

Abstract
Objective
The tumor microenvironment frequently displays abnormal cellular metabolism, which contributes to aggressive behavior. Metformin inhibits mitochondrial oxidative phosphorylation, altering metabolism. Though the mechanism is unclear, epidemiologic studies show an association between metformin use and improved outcomes in head and neck squamous cell carcinoma (HNSCC). We sought to determine if metformin alters metabolism and apoptosis in HNSCC tumors.
Study Design
Window of opportunity trial of metformin between diagnostic biopsy and resection. Participants were patients with newly diagnosed HNSCC. Fifty patients were enrolled, and 39 completed a full‐treatment course. Metformin was titrated to standard diabetic dose (2,000 mg/day) for a course of 9 or more days prior to surgery.
Methods
Immunohistochemistry (IHC) for the metabolic markers caveolin‐1 (CAV1), B‐galactosidase (GALB), and monocarboxylate transporter 4 (MCT4), as well as the Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) apoptosis assay and Ki‐67 IHC, were performed in pre‐ and postmetformin specimens. Exploratory mass spectroscopy imaging (MSI) to assess lactate levels also was performed in three subjects.
Results
Metformin was well tolerated. The average treatment course was 13.6 days. Posttreatment specimens showed a significant increase in stromal CAV1 (P < 0.001) and GALB (P < 0.005), as well as tumor cell apoptosis by TUNEL assay (P < 0.001). There was no significant change in stromal MCT4 expression or proliferation measured by Ki67. Lactate levels in carcinoma cells were increased 2.4‐fold postmetformin (P < 0.05), as measured by MSI.
Conclusion
Metformin increases markers of reduced catabolism and increases senescence in stromal cells as well as carcinoma cell apoptosis. This study demonstrates that metformin modulates metabolism in the HNSCC microenvironment.
Level of Evidence
4. Laryngoscope, 127:1808–1815, 2017
Keywords: Head and neck cancer, squamous cell carcinoma, metformin, tumor metabolism
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common type of cancer.1 Five‐year recurrence is as high as 50% despite advances in treatment.2 Moreover, current strategies have significant side effects. Current research efforts are focusing on novel antineoplastic agents such as immunotherapy or metabolic modulators.3 Metformin (N, N‐dimethylbiguanide) alters cellular metabolism; in epidemiologic studies of patients with type 2 diabetes mellitus (DM2), it is associated with improved outcomes and decreased incidence of HNSCC.4, 5, 6, 7
Metformin is safe, well tolerated, well characterized, and is the most commonly prescribed drug for DM2.8, 9, 10 It accumulates 100‐ to 500‐fold in mitochondria and directly inhibits mitochondrial oxidative phosphorylation (OXPHOS) complex I (NADH:ubiquinone oxidoreductase) decreasing Adenosine Triphosphate (ATP) generation.11, 12, 13, 14 It thereby activates the energy sensor AMP‐activated protein kinase (AMPK), which shifts the cell toward an energy‐sparing state with increased glycolysis, increased lactate production, and reduced activity of the citric acid cycle.9, 15, 16 These metabolic effects may directly impact HNSCC carcinoma cells. Metformin has been shown to induce cancer cell apoptosis and to reduce tumor size in mouse models of HNSCC.17, 18, 19 Metformin also may impact the stromal component of the tumor in addition to the carcinoma cells.
The tumor microenvironment (TME) is composed of carcinoma cells and a number of stromal‐supporting cells, such as fibroblasts, macrophages, and other immune cells. Stromal cells provide nutrients for cancer cells, promoting resistance to apoptosis and enhancing proliferation, invasion, and metastasis.20, 21, 22, 23 This relationship between stromal and cancer cells relies on linked metabolism between tumor compartments, or multicompartment metabolism (MCM), to meet the high bioenergetic needs of rapid tumor growth.24, 25, 26 In HNSCC, proliferative cancer cells show an increased uptake of mitochondrial fuels and OXPHOS, whereas nonproliferative stromal cells have high rates of glycolysis associated with the export of monocarboxylates.27, 28, 29 Low stromal caveolin‐1 (CAV1) expression commonly is found in cancer including HNSCC, and is a marker of MCM, high fibroblast glycolysis, and the cancer‐associated fibroblast phenotype.20, 30, 31, 32 Metformin preferentially targets cells with altered glycolysis; thus, cancer associated fibroblasts (CAFs) may be more susceptible to metformin. Loss of CAV1 in CAFs induces the upregulation of monocarboxylate transporter 4 (MCT4), which is an exporter of glycolytic byproducts.30 These byproducts can be used for OXPHOS in proliferative cancer cells.27 No clinical trials have been performed to assess the effect of drugs on markers of the metabolic profile of human tumors. Hence, the purpose of the study was to assess the effect of metformin on markers of tumor metabolism.
MATERIALS AND METHODS
Trial Design
The clinical trial was registered on ClinicalTrials.gov., Identifier NCT02083692. Our institutional review board approved this trial. Eligible patients were HNSCC patients with a biopsy of their primary lesion and plan for definitive surgical resection, with a window of at least 9 days between biopsy and surgery for metformin treatment. Primary lesions were biopsied in the office or on operative endoscopy; when biopsies had already been performed elsewhere, the tissue blocks were obtained for analysis. Following biopsy‐proven diagnosis of HNSCC, metformin was started at 500 mg/day and increased to 1,000 mg twice daily by day 6.
The primary endpoint was immunohistochemistry (IHC) staining for CAV1 and MCT4 in the tumor stroma from pre‐ to postmetformin specimens. Secondary endpoints were IHC for GALB and Ki‐67 IHC and tumor cell apoptosis by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. We performed an exploratory analysis by mass spectroscopy imaging (MSI) comparing premetformin and postmetformin carcinoma regions in three subjects.
Immunohistochemistry
Two tissue samples were prepared for IHC analysis for each patient: one a premetformin and one postmetformin. Tissue samples were fixed in neutral buffered formalin and then embedded in paraffin; samples were sectioned at 4‐micrometer thickness, then dewaxed and rehydrated through graded ethanols.
Antigen retrieval was performed in 10‐mM citrate buffer, pH 6.0, for 10 minutes using a pressure cooker. Sections were cooled, blocked for endogenous peroxidase with 3% H2O2, and blocked for endogenous biotin using the DakoCytomation Biotin Blocking System (Dako, Carpintera, CA). Sections were next incubated at room temperature with 10% goat serum for 30 minutes and then incubated at 4oC with primary antibodies for CAV1, GALB, MCT4, and Ki‐67 (Santa Cruz Biotechnologies, Santa Cruz, CA).
Primary antibody binding was detected by biotinylated species‐specific secondary antibody (Vector Labs, Burlingame, CA), followed by a streptavidin‐horseradish peroxidase conjugate (Dako). Immunoreactivity was revealed with 3,3′ diaminobenzidine (Dako). All sections were counterstained with hematoxylin.
Apoptotic cells were identified using the TUNEL‐based ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore, EMD Millipore, Darmstadt, Germany).
Histochemical grading was performed by two blinded pathologists. For CAV1, GALB, and MCT4, strength of staining in the tumor stroma and for Ki‐67 in carcinoma cells was reported on a continuous percentage scale (eg 5% positive staining). For TUNEL, number of nuclei with TUNEL staining per high‐power field (HPF) was averaged over five HPFs in each specimen, and the mean was reported. For each sample, scores from two pathologists were averaged to yield a final score for statistical analysis.
Specimens from 12 patients with surgically treated HNSCC who had biopsy and resection tissues available for analysis also were identified as controls. These controls were stained for CAV1, BGAL, and TUNEL, and scored by two blinded pathologists. Their results were averaged and compared in the same way as the specimens from experimental patients.
Collection of Tumor Specimens for Mass Spectrometry Imaging
Specimens were collected from the tumor and processed, as previously described.33, 34, 35 Briefly, samples were wrapped in aluminum foil, snap‐frozen in liquid nitrogen, and then stored at −80oC.
Gold‐coated matrix‐assisted laser desorption/ionization (MALDI) plates were precoated with α‐cyano‐4‐hydroxycinnamic acid (CHCA) using an automated sprayer (TM Sprayer, HTX Technologies, Carrboro, NC). CHCA was prepared as 5 mg/mL in 90% acetonitrile, and was sprayed at 0.15 mL/min at 100°C and 700 mm/min plate velocity. Nitrogen was used as the nebulization gas and was set to 10 psig. Four passes were deposited at 2‐mm spacing, alternating horizontal and vertical positions between passes with a 1‐mm offset for the second passes in each direction. Sections from fresh‐frozen tumor tissue were obtained at 12‐μm thickness in a cryostat (Leica Biosystems, Buffalo Grove, IL). The sections were directly thaw‐mounted onto the precoated target plates. The tissues were subsequently postcoated with 9‐aminoacridine matrix, prepared at 5 mg/mL in 90% methanol, and applied using the same program as for CHCA on the TM Sprayer (HTX Technologies).
Serial sections were obtained stained with hematoxylin and eosin (H&E). Carcinoma regions of interest (ROIs) were determined via histological evaluation of the H&E slides by the head and neck pathologists. The tissue specimens were analyzed using a MALDI Solarix 9.4 T FT‐ICR mass spectrometer (Bruker Daltonics, Billerica, MA). Spectra were acquired for several tissue morphological regions (carcinoma, stroma, and tumor‐associated stroma). Spectra were acquired in negative ion mode from m/z 50 to 200, and instrument parameters were optimized to detect lower molecular weight metabolites, including lactate. Data were acquired in an ordered array spaced at 100 μm within the ROIs. The laser diameter was 15 μm, and the laser motion was set to raster within a 75‐μm area, for a total of 2,000 laser shots per pixel. Standard lactate was run prior to the tissues to evaluate instrument performance and mass accuracy, which was always better than 2 parts per million. Average spectra for each region of interest were exported into mMass,36 and spectra from premetformin ROIs were compared to postmetformin.
Statistical Analysis
Strength‐of‐staining scores were compared between pre‐ and postmetformin samples by a paired, two‐tailed Student t test. Linear regression to assess strength of associations between continuous variables was performed. Significant P values were considered less than 0.05.
RESULTS
Patients
Fifty patients were enrolled. Average age was 62.3 (range 35–80) in 37 men and 13 women. Nine patients enrolled in the study and then decided not to take metformin; two others had less than 9 days between their biopsy and definitive resection. Of the 39 patients who took at least 9 days of metformin, the range was 9 to 24 days and the mean was 13.6 days.
Most common subsites of disease were oropharynx (26 subjects, 52%) and oral cavity (17 subjects, 34%), with tumors also in the larynx, hypopharynx, and skin (Table 1). Of 26 patients with oropharyngeal HNSCC, 23 (88%) were positive for human papillomavirus (HPV). Staging and additional demographic and pathologic data are shown in Table 1.
Table 1.
Demographics and Pathologic Characteristics.
| Demographics (n = 50) | |||
|---|---|---|---|
| Age | 62.3 (35–80) | ||
| Gender | 37 male/13 female | ||
| Smoker (greater than 10 pack‐years) | 29 (58%) | ||
| Mean length of follow‐up (days) | 388 (4–870) | ||
| Days on metformin | 10.96 (0–24) | ||
| Subsite | |||
| Oral cavity | 17 (34%) | Oropharynx | 26 (52%) |
| Oral tongue | 5 | Tongue base | 8 |
| Floor of mouth | 4 | Tonsil | 15 |
| Gingiva | 4 | Soft palate | 2 |
| RMT | 2 | PPW | 1 |
| Hard palate | 2 | p16 positive | 23/26 (88%) |
| Larynx | 4 (8%) | Hypopharynx | 1 (2%) |
| Glottis | 2 | Skin | 2 (4%) |
| Supraglottis | 2 | ||
| T stage | N stage | ||
| Tis | 1 (2%) | N0 | 18 (36%) |
| T1 | 9 (18%) | N1 | 5 (10%) |
| T2 | 24 (48%) | N2a | 6 (12%) |
| T3 | 5 (10%) | N2b | 16 (32%) |
| T4a | 11 (22%) | N2c | 1 (2%) |
| T4b | 0 (0%) | N3 | 4 (8%) |
| Path parkers | Differentiation | ||
| ECE | 13 (26%) | in situ | 1 (2%) |
| Positive margins | 4 (8%) | Well | 5 (10%) |
| PNI | 20 (40%) | Moderate | 21 (42%) |
| LVI | 18 (36%) | Poor | 22 (44%) |
ECE = extracapsular extension; LVI = lymphovascular invasion; N = node; PNI = perineural invasion; PPW = posterior pharyngeal wall; RMT = retromolar trigone; T = tumor.
Adverse Events
Fourteen patients reported metformin‐related adverse events, with five patients reporting more than one symptom (Table 2). Toxicities were graded using National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. The most common toxicity was grade 1 diarrhea, occurring in 10 patients. Two patients experienced grade 2 toxicities: one with grade 2 diarrhea and hemorrhoids, and one with grade 2 nausea. No patients experienced grade 3 or grade 4 toxicities. No patients had perioperative glucose abnormalities or lactic acidosis.
Table 2.
Toxicity in Metformin in HNSCC Clinical Trial as per NCI CTCAEv4.0 Scoring.
| Metformin‐Related Toxicity | ||
|---|---|---|
| Toxicity | Grade | n |
| Diarrhea | 1 | 10 |
| Nausea | 1 | 2 |
| Fatigue | 1 | 1 |
| Weakness | 1 | 1 |
| Dizziness | 1 | 1 |
| Increased ALT | 1 | 1 |
| Diarrhea | 2 | 1 |
| Nausea | 2 | 1 |
| Hemorrhoids | 2 | 1 |
ALT = alanine aminotransferase; CTCAE = Common Terminology Criteria for Adverse Events; HNSCC = head and neck squamous cell carcinoma; NCI = National Cancer Institute.
Immunohistochemistry
Of the 39 patients with a full‐treatment course, 33 had evaluable samples for both pre‐ and posttreatment specimens; the remaining six patients had a premetformin sample that either was unavailable from an outside institution or was of inadequate quantity for immunohistochemistry staining. Thirty‐one showed increased CAV1 staining after metformin, whereas two had a decrease. Of the patients completing a full‐treatment course, average staining intensity of stromal CAV1 was 25.7% on pretreatment specimens and increased to 62.8% on posttreatment specimens (P < 0.001) (Fig. 1A). There was no difference in CAV1 staining between biopsy and resection specimens for 12 control patients (41.1% vs. 41.4%, P = 0.97). No associations were noted between CAV1 and TUNEL or between CAV1 and GALB staining patterns. There was no significant change in stromal MCT4 after metformin (P = 0.44).
Figure 1.
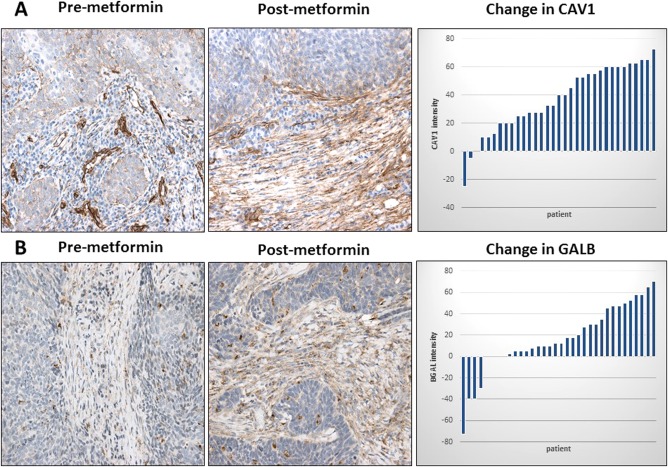
(A–B). Effect of metformin on stromal CAV1 and GALB expression in HNSCC. CAV1 and GALB immunostaining were performed on paired premetformin (A and B, respectively) and postmetformin HNSCC samples, and a representative example is shown. Note that there is a postmetformin increase in CAV1 and GALB stromal staining. Original magnification: 40×. Waterfall plot of the change in CAV1 and GALB intensity between pre‐ and postmetformin samples also is shown. CAV1 = caveolin‐1; GALB = beta galactosidase; HNSCC = head and neck squamous cell carcinoma.
Average staining intensity of stromal GALB, which is a marker of senescence, increased from 32.4% to 50.2% after treatment with metformin (P = 0.005) (Fig. 1B). Only five subjects had tumors with reduced GALB staining after metformin. There was no difference in BGAL staining between biopsy and resection specimens for 12 control patients (40.4% vs. 37.9%, P = 0.84).
There was increased carcinoma cell apoptosis upon metformin exposure. Pretreatment specimens from patients receiving a full‐treatment course had an average of 4.53 apoptotic nuclei per HPF, and increased to 12.6 per HPF on posttreatment specimens on TUNEL assay (P < 0.001) (Fig. 2). Seven subjects had reduced TUNEL staining upon metformin exposure. There was no difference in TUNEL staining between biopsy and resection specimens for 12 control patients (4.20 vs. 9.06, P = 0.59). There was no significant change in the Ki67 proliferation assay between pretreatment and posttreatment specimens (P = 0.76).
Figure 2.
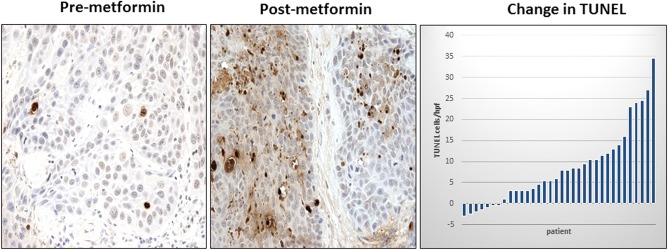
Effect of metformin on TUNEL staining in HNSCC. TUNEL was performed on paired premetformin and postmetformin HNSCC samples, and a representative example is shown. Note that there is a postmetformin increase in TUNEL staining in carcinoma cells. Original magnification: 40×. Waterfall plot of the change in GALB intensity between pre‐ and postmetformin samples also is shown. HNSCC = head and neck squamous cell carcinoma; TUNEL = terminal deoxynucleotidyl transferase dUTP nick end labeling.
There was an association between number of days on metformin and higher GALB expression (r2 = 0.129, P < 0.05). Also, higher GALB induction was associated with higher apoptosis in carcinoma cells by TUNEL staining (r2 = 0.239, P < 0.05). Lack of GALB stromal upregulation upon metformin exposure was associated with perineural invasion when all patients were considered (P < 0.05).
The observed changes in stromal CAV1, stromal GALB, and carcinoma cell TUNEL were not different between HPV‐positive and HPV‐negative tumors (P = 0.21, 0.74, 0.22, respectively).
Mass Spectroscopy Imaging
MSI was performed on samples from three subjects to compare lactate levels in carcinoma regions between premetformin and postmetformin samples. At least three carcinoma regions were delineated to perform MSI (Fig. 3A). Lactate levels increased 2.4‐fold in carcinoma cells postmetformin (P < 0.05) (Fig. 3B). Lactate is the end product of glycolysis or metabolism of glucose in the cytosol. Lactate production increases when mitochondrial OXPHOS activity is reduced, which is a mechanism of action of metformin.
Figure 3.
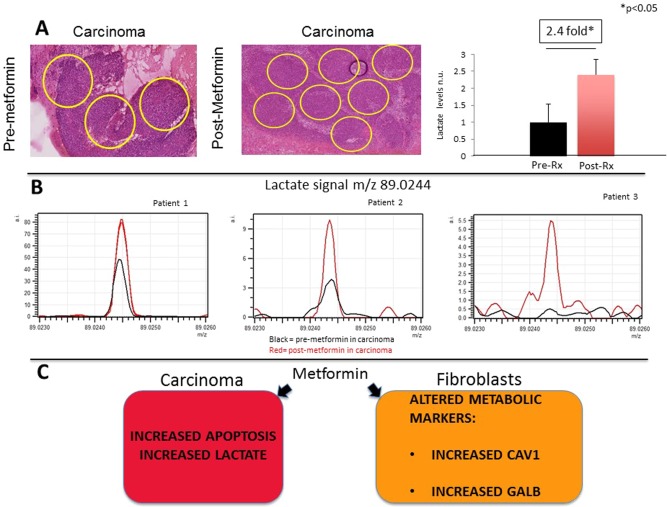
(A–C) Mass spectroscopy imaging (MSI) comparing carcinoma regions premetformin and postmetformin. Hematoxylin and eosin staining was performed on HNSCC samples, and carcinoma regions were marked in pre‐ and postmetformin samples. Original magnification: 40× (A). Carcinoma cells have a 2.4‐fold reduction in lactate levels postmetformin (post‐rx) compared to premetformin (pre‐rx) in all samples (P < 0.05). Lactate levels were measured by MSI in the carcinoma regions pre‐ and postmetformin, and results were compared for each patient (B). Note that the black lines are the tracings for the premetformin samples and the red lines for the postmetformin samples. Effects of metformin on the tumor microenvironment of HNSCC (C). The current clinical trial demonstrates in HNSCC that metformin has anticancer activity. Metformin in carcinoma cells increases apoptosis, as measured by terminal deoxynucleotidyl transferase dUTP nick end labeling, and increases lactate levels. Metformin also alters metabolic markers in stromal cells. The tumor stroma after metformin exposure has increased CAV1 and GALB expression, which are markers of reduced catabolism and increased senescence. CAV1 = caveolin‐1; GALB = beta galactosidase; HNSCC = head and neck squamous cell carcinoma; MSI = mass spectroscopy imaging; rx = treatment.
DISCUSSION
The current trial demonstrates that metformin has anticancer effects in HNSCC by inducing apoptosis and altering stromal markers of metabolism and senescence with increased CAV1 and GALB expression. This is the first trial to study the anticancer effects of metformin in HNSCC and is among few clinical trials in other tumor types to have assessed its effects on apoptosis.37, 38, 39 The presence of a mechanistic link between these effects will require further study; however, we suspect that alterations in tumor metabolism may disrupt cancer cell metabolism as well as interrupt metabolic systems between the tumor cell and stromal cells, such as multicompartment metabolism (MCM).
Resistance to apoptosis is a hallmark of cancer cells and is fundamental to carcinogenesis.40 In this study, the TUNEL assay showed a significant increase in apoptosis after treatment (P < 0.01). Metformin inhibits mitochondrial complex I, which decreases ATP production and increases activation of AMPK increasing glycolysis and lactate production, inducing metabolic stress. The MSI performed in a limited subset of tumors did demonstrate increased lactate production spatially located within cancer cells after metformin therapy. This supports the notion that the doses of metformin in the trial can directly impact tumor metabolism. In vitro and in vivo HNSCC models have shown that metformin also inhibits tumor cell proliferation and induces apoptosis.8, 13, 25, 41, 42, 43, 44, 45, 46, 47 Further, hypoxia seen in HNSCC tumors may make some cell populations more susceptible to the effect of metformin.27, 48, 49, 50 The results of our study suggest for the first time that such effects can be seen in human HNSCC.
Support from stromal cells is required for cancer cell survival, and most TME studies have focused on prosurvival growth factors and cytokines; however, metabolic interactions in the TME also may drive resistance to apoptosis in carcinoma cells.51 This trial attempted to assess impact of the metabolic changes induced by metformin on tumor–stroma interactions by quantifying the expression of CAV1, MCT4, and GALB on cancer‐associated fibroblasts. CAV1 is the principal structural protein coating caveolae in the plasma membrane; it functions as a scaffolding protein.30 Downregulation of CAV1 in CAFs induces signaling through transforming growth factor beta, nuclear factor kB, and hypoxia inducible factor, increasing catabolism, which results in increased tumor aggressiveness, invasion, and metastasis.30, 32, 52 Loss of CAV1 in CAFs is sufficient to drive the glycolytic phenotype in the stromal compartment and mitochondrial metabolism in cancer cells, and is a marker of MCM.10, 30 It has been proposed that metformin should be an effective agent in tumors with MCM,53 and it has been shown to restore CAV1 expression in CAFs in vitro.54 Further, GALB expression in stromal cells is a marker of senescence.55, 56, 57 Senescence also is associated with reduced catabolism and increased anabolism.58, 59, 60 GALB expression is induced by Cav1, resulting in senescence and reduced glycolysis.55, 61, 62 Fibroblasts with high GALB reduce tumorigenesis in SCC.30, 63
Monocarboxylate transporter 4 (MCT4) is an exporter of glycolytic byproducts such as pyruvate and lactate.30 Low stromal staining for CAV1 and high MCT4 are markers of aggressive disease in HNSCC, and high stromal MCT4 predicts poor outcomes for patients across many types of cancer.27, 30, 64 In experimental models, manipulating CAV1 alters the expression of MCT4. Our study showed no effect on MCT4 expression, which may be due to a variety of factors but could be due to the short treatment window or drug dosing.
This trial demonstrates that standard metformin doses (1,000 mg twice daily) have potentially beneficial pharmacodynamic effects on HNSCC tumors. There is controversy concerning whether higher doses of metformin should be used in oncology clinical trials.8, 14, 37, 65, 66 Those who favor using higher doses of metformin base this on the fact that, in many in vitro and in vivo experimental cancer models, higher doses are required to demonstrate anticancer activity.8, 14, 66 The dose used here is lower than the highest recommended dose in patients with diabetes mellitus (2,500 mg daily), and no patient experienced severe toxicity (grade 3 NCI CTCAEv4.0). The low observed toxicity is consistent with previous clinical trials in diabetes mellitus and in oncology and the pharmacovigilance data, which has detected very little toxicity despite common use of metformin47; thus, this may be a reasonable dose for phase II clinical trials. However, higher doses may be more efficacious and will need to be evaluated.
Metformin or similar antimetabolic drugs may have clinical impact on treatment of HNSCC. Metformin has been shown to synergize with the effect of radiotherapy and drugs that cause DNA damage and oxidative stress.42, 67 In addition to targeting tumor metabolism, metformin also may impact immune interaction in the microenvironment. Immune exhaustion, a state in which T cells become functionally inept, is a common feature of many tumors, including HNSCC.68, 69 Similarly to immune checkpoint inhibitors such as pembrolizumab, metformin has been shown to reverse immune exhaustion.70, 71 Interestingly, metformin has been shown to potentiate the anticancer effects of other drugs, including antimetabolites and tyrosine kinase inhibitors.72, 73
CONCLUSION
In sum, this pilot trial shows that metformin is safe and has biological activity in HNSCC. It also shows that conventional antidiabetic doses of metformin are sufficient to induce apoptosis of carcinoma cells and to alter markers of stromal metabolism consistent with reduced catabolism and increased senescence. Future clinical trials will need to be performed to test if metformin is clinically beneficial and whether other metabolic modulators are effective in HNSCC.
Acknowledgments
Joseph Curry, MD, and Ubaldo Martinez‐Outschoorn, MD, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Tingting Zhan, PhD, and Benjamin Leiby, PhD, assisted with statistical analyses.
Portions of this clinical trial were presented at the American Head and Neck Society International Meeting in Seattle, Washington, U.S.A., July 17, 2016.
This work was funded by Young Investigator Award 2014, American Academy of Otolaryngology and American Head and Neck Society, award ID 314313; and by the National Cancer Institute of the National Institutes of Health, award numbers K08 CA175193‐01A1 and 5P30CA056036‐17. Funding was used to provide material support for laboratory testing. The authors have no other funding, financial relationships, or conflicts of interest to disclose.
Correction added on June 29, 2019 after first online publication: The copyright line has been updated.
BIBLIOGRAPHY
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Kawecki A, Krajewski R. Follow‐up in patients treated for head and neck cancer. Memo 2014;7:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bann DV, Deschler DG, Goyal N. Novel immunotherapeutic approaches for head and neck squamous cell carcinoma. Cancers (Basel) 2016;8. pii: E87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skinner HD, McCurdy MR, Echeverria AE, et al. Metformin use and improved response to therapy in esophageal adenocarcinoma. Acta Oncol 2013;52:1002–1009. [DOI] [PubMed] [Google Scholar]
- 5. Sandulache VC, Hamblin JS, Skinner HD, Kubik MW, Myers JN, Zevallos JP. Association between metformin use and improved survival in patients with laryngeal squamous cell carcinoma. Head Neck 2014;36:1039–1043. [DOI] [PubMed] [Google Scholar]
- 6. Yen YC, Lin C, Lin SW, Lin YS, Weng SF. Effect of metformin on the incidence of head and neck cancer in diabetics. Head Neck 2015;37:1268–1273. [DOI] [PubMed] [Google Scholar]
- 7. Rego DF, Pavan LM, Elias ST, De Luca Canto G, Guerra EN. Effects of metformin on head and neck cancer: a systematic review. Oral Oncol 2015;51:416–422. [DOