutinib (Imbruvica, MW=441; AbbVie, Inc., Lake Bluff, Illinois, USA) is a kinase inhibitor indicated for the treatment of patients with mantle cell lymphoma. Ibrutinib is currently dosed at 560 mg orally, once daily, with or without food. When taken with food, ibrutinib exposure increased approximately two-fold. It is likely a BCS class II molecule and has a large Vdss/F of 10 000 l with 97.3% bound to plasma proteins. The PK had a linear dose response, and no apparent accumulation was observed following multiple doses.
In the human mass balance study, ∼90% of the 14C dose was recovered, among which 80.6 and 7.8% was recovered in feces and urine, respectively. Only 1% parent drug was recovered in urine. Although the absolute BA of ibrutinib has not been evaluated, it is likely low because of extensive first-pass metabolism. Ibrutinib is mainly metabolized by CYP3A4 and to a lesser extent by CYP2D6. An active metabolite, PCI-45227, was identified. However, its contribution to efficacy is expected to be low as it has a lower plasma exposure (7%) and a 15-fold higher IC50 compared with ibrutinib. Consistent with these observations, a population PK analysis indicated that ibrutinib exposure was not altered in patients with CLcr more than 25 ml/min. Thus, the FDA deemed no dedicated RI study necessary, but it was cautioned that no data are available for severe RI or dialysis patients. These data clearly indicated that renal excretion has minimal direct impact on the PK of ibrutinib. However, it was not clear whether RI could impact ibrutinib PK through secondary mechanisms (e.g. by elevating levels of uremic toxins and thereby decreasing first-pass effect and/or metabolism kinetics).
DDI studies showed that ketoconazole increased and rifampicin decreased ibrutinib exposures significantly. In addition, preliminary data that were available from a dedicated HI study by the time of NDA showed a six-fold increase in ibrutinib exposure in patients with moderate HI. E–R analysis showed that there was no correlation of PK exposure with toxicity across two pivotal studies at doses up to 840 mg (approved dose 560 mg).
In summary, it appeared that not carrying out a RI study as a PMR was supported by a combination of data, which included: (i) a mass balance showing little renal excretion; (ii) a population PK analysis showing no effect of renal function at CLcr more than 25 ml/min; (iii) DDI and hepatic dysfunction data allowing extrapolation for a potential uremic toxin effect on hepatic metabolism; (iv) a favorable therapeutic window as no MTD was reached in phase I studies; and (v) E–S analysis that showed no association of PK-AE at doses up to 840 mg (approved dose=560 mg).
Trametinib
Trametinib dimethyl sulfoxide (Mekinist, MW=694; Novartis, East Hanover, New Jersey, USA) is an inhibitor of the mitogen-activated extracellular signal-regulated kinase 1 and 2 indicated for the treatment of patients with nonresectable or metastatic melanoma with BRAFV600E or BRAFV600K mutations. The recommended dose is 2 mg oral daily at least 1 h before or 2 h after food. It is likely a BCS class II molecule with 72.3% absolute oral BA and a median Tmax of 1.5 h. The V/F was estimated to be 1060 l with 97% bound to plasma proteins. On the basis of a population PK analysis, the CL/F and T1/2 were 4.9 l/h and 3.9–4.8 days, respectively. The PK exposure appeared to be linear with respect to dose with a six-fold accumulation with multiple doses.
The metabolism of trametinib is mainly through non-CYP450 pathways, including deacetylation, hydroxylation, and glucuronidation. In plasma, metabolites constitute less than 25% of drug-related material. No clinically significant DDIs have been identified. A trametinib metabolite, M5, has similar potency as the parent drug, but represents only 9.3% of the parent drug concentration in plasma.
Following oral administration of 14C-trametinib, more than 80% of excreted radioactivity was recovered in the feces whereas less than 20% of excreted radioactivity was recovered in the urine. Only less than 0.1% of the dose was excreted as the parent drug in urine. Considering the BA of 72.3%, renal excretion accounted for less than 27.7% of the absorbed dose. As the amount of parent drug in urine is negligible, the data suggest that RI should have limited direct impact on trametinib PK. A population PK analysis with data from patients with mild and moderate RI indicated no effect of RI on the systemic exposure of trametinib. Therefore, no dose adjustment is indicated for patients with mild to moderate RI, whereas an appropriate dose was not established for severe RI. To address the potential impact of hepatic impairment (including a secondary effect because of RI), a PMR for a dedicated HI study was issued.
In summary, it appeared that not carrying out a RI study as a PMR was supported by the a combination of data, which included: (i) a low amount of parent drug is in urine in the mass balance study: (ii) a high BA indicating a low impact if RI increased absorption; (iii) a population PK analysis indicating no effect of renal function on CL: (iv) an E–R analysis showing no relationship with progression-free survival or overall response rate, no relationship with safety; and (v) a favorable therapeutic window with an approved dose 2 mg that is 33% lower than the 3 mg MTD.
Axitinib
Axitinib (Inlyta, MW=386; Pfizer, New York City, New York, USA) is a kinase inhibitor indicated for the treatment of advanced renal cell carcinoma. Axitinib is administered orally twice a day at 5 mg with or without food. It is a BCS class II compound with an absolute oral BA of 58%. Axitinib binds extensively to plasma proteins (>99% bound), shows linear PK with respect to dose, and shows minimal accumulation with repeated dosing.
Axitinib is metabolized primarily by CYP3A4/5 and to a lesser extent by CYP1A2, CYP2C19, and UGT1A1. N-glucuronide and sulfoxide metabolites are the two major metabolites found in circulation, but their pharmacological activity is negligible. DDI studies with axitinib as a target showed that CYP3A4 inhibition and induction resulted in a clinically meaningful increase and decrease in plasma axitinib exposure, respectively. A human mass balance study showed that 64% of the 14C-axitinib dose was recovered, with 41 and 23% in feces and urine, respectively; intact drug was undetectable in urine.
Although a dedicated RI study was not carried out with axitinib, no starting dose adjustment is indicated for patients with pre-existing mild to severe RI. This appeared to be supported by a population PK analysis with a wide range of renal function, including 64 patients with moderate RI, five patients with severe RI, and one patient with ESRD. Enrollment of patients with RI to the axitinib clinical studies was possible because of the high prevalence of RI in the target patient population with renal cell carcinoma who had failed previous anti-cancer therapy. Indeed, among all the drugs reviewed in Table 2, axitinib is the only one with: ‘No starting dose adjustment is needed’, in the USPI for patients with mild to severe RI. Axitinib was also the only drug listed in Table 2 with no clinical pharmacology study as a PMR upon the initial NDA review.
In summary, a combination of data provided support for not carrying out a PMR RI study, including (i) little intact drug on the mass balance study and (ii) population PK analysis showing no renal covariate effect. Although E–R analysis indicated a dose-related increase for hypertension, fatigue, and diarrhea, the starting dose was approved with an upward titration (from 5 to 10 mg) to the MTD on the basis of patient tolerability, thus providing support for patient safety.
Cabozantinib
Cabozantinib (S)-malate (Cometriq, MW=636; Exelixis, South San Francisco, California, USA) is an inhibitor of tyrosine kinases and is indicated for the treatment of patients with progressive, metastatic medullary thyroid cancer. The recommended daily dose is 140 mg administered orally without food. It is likely a BCS class II compound on the basis of the assessment report by EMA (no official BCS classification/designation from the FDA). The absolute BA of cabozantinib was not determined. Cabozantinib binds extensively to plasma proteins (>99.7% bound), with limited distribution in RBC. On the basis of population PK analysis, the CL/F and V/F were 4.4 l/h and 349 l, respectively. The PK is approximately linear, with four- to five-fold accumulation after repeated doses.
Cabozantinib is a substrate of CYP3A4 and an inhibitor of P-gp. Strong CYP3A4 inhibitors can cause clinically significant increases in cabozantinib exposure when administered concomitantly. Some metabolites with much lower potency were reported. The parent drug accounted for 27.2% of the total drug substances in circulation and it was concluded that the metabolites do not contribute significantly toward the overall pharmacology.
In a human mass balance study, 81% of the 14C-cabozantinib dose was recovered after a single dose, with 54 and 27% recovered in feces and urine, respectively 16. However, the percentage of intact drug in the urine is unknown (Table 2). Cabozantinib is a victim of DDI, which allows extrapolation, such that uremic toxins could play a role in decreasing the rate of metabolism and/or excretion. Data on the impact of impaired hepatic function on the PK of cabozantinib were not available at the time of the initial NDA filing.
Results from a population PK analysis indicated no apparent increase in PK exposures in patients with mild or moderate RI. The exact numbers of patients with varying degrees of RI were not provided in the SBA (Table 4), but seemed sufficiently large for patients with normal and mildly or moderately impaired renal function. E–R analyses showed that PK exposure was not correlated to progression-free survival, but the first dose modification for adverse effects and QTc prolongation 9 were associated with time.
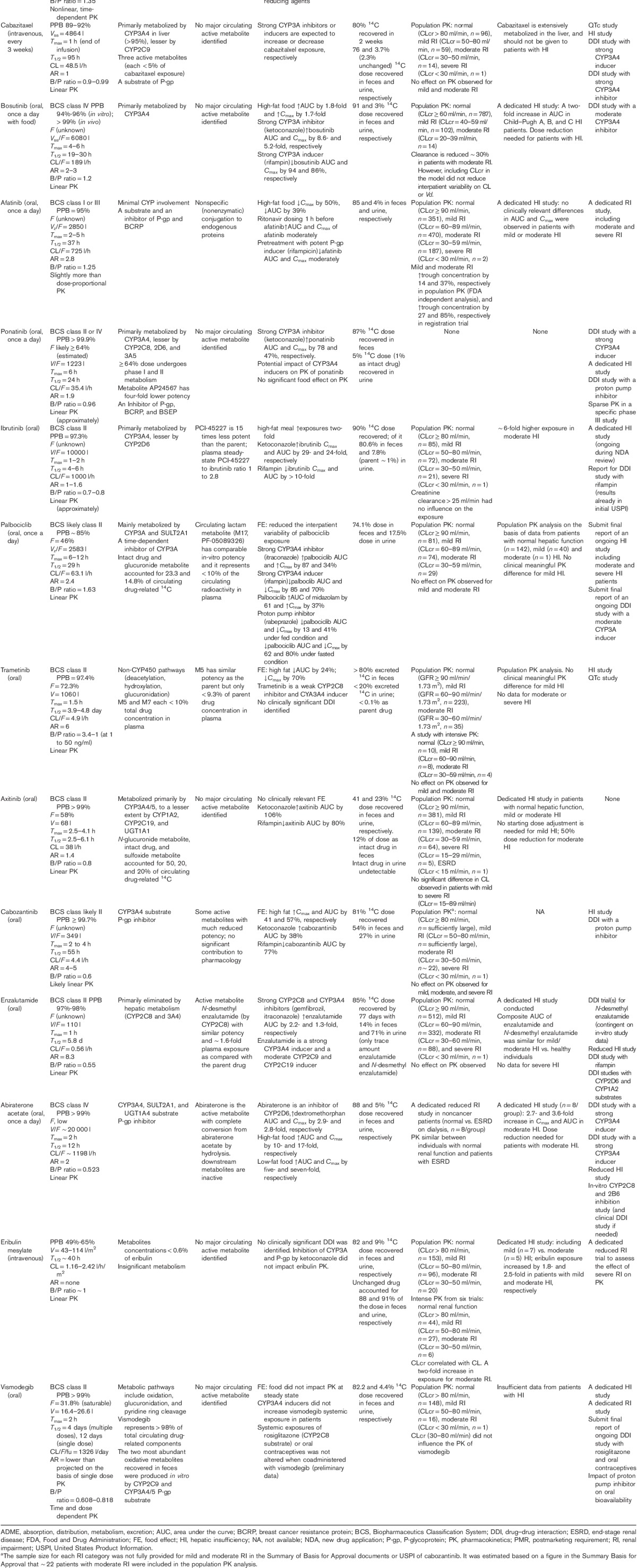
Table 4.
Clinical pharmacology characteristics for the 15-selected small molecule oncology drugs approved by the Food and Drug Administration during the survey period that did not perform a renal impairment study as a part of the new drug application or as a postmarketing requirement study
The USPI states that no dose adjustment is recommended for patients with mild or moderate RI. Notably, a dedicated HI study was requested as a PMR, the results of which could serve to assess the impact of altered drug-metabolizing enzyme/transporter function on the PK of cabozantinib.
In summary, support for not carrying out a PMR RI study for cabozantinib appeared to be primarily driven by the findings of the population PK analysis showing no increase of exposure in patients with RI.
Enzalutamide
Enzalutamide (Xtandi, MW=464; Medivation, Inc., South San Francisco, California, USA) is an androgen receptor inhibitor indicated for the treatment of patients with metastatic castration-resistant prostate cancer. It is administered orally 160 mg once a day with or without food. Enzalutamide binds extensively to plasma proteins (>97%), shows poor penetration into red blood cells, and is likely to have high oral absolute BA (BCS class II). Enzalutamide is primarily eliminated by hepatic metabolism (CYP2C8 and 3A4). The active metabolite N-desmethyl enzalutamide had similar potency as the parent drug, and showed plasma concentrations ∼1.6-fold that of the parent drug.
Enzalutamide is a strong inducer of CYP3A4 and a moderate inducer of CYP2C9 and CYP2C19; thus, substrates of such enzymes with narrow therapeutic windows should be avoided. In addition, enzalutamide is a substrate of CYP2C9 and CYP3A4; thus, strong CYP2C9 and CYP3A4 inhibitors can increase the exposure of enzalutamide. In theory, HI secondary to RI could result in decreased hepatic enzyme activities, elevated enzalutamide exposures, and complicated DDI with concomitant medications.
Results from the human mass balance study indicate that 85% of the 14C dose was recovered in 77 days following a single oral dose of enzalutamide. Fecal and urinary elimination accounted for 14 and 71% of the recovered dose, respectively. Although renal excretion of total radioactivity was a major route for dose elimination, only trace amounts of enzalutamide and N-desmethyl enzalutamide were detected in urine. Population PK analysis (on the basis of the parent concentration) showed that there was a small increase in enzalutamide exposure in patients with mild and moderate RI. However, this effect was not considered to be clinically relevant.
A dedicated HI study was carried out to support the NDA. The results showed that the composite AUC of enzalutamide plus N-desmethyl enzalutamide was similar for mild or moderate HI compared with patients with normal hepatic function. A second reduced HI study was requested as a PMR to assess the impact of severe HI on the PK of enzalutamide.
The renal route of elimination appears to be significant only for inactive metabolites of enzalutamide. The population PK analysis indicated only small changes in PK of parent drug with RI. Although enzalutamide is a substrate of CYP enzymes (PK could thus be altered because of the decreased activity of these enzymes through uremic toxins or hepatic impairment), there was no impact of hepatic impairment on the AUC of enzalutamide and the active N-desmethyl metabolite. Estrogen receptor analysis showed no relationship between exposure and overall survival, or safety at the approved dose of 160 mg. There were no clinically meaningful E–R relationships for fatigue, flushing, headache, or hypertension within the limited exposure range for 160 mg/day. In addition, enzalutamide has a favorable therapeutic window, with the approved dose of 160 mg being 33% lower than the 240 mg MTD identified in early clinical trials. For patients with calculated CLcr of at least 30 ml/min, no enzalutamide dose adjustment was necessary and no further RI assessment was requested as a PMR.
In summary, the rationale for not carrying out a PMR RI study for enzalutamide appears to be supported by a combination of data including (i) trace amounts of parent and active metabolite in the urine in the mass balance study; (ii) a population PK analysis showing no relevant effect of RI on CL; (iii) no effect of mild or moderate HI on PK suggesting no uremic toxin effect on metabolism or excretion; and (iv) a favorable therapeutic window with no E–S relationship at the approved dose, and with the approved dose 33% lower than the MTD.
Palbociclib
Palbociclib (Ibrance, MW=448; Pfizer, New York City, New York, USA) is a kinase inhibitor indicated in combination with letrozole for the treatment of postmenopausal women with estrogen receptor-positive and human epidermal growth factor receptor 2-negative advanced breast cancer. The V/F was estimated to be 2583 l with 85.3% plasma protein binding (concentration range: 0.5–5 µg/ml). The geometric mean CL/F was 63.1 l/h and the mean plasma T1/2 was 29 h in patients. The recommended dose is 125 mg oral, once daily with food for 21 consecutive days, followed by 7 days off treatment to comprise a complete cycle of 28 days. The PK exposure appeared dose linear, with a 2.4-fold accumulation at steady state. The BCS classification of palbociclib was masked in the FDA’s ‘SBA’ document, but according to EMA’s assessment, palbociclib was considered a BCS class II compound.
The metabolism of palbociclib is mainly through hepatic metabolism with minimal renal elimination. In vitro, palbociclib is metabolized by CYP3A4 and SULT2A1. Following an oral administration of 14C-palbociclib, the primary metabolic pathways involved oxidation and sulfonation, with acylation and glucuronidation contributing as minor pathways. Palbociclib is extensively absorbed, metabolized, and excreted in feces (74.1%) and urine (17.5%), respectively. The urinary and fecal recovery of unchanged palbociclib was ∼6.9 and 2.3% of the administered dose, respectively. In plasma, palbociclib and a major glucuronide conjugate (pharmacological activity not assessed) accounted for 23 and 14.8% of circulating drug-derived entities, respectively. PF-05089326, a lactam metabolite of palbociclib, showed comparable pharmacological potency as the parent drug, but only accounted for less than 3% of drug-related radioactivity in plasma. A full in-vivo characterization of the metabolite was not performed. In DDI studies, coadministration of itraconazole, a potent CYP3A inhibitor, increased plasma palbociclib AUC by 87% and coadministration of rifampin, a potent CYP3A inducer, decreased palbociclib AUC by 85%.
Palbociclib has also been shown to inhibit CYP3A activity in a time-dependent manner. When coadministered with the anesthetic midazolam, a sensitive CYP3A4 substrate, palbociclib induced a 61% increase in the plasma midazolam AUC compared with the administration of midazolam alone.
A population PK analysis from patients with mild and moderate RI indicated no effect of RI on the exposure of palbociclib, thus providing support that dose adjustment for patients with mild to moderate RI is not required. An appropriate dose of palbociclib was not established for patients with severe RI. On the basis of a population PK analysis, mild HI had no effect on palbociclib exposure, suggesting that a uremic toxin effect on metabolism and/or excretion may not translate to a clinical effect on palbociclib PK. A clinical HI study was ongoing during the NDA filing, with a final study report requested as a PMR by FDA. E–R analysis indicated that PK exposure was associated with a greater reduction in the absolute neutrophil count at the approved starting dose (MTD).
In summary, the rationale for not carrying out a PMR RI study appears to be supported by a combination of data including (i) a low percentage of intact drug in the urine in the mass balance study and (ii) the results of the population PK analysis showing no suggested dose modifications for mild to moderate RI.
Table 4 reviews the characteristics of the supportive clinical pharmacology data for the 11 drugs that did not carry out a RI study as a part of the NDA or as a PMR study.
Discussion
Recent advances in renal impairment assessments
Assessing the impact of RI on drug PK to inform dose selection for patients with CKD has been an important aspect of clinical pharmacology during drug development 17–20. Our current understanding that RI could impact the absorption and disposition of drugs with minimal renal elimination has been based on nonclinical research 5,7 and clinical observation 4,6. It is generally agreed that accumulating uremic toxins and inflammation, as consequences of CKD-induced RI, might alter the expression and/or activity of drug-metabolizing enzymes, transporters, and drug-binding proteins in vivo.
Dose adjustment in patients with RI has been documented previously for SM oncology drugs. The majority of these drugs are mainly eliminated by the kidney 18,21–24. In a special issue of the Journal of Clinical Pharmacology in February 2012 25, major aspects of RI in drug development were discussed. It included reviews of the evolving concepts, mechanism, methods, and regulatory requirements on assessing the impact of RI in drug development, as well as dose adjustment in RI patients, and special considerations of hemodialysis 8,26–37.
We believe that the knowledge obtained from the recently approved SM oncology drug studies allows for a better understanding of the impact of RI on the PK of drugs with minimal renal excretion. The studies also underscore the importance of the regulatory considerations and how to efficiently assess the impact of RI on drug PK. This information is critical for drug labeling to better inform health care providers on how to treat their patients. The current FDA and EMA guidance documents on assessing the impact of RI on PK recommend carrying out at least a reduced PK study for the majority of SMs, irrespective of therapeutic class 2,3. Although the EMA guidelines do allow, with justification, a population PK analysis of sparse PK data if a conventional study is not feasible, the FDA does not have specific language in this respect. Our analysis of the FDA review documents for drug approvals between 2010 and the first quarter of 2015 suggests that special considerations be made for SM oncology drugs, taking into account the benefit/risk in patient populations and the totality of the relevant data with respect to how RI might impact PK.
Practical challenges to enroll renal impairment patients
RI assessment has been a practical challenge for SM oncology drugs. For dedicated RI studies, cancer patients with RI (especially moderate and severe RI) willing to participate in the studies are limited. On the basis of consultations with major contract research organizations carrying out RI studies in cancer patients, the estimated enrollment rate for patients with severe RI is extremely low. Although RI studies with SM oncology drugs can be carried out in individuals without cancer, there are often ethical and safety concerns when a therapeutic dose or multiple doses are needed. Furthermore, noncancer individuals may not be appropriate for such studies because of various reasons (e.g. reprotoxicity, clastogenicity, carcinogenicity, etc.), where the benefit/risk might be different in patients with cancer.
Regulatory guidance documents set the expectation that nearly all SMs in clinical development will need to be assessed in RI patients. These studies would have to be carried out during earlier stages of clinical development before the safety of the investigational drug has been assessed thoroughly in large numbers of patients to identify important but less frequent AEs if they are to provide information to inform later stage trial dosing and the development of eligibility criteria. When RI has a marked impact on the PK of an investigational drug with a narrow therapeutic index, there is a safety risk to study participants. Furthermore, requiring clinical RI studies for nearly all SMs in development will decrease the availability of an already limited patient population. In our opinion, this valuable resource of patient population should be prioritized on the basis of available PK and safety data as well as the clinical need.
Practically, pharmaceutical companies may be conservative about enrolling patients with moderate or severe RI in later stage trials because of the risk of ‘contaminating’ the safety and/or efficacy results, despite the totality of information, suggesting a lack of RI impact on PK. In addition, regulatory agencies may have concerns over ensuring adequate safety measures for enrolling severe RI patients in phase II or III studies. Typically, late-stage clinical studies consist of data collected from patients with moderate and severe RI as a result of protocol deviation or because of renal function deterioration during the course of the study. Consequently, RI data from late-stage clinical trials may have limited value in informing the drug label at the initial approval for a SM oncology drug.
As summarized in Table 5, when no clinically meaningful PK difference was observed between patients with moderate RI and normal/typical renal function on the basis of the totality of data, a dedicated RI study was not required (e.g. vemurafenib, cabazitaxel, bosutinib, ibrutinib, trametinib, cabozantinib, and enzalutamide). In our opinion, patients with mild and moderate (along with potentially severe) RI should be considered for enrollment in late-stage clinical trials if renal excretion is determined to be a minor route of elimination during early clinical development. Obviously, sufficient safety measures would need to be in place; for example, using a stepwise approach to first enroll and evaluate patients with mild RI before enrolling patients with moderate RI. The target number of patients with RI to assess the impact on PK depends on the size of the trial, the prevalence of RI among the target disease population, as well as the PK variability and confidence in the results. In addition, factors such as performance status, other comorbidities, concomitant medications, etc., in the RI patient population would also have to be considered. Taking a cautious approach during clinical development by enrolling patients with RI in late-stage clinical studies could provide helpful information to adequately assess the impact of RI on PK and safety, and improve the quality of information in the label.
Table 5.
Summary of the totality of clinical pharmacology data for the 11 drugs that did not perform a new drug application or postmarketing renal impairment study
Key clinical data and analysis that inform the risk of renal impairment
The draft guidance issued by the FDA in 2010 provided a modified decision tree for conducting a RI study 2, according to which a dedicated RI study is generally required irrespective of the extent of renal excretion, acknowledging that drugs eliminated by nonrenal pathways can have their rate and extent of oral ADME altered by uremic toxins. Nonetheless, considering the totality of data from other studies, including human mass balance, absolute oral BA, population PK, HI, DDI, active metabolites, and E–R analyses, etc., allows for a rational and alternative approach to adequately assess the impact of renal function on drug PK and safety, thereby justifying the dose selection for patients with RI, and informing the need for a dedicated RI study.
Mass balance
Urinary dose recovery, as the intact drug and/or active metabolites, remains the primary basis to assess the potential impact of RI on PK when considering the route of elimination. Vemurafenib, cabazitaxel, ibrutinib, trametinib, cabazitaxel, and ponatinib serve as good examples. All of these drugs showed low renal excretion of total radioactivity (≤5%) or intact drug after a single oral dose, suggesting limited renal involvement in drug elimination. These findings were critical for dose selection for patients with RI and supported the sponsors’ decision to not carry out a dedicated RI study. However, bosutinib had low but questionable urinary dose recovery because of a dosing error in the human mass balance study and the sponsor volunteered to carry out a dedicated RI study before the NDA filing. In general, we found that mass balance studies were not the sole or the primary element for decision-making for postmarketing RI trials, but were combined with other data, usually population PK analyses, as in the case of axitinib, cabazitaxel, palbociclib, and vemurafenib (Table 4).
Absolute bioavailability
We noted that for the majority of drugs, absolute BA was not assessed. A proposed best practice is to assess the absolute BA of a drug early in development to facilitate interpretation of mass balance study data on the route of elimination and the potential need for a RI study. The importance of the absolute BA may be shown in the two following example scenarios: (i) if a drug has a minimal fraction absorbed (e.g. 10% absolute oral BA), 5% dose by renal excretion accounts for 50% of the absorbed dose, and RI is more likely to directly impact the PK, and (ii) if a drug has a reasonable fraction absorbed (e.g. 50%), 5% dose by renal excretion is 10% of an absorbed dose, and supports that RI would have a minor direct impact on PK.
Among the drugs in Table 2, cabazitaxel is administered intravenously with 100% BA. The urinary 14C dose recovery of 3.7% indicated negligible impact of RI on PK. For some orally administered drugs with determined absolute oral BA (e.g. trametinib) or likely high oral BA such as ponatinib (estimated to be at least 65% according to NDA documents), the impact of RI on PK was assessed with better confidence on the basis of the urinary dose recovery data.
Unfortunately, the absolute oral BA was not determined for most orally dosed SM oncology drugs covered in this review (Table 2). Consequently, a definitive conclusion on the relative importance of renal elimination on PK generally cannot be made with only human mass balance data. In these cases, further support and conclusions of the impact of RI on the drug are obtained by other means, typically a population PK analysis (Tables 2, 4, and 5).
Model-based approaches – population pharmacokinetics, exposure–response, and physiological-based pharmacokinetics
Population PK has been an important tool to assess the impact of RI on PK. The estimated CLcr on the basis of serum creatinine determinations has been the measure of renal function used in the population PK analysis for the SM oncology drugs discussed in this review. This reflects the clinical preference for a calculated CLcr over the alternatives such as measured or estimated glomerular filtration rate (GFR), especially in clinical trials that are not designed for RI assessment. In addition, baseline CLcr was usually used in such analysis despite possible deteriorations/fluctuations in CLcr during clinical studies. Incorporation of CLcr as a time-varying covariate has been challenging because of infrequent CLcr determination, sparse PK data, high PK variability, factors that confound using serum creatinine as a surrogate for renal function (e.g. concomitant medicines that inhibit OCT2 or MATE1 transporters), and/or technical difficulties in quantifying the effect in population PK modeling. A total of 21 out of the 29 SM oncology drugs approved by the FDA during our survey period included population PK analysis in the initial NDAs (Table 2). Among the remaining eight drugs, ponatinib had a waiver for a dedicated RI study on the basis of mass balance study data as well as likely high absolute oral BA; abiraterone, vandetinib, carfilzomib, and ruxolitinib had a dedicated RI study before the NDA to inform the adjustment for patients with RI; pomalidomide and deferiprone had a dedicated RI study as PMR; and regorafenib had population PK as a PMR to further assess the risk of RI. When a population PK analysis was carried out to assess the impact of RI on PK, no dedicated RI study was carried out (with the NDA submission or as a PMR) in 13 (62%) of 21 of instances. It is noteworthy that bosutinib had a dedicated RI study, but no data were available when the NDA was reviewed. On the basis of its USPI, the population PK analysis appeared to be sufficient to inform the bosutinib dose selection for patients with CLcr of at least 25 ml/min.
Population PK analysis enables an integrated and quantitative analysis across available clinical PK data. Population PK is often expected to be confirmatory of mass balance study results with respect to the relative contribution of urinary excretion. However, sometimes, it could suggest the presence or the lack of impact of RI on drug PK that is not consistent with the urinary dose recovery data from a mass balance study. For example, eribulin is administered intravenously and the mass balance study indicated minimal renal contribution to eribulin elimination (9%). However, population PK analysis indicated a clear correlation between CLcr and eribulin CL that was determined to be of clinical significance. As a result, a lower starting dose of eribulin is recommended for patients with moderate RI. Another example is cabozantinib with 27% urinary dose recovery of total radioactivity and with unknown absolute oral BA 16. Population PK analysis showed no PK difference associated with RI, and thus no dose adjustment is recommended for patients with mild or moderate RI.
The sample size requirement for population PK analysis to inform dose selection for varying degrees of RI has not been clearly defined. On the basis of the FDA review documents, the RI sample size of population PK analyses was available for some of the drugs of interest (Table 4). Among these drugs, the number of patients with mild and moderate RI ranged from 16 to 470 and from 1 to 187, respectively. In most cases, the number of patients with severe RI or ESRD has been very limited (n=1 for vemurafenib, cabozantinib, vismodegib, and enzalutamide, and n=2 for afatinib), and was considered insufficient to inform dose selection for patients with severe RI in most USPIs. One exception was axitinib (n=5 for severe RI), for which no dose adjustment is necessary for patients with severe RI. No ESRD patients were included for population PK analyses for all drugs except axitinib (n=1). The clinical meaningful threshold of change in PK exposures may differ among drugs; therefore, the findings of the RI in population PK analysis must be integrated with E–R analysis to further define the clinical relevance of any expected change in PK on efficacy or safety (Table 5).
E–R analysis has become a fundamental component of drug development, NDAs, and regulatory reviews. During FDA review, exposure–efficacy, and E–S are critical components for determination of the optimal dose or justification of the label dose 9. E–R analyses are an important component in assessing the totality of data for the decision as to the need for a RI study during clinical development or as a PMR study. For example, if a drug has some degree of renal CL (based on mass balance study) and a steep E–S curve (a small change in PK produces toxicity), then a RI study may be warranted to better describe the dose across varying degrees of RI. Conversely, if a drug shows a statistically significant effect of RI on population PK analysis, but the degree of PK exposure increase with RI is shown to be of no clinical relevance to E–S analysis, a RI study or dose adjustment for RI patients may not be necessary.
Recently, physiologically based pharmacokinetic (PBPK) modeling and simulation have been applied to describe the observed PK data of nonrenally cleared drugs (i.e. sildenafil, telithromycin, and repaglinide) in patients with varying degrees of renal function 36. Within the chronological window of this review on SM oncology drugs, there were no instances of PBPK analysis; however, PBPK modeling is an attractive modeling technique and is increasingly being used during drug discovery, development, and regulatory decision-making. In addition, PBPK models incorporate intrinsic (e.g. genetics, demographics, diseases) and extrinsic factors (e.g. DDIs), and thus have the potential to allow integrated and mechanistic understanding of the effect of varying renal functions on PK of SM oncology drugs in a variety of clinical circumstances 38–41. As this technique is further developed, it is likely that PBPK will have more applications in drug development and regulatory decisions for SM oncology drugs.
Hepatic impairment data
The 2010 draft FDA guidance on RI assessment is largely based on the scientific observations that some drugs with minimum renal elimination have significantly different PK exposure in patients with RI. This has been attributed to elevated uremic toxins, which in turn cause secondary changes in the expression and/or the activity of drug metabolic enzymes and transporters; thus, the outcome is the same as a decreased expression of these enzymes or transporters because of HI. Therefore, a HI study could theoretically serve as a surrogate for a reduced RI PK study. The potential utility and interpretation of HI study results would be that if HI data indicate no change in PK exposure in patients with impaired hepatic function, then one can conclude that altered PK exposure in RI patients because of such secondary effects of elevated uremic toxins is unlikely. For example, vemurafenib showed negligible (0.97%) dose recovery in urine, but because the absolute oral BA was not determined, the relative contribution of renal elimination to the overall drug disposition was unclear. Population PK analysis suggested no apparent effect of mild (n=94) or moderate (n=11) RI on the PK of vemurafenib. In addition, population PK analysis showed no meaningful impact of mild (n=58), moderate (n=27), or severe (n=3) HI on the PK of vemurafenib. Combining the population PK analysis with both RI and HI, no dose adjustment is recommended for mild and moderate RI in the USPI of vemurafenib.
Drug–drug interaction data
The strategy of using of DDI data is similar to that described above for HI, which surrounds the concept and observations that uremic toxins may cause secondary changes in the expression and/or the activity of drug-metabolizing enzymes and transporters.
Therefore, if the DDI assessments indicate a low likelihood of DDI, it helps in alleviating concerns that uremic toxins in RI may impact PK. Conversely, if an enzyme/transporter inhibitor such as ketoconazole had an effect on PK, RI could therefore result in alteration of the drug’s PK behavior. Thus, the lack of a DDI from an enzyme/transporter inhibitor/inducer would provide support that uremic toxins are unlikely to impact the drug’s PK by the tested enzyme/transporter, and support the strategy of not carrying out a dedicated RI study. For example, vismodegib was tested in DDI studies, which indicated that vismodegib PK is not altered when coadministered with strong chemical inhibitors of CYP enzymes of interest; therefore, it is highly unlikely that the presumed less potent effect of uremic toxins would impact PK. We believe that the addition of DDI findings to the totality of data strengthens the decision-making for carrying out a RI study.
Therapeutic window
Similar to that described for E–R analysis, the therapeutic window is a required element to be integrated into the totality of clinical data for determination of the strategy for a RI study during clinical development or as a PMR study. For example, enzalutamide and trametinib proposed label doses were ∼33% lower than the MTD identified in early clinical studies. Neither drug was required to carry out a RI study, which was supported by clinical pharmacology data. The knowledge of the MTD and therapeutic window provided a comfort margin for the safety of the approved dose. For axitinib, although the MTD of 5 mg twice daily was defined in early clinical trials, knowledge of the therapeutic window and interpatient variability allowed for an individual dose titration from 5 mg, twice daily, up to 10 mg, twice daily, irrespective of the renal function to maximize its efficacy 9.
Active metabolites
The presence of active metabolites and how RI impacts the systemic exposure of these metabolites should also be assessed. In theory, RI could either slow down the formation and/or the elimination of the metabolites depending on the formation kinetics and disposition properties of the metabolites.
It can be extremely challenging to characterize the impact of RI on the PK of multiple active moieties as the formation and elimination kinetics can be governed by different mechanisms and could be differentially impacted by RI. If a drug has multiple active metabolites that are mainly excreted by the kidney, a detailed assessment of the impact of RI on the PK of the intact drug as well as the active metabolites is likely warranted. A classic example is capecitabine 42. The PK exposures of capecitabine and 5-fluorouracil (the active metabolite) did not change significantly in patients with varying degrees of renal function. However, two other toxic metabolites, 5′-doxifluridine and α-fluoro-β-alanine, showed 23 and 109%, increases, respectively, in steady-state AUC for a 50% reduction in CLcr. Further analysis indicated higher incidences of grade 3/4 adverse effects with increasing 5′-doxifluridine AUC. On the basis of the results, a lower starting capecitabine dose was recommended.
Among the recently approved SM drugs, five had active metabolites identified. These included enzalutamide, ibrutinib, cabozantinib, palbociclib, and trametinib (Table 4). More detailed descriptions of the role of the active metabolites of these drugs are provided in the discussion for the individual drugs below. In the case of ibrutinib and cabozantinib, the lower potency of their metabolites appeared to play a small role in their pharmacology and thus decision-making in RI. Although comparable potency of the metabolites to the parent was noted for palbociclib and trametinib, the low circulating levels of these metabolites were likely of little clinical relevance. Enzalutamide is worthy of additional discussion. Although the active metabolite d-desmethyl enzalutamide had higher plasma exposures than the parent drug (1.6-fold), it appeared unnecessary to study the impact of RI on its PK. This was because N-desmethyl enzalutamide is unlikely excreted in the urine as suggested by the human mass balance data that showed only trace amounts of N-desmethyl enzalutamide in urine despite the high urinary 14C dose recovery (≥71%). An HI study showed comparable exposures of enzalutamide and N-desmethyl enzalutamide in patients with normal or mildly/moderately impaired hepatic function. The population PK analysis was thus limited to the parent drug only, but suggested that enzalutamide CL/F was not impacted by RI. As a result, no dose adjustment was deemed necessary for patients with CLcr of at least 30 ml/min on the basis of PK and safety information.
Regulatory considerations
As SM oncology drugs do not belong to the three categories exempted from the FDA’s requirement for a dedicated RI study (Fig. 1), it could be interpreted that a dedicated RI study be carried out before the NDA or issued as a PMR for most of the SM oncology drugs approved since 2010. Notably, only eight (28%) of the 29 drugs had results from a dedicated RI study included in their initial NDAs and 11 (38%) additional drugs had a dedicated RI study included in their list of PMRs (Table 2). A total of 10 drugs had neither a dedicated RI study before the NDAs nor RI study as PMR. This apparent difference between regulatory requirement and practice deserves further review and discussion.
On the basis of our survey, the decision tree provided in the FDA draft guidance appears to be conservatively oversimplified as the requirement for carrying out a dedicated RI study is not based on all relevant clinical pharmacology data and integrated analyses − the approach that we propose here.
Although it has been generally accepted that RI can impact hepatic enzyme and transporter levels/activities through elevated uremic toxins, it is important to recognize that a dedicated RI study may not be the best way to assess the secondary effects of RI. Practically, when a RI study is carried out, neither the levels of uremic toxins nor the hepatic enzyme/transporter activities are monitored. In addition, patients with signs of HI or poor physiological conditions (e.g. ECOG≥2) are usually excluded from RI studies. With the small sample size of such RI studies, it is possible that the results from these RI studies do not represent the target population of interest.
It should be noted that the definition and classification of RI vary between regulatory agencies. A comparison of the different RI categories among the agencies is provided in Table 6. Although acceptable correlations exist between GFR and CLcr, and between measured and estimated CLcr, these measurements differ, especially in the lower end of renal function 33. In addition, the cutoff values for each category of renal function measurement are different. The recommended cutoff values from the FDA and the EMA are not specific for oncology drugs, whereas the National Cancer Institute guidance provides more clinically relevant cutoffs for oncology drug development. It is also noteworthy that the EMA classifications of renal function from 2004 were revised in 2014 to abandon the use of body size-adjusted GFR (in ml/min/1.73 m2) to absolute GFR (in ml/min) as the latter was believed to better reflect CL of renally filtrated drugs.
Table 6.
Comparison of definition and classification of renal impairment by various health authorities
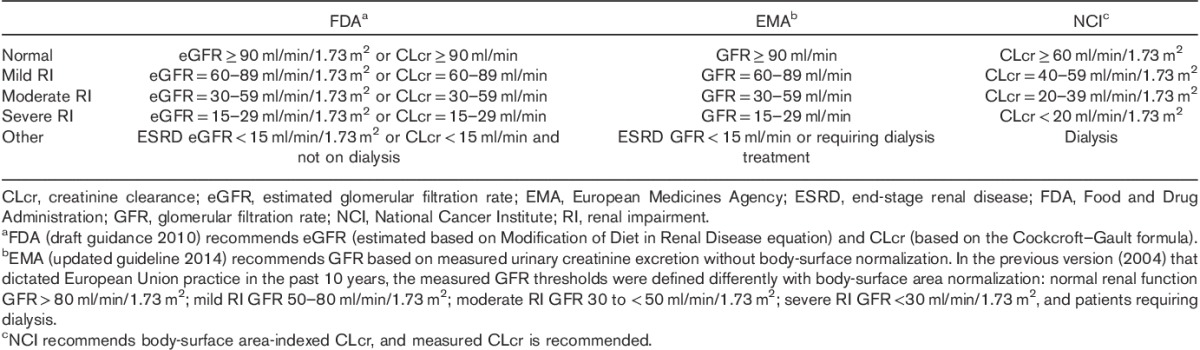
For the drugs in Table 3 that have RI classification mentioned in their initial USPIs, the FDA-recommended categorization (or new EMA-recommended categories since 2014) was used for 14 drugs (i.e. vemurafenib, ceritinib, afatinib, palbociclib, trametinib, axitinib, crizotinib, dabrafenib, idelalisib, lenvatinib, regorafenib, ruxolitinib, enzalutamide, and abiraterone); the EMA-recommended categorization (cutoff values before the updated 2014 guideline, Table 5 footnotes) was used for seven drugs (i.e. cabazitaxel, eribulin, vismodegib, carfilzomib, olaparib, panobinostat, and vandetinib). This may not impact the population PK and/or mathematical regression analysis in which CLcr or GFR was used as continuous variables. However, using different cutoff values could have implications in the result interpretation and dose adjustment in practice. The renal function defined as ‘normal’ in the FDA and the EMA guidelines may be too high to represent the target treatment population depending on the demographics of the disease. The different cutoffs for mild, moderate, and severe RI could cause confusion in dose adjustment by RI category and need to be specified in prescribing information 43.
In addition, sponsors seemed to use a single set of RI categorization in population PK analysis to support both NDA and EMA for a drug (e.g. ibrutinib and cabozantinib with EMA cutoff values in previous RI guideline, and bosutinib with the National Cancer Institute categorization). This was not a problem when renal function was treated as a continuous variable (e.g. bosutinib, ibrutinib), but reanalysis/recategorization was needed by the FDA when dose recommendation in USPI was provided by RI category per FDA guidance (e.g. cabozantinib).
An integrated approach for assessing the impact of renal impairment on pharmacokinetics
The 2010 draft FDA guidance recommends a RI study for all nonrenally eliminated drugs with a few exceptions. The questions remain on how to best understand the need for a dedicated RI study in SM oncology development and how to determine the true impact of RI on PK. In this respect, the current decision tree in the 2010 draft FDA guidance (Fig. 1) provides high-level guidance; however, it remains unclear how exactly a dedicated RI study can help address the regulatory concern on PK alterations because of changes in drug metabolism/transport secondary to RI.
Here, we propose an integrated approach to assess the impact of RI on PK during clinical development of SM oncology drugs. In our proposal, which requires the parenteral administration of the drug, the direct contribution of renal function on the urinary excretion of the drug would be based on the urinary 14C dose recovery determined from the human mass balance study with determination of the intact drug and major active metabolites and data on absolute BA, a cutoff value of less than 30% urinary recovery of the absorbed dose as active components would suggest minimal direct involvement of the kidney, which is consistent with the current guidelines. Results from the mass balance/BA study (or studies) would be integrated with available clinical trial data. A number of patients with mild to moderate RI would need to be enrolled in the clinical trials. Approximately 10 patients with moderate RI might provide informative PK data; however, this number is contingent on the amount of PK data collected per patient, the degree of impact of RI on CL, the interpatient variability, and the therapeutic window. Statistical inference and confidence intervals would be determined for the population PK analysis for the assessment. If the data consistently show a lack of impact of RI on drug PK, it would strongly support the notion that moderate RI has negligible clinical impact on drug PK. Analysis of possible secondary effects of RI on other elimination pathways would not be a core component of the RI assessment. Instead, the impact of HI on PK and DDI studies would be evaluated independently of RI for drugs primarily eliminated through nonrenal pathways.
Conclusion
Our review of FDA documents during the survey period (2010 through the first quarter of 2015) allowed us to better understand how the emerging knowledge of RI and uremic toxins and their effects on drug PK has impacted clinical drug development and regulatory assessment of SM oncology drugs. Overall, the strategy of the FDA review to assess the need for carrying out RI studies during clinical oncology development or as a PMR study incorporates the integration of the totality of data, including mass balance, absolute BA, DDI, hepatic dysfunction, population PK, E–R (safety-efficacy) analysis, and the therapeutic window for optimal guidance and determination of the optimal dose for the RI specific population. We propose further discussion of the ‘totality of data’ approach in future discussions of the guidance document for PK studies in oncology patients with RI.
Acknowledgements
Genentech Inc. provided support to Anshin Biosolutions for support of third-party assistance in editing this manuscript.
This survey received funding from Genentech Inc., a member of the Roche Group, South San Francisco, California, USA.
Conflicts of interest
All the authors are employees and stockholders in their respective companies.
References
- 1.US Food and Drug Administration. Guidance for industry: pharmacokinetics in patients with impaired renal function – study design, data analysis and impact on dosing and labeling; 1998. Available at: http://www.fda.gov/downloads/Drugs/Guidance Compliance Regulatory Information/Guidances/ucm072127.pdf. [Accessed 14 May 1998].
- 2.US Food and Drug Administration. Guidance for industry: pharmacokinetics in patients with impaired renal function – study design, data analysis, and impact on dosing and labeling; 2010. Available at: http://www.fda.gov/downloads/Drugs/…/Guidances/UCM204959.pdf. [Accessed 10 March 2010].
- 3.European Medicines Agency. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function; 2015. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/02/WC500200841.pdf. [Accessed 17 December 2015].
- 4.Huang SM, Temple R, Xiao S, Zhang L, Lesko LJ. When to conduct a renal impairment study during drug development: US Food and Drug Administration perspective. Clin Pharmacol Ther 2009; 86:475–479. [DOI] [PubMed] [Google Scholar]
- 5.Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther 2008; 83:898–903. [DOI] [PubMed] [Google Scholar]
- 6.Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, et al. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia – the good, the bad, and the ugly. Kidney Int 2005; 67:1216–1233. [DOI] [PubMed] [Google Scholar]
- 7.Sun H, Frassetto L, Benet LZ. Effects of renal failure on drug transport and metabolism. Pharmacol Ther 2006; 109 (1–2):1–11. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Xu N, Xiao S, Arya V, Zhao P, Lesko LJ, et al. Regulatory perspectives on designing pharmacokinetic studies and optimizing labeling recommendations for patients with chronic kidney disease. J Clin Pharmacol 2012; 52 (Suppl):79S–90S. [DOI] [PubMed] [Google Scholar]
- 9.Lu D, Lu T, Stroh M, Graham RA, Agarwal P, Musib L, et al. A survey of new oncology drug approvals in the USA from 2010 to 2015: a focus on optimal dose and related postmarketing activities. Cancer Chemother Pharmacol 2016; 77:459–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong G, Hayen A, Chapman JR, Webster AC, Wang JJ, Mitchell P, et al. Association of CKD and cancer risk in older people. J Am Soc Nephrol 2009; 20:1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Launay-Vacher V, Oudard S, Janus N, Gligorov J, Pourrat X, Rixe O, et al. Prevalence of renal insufficiency in cancer patients and implications for anticancer drug management: the renal insufficiency and anticancer medications (IRMA) study. Cancer 2007; 110:1376–1384. [DOI] [PubMed] [Google Scholar]
- 12.Humphreys BD, Soiffer RJ, Magee CC. Renal failure associated with cancer and its treatment: an update. J Am Soc Nephrol 2005; 16:151–161. [DOI] [PubMed] [Google Scholar]
- 13.US Food and Drug Administration. Hematology/oncology (cancer) approvals & safety notifications; 2016. Available at: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm. [Accessed 9 May 2017].
- 14.Graham RA, Hop CE, Borin MT, Lum BL, Colburn D, Chang I, et al. Single and multiple dose intravenous and oral pharmacokinetics of the hedgehog pathway inhibitor vismodegib in healthy female subjects. Br J Clin Pharmacol 2012; 74:788–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu T, Wang B, Gao Y, Dresser M, Graham RA, Jin JY. Semi-mechanism-based population pharmacokinetic modeling of the hedgehog pathway inhibitor vismodegib. CPT Pharmacometrics Syst Pharmacol 2015; 4:680–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lacy S, Hsu B, Miles D, Aftab D, Wang R, Nguyen L. Metabolism and disposition of cabozantinib in healthy male volunteers and pharmacologic characterization of its major metabolites. Drug Metab Dispos 2015; 43:1190–1207. [DOI] [PubMed] [Google Scholar]
- 17.Lam YW, Banerji S, Hatfield C, Talbert RL. Principles of drug administration in renal insufficiency. Clin Pharmacokinet 1997; 32:30–57. [DOI] [PubMed] [Google Scholar]
- 18.Superfin D, Iannucci AA, Davies AM. Commentary: oncologic drugs in patients with organ dysfunction: a summary. Oncologist 2007; 12:1070–1083. [DOI] [PubMed] [Google Scholar]
- 19.Tett SE, Kirkpatrick CM, Gross AS, McLachlan AJ. Principles and clinical application of assessing alterations in renal elimination pathways. Clin Pharmacokinet 2003; 42:1193–1211. [DOI] [PubMed] [Google Scholar]
- 20.Verbeeck RK, Musuamba FT. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur J Clin Pharmacol 2009; 65:757–773. [DOI] [PubMed] [Google Scholar]
- 21.Badros AZ, Vij R, Martin T, Zonder JA, Kunkel L, Wang Z, et al. Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety. Leukemia 2013; 27:1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonate PL, Cunningham CC, Gaynon P, Jeha S, Kadota R, Lam GN, et al. Population pharmacokinetics of clofarabine and its metabolite 6-ketoclofarabine in adult and pediatric patients with cancer. Cancer Chemother Pharmacol 2011; 67:875–890. [DOI] [PubMed] [Google Scholar]
- 23.Galsky MD, Camacho LH, Chiorean EG, Mulkerin D, Hong DS, Oh WK, et al. Phase I study of the effects of renal impairment on the pharmacokinetics and safety of satraplatin in patients with refractory solid tumors. Ann Oncol 2012; 23:1037–1044. [DOI] [PubMed] [Google Scholar]
- 24.Tomkinson H, Kemp J, Oliver S, Swaisland H, Taboada M, Morris T. Pharmacokinetics and tolerability of zibotentan (ZD4054) in subjects with hepatic or renal impairment: two open-label comparative studies. BMC Clin Pharmacol 2011; 11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfister M, Nolin TD, Arya V. Optimizing drug development and use in patients with kidney disease: opportunities, innovations, and challenges. J Clin Pharmacol 2012; 52 (Suppl):4S–6S. [DOI] [PubMed] [Google Scholar]
- 26.Churchwell MD. Use of an in vitro model of renal replacement therapy systems to estimate extracorporeal drug removal. J Clin Pharmacol 2012; 52 (Suppl):35S–44S. [DOI] [PubMed] [Google Scholar]
- 27.Fissell R, Schulman G, Pfister M, Zhang L, Hung AM. Novel dialysis modalities: do we need new metrics to optimize treatment? J Clin Pharmacol 2012; 52 (Suppl):72S–78S. [DOI] [PubMed] [Google Scholar]
- 28.Hariharan S, Madabushi R. Clinical pharmacology basis of deriving dosing recommendations for dabigatran in patients with severe renal impairment. J Clin Pharmacol 2012; 52 (Suppl):119S–125S. [DOI] [PubMed] [Google Scholar]
- 29.Joy MS. Impact of glomerular kidney diseases on the clearance of drugs. J Clin Pharmacol 2012; 52 (Suppl):23S–34S. [DOI] [PubMed] [Google Scholar]
- 30.Meibohm B, Zhou H. Characterizing the impact of renal impairment on the clinical pharmacology of biologics. J Clin Pharmacol 2012; 52 (Suppl):54S–62S. [DOI] [PubMed] [Google Scholar]
- 31.Naud J, Nolin TD, Leblond FA, Pichette V. Current understanding of drug disposition in kidney disease. J Clin Pharmacol 2012; 52 (Suppl):10S–22S. [DOI] [PubMed] [Google Scholar]
- 32.Riggs MM, Peterson MC, Gastonguay MR. Multiscale physiology-based modeling of mineral bone disorder in patients with impaired kidney function. J Clin Pharmacol 2012; 52 (Suppl):45S–53S. [DOI] [PubMed] [Google Scholar]
- 33.Steffl JL, Bennett W, Olyaei AJ. The old and new methods of assessing kidney function. J Clin Pharmacol 2012; 52 (Suppl):63S–71S. [DOI] [PubMed] [Google Scholar]
- 34.Tortorici MA, Cutler D, Zhang L, Pfister M. Design, conduct, analysis, and interpretation of clinical studies in patients with impaired kidney function. J Clin Pharmacol 2012; 52 (Suppl):109S–118S. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Boulton DW, Pfister M. A pharmacometric approach to quantify the impact of chronic kidney disease and hemodialysis on systemic drug exposure: application to saxagliptin. J Clin Pharmacol 2012; 52 (Suppl):126S–133S. [DOI] [PubMed] [Google Scholar]
- 36.Zhao P, Vieira Mde L, Grillo JA, Song P, Wu TC, Zheng JH, et al. Evaluation of exposure change of nonrenally eliminated drugs in patients with chronic kidney disease using physiologically based pharmacokinetic modeling and simulation. J Clin Pharmacol 2012; 52 (Suppl):91S–108S. [DOI] [PubMed] [Google Scholar]
- 37.Zuppa AF. Understanding renal replacement therapy and dosing of drugs in pediatric patients with kidney disease. J Clin Pharmacol 2012; 52 (Suppl):134S–140S. [DOI] [PubMed] [Google Scholar]
- 38.Ferl GZ, Theil FP, Wong H. Physiologically based pharmacokinetic models of small molecules and therapeutic antibodies: a mini-review on fundamental concepts and applications. Biopharm Drug Dispos 2016; 37:75–92. [DOI] [PubMed] [Google Scholar]
- 39.Huang SM, Rowland M. The role of physiologically based pharmacokinetic modeling in regulatory review. Clin Pharmacol Ther 2012; 91:542–549. [DOI] [PubMed] [Google Scholar]
- 40.Rowland M. Physiologically-based pharmacokinetic (PBPK) modeling and simulations principles, methods, and applications in the pharmaceutical industry. CPT Pharmacometrics Syst Pharmacol 2013; 2:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N. physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos 2015; 43:1823–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poole C, Gardiner J, Twelves C, Johnston P, Harper P, Cassidy J, et al. Effect of renal impairment on the pharmacokinetics and tolerability of capecitabine (Xeloda) in cancer patients. Cancer Chemother Pharmacol 2002; 49:225–234. [DOI] [PubMed] [Google Scholar]
- 43.Beumer JH, Ding F, Tawbi H, Lin Y, Viluh D, Chatterjee I, et al. Effect of renal dysfunction on toxicity in three decades of cancer therapy evaluation program-sponsored single-agent phase I studies. J Clin Oncol 2016; 34:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]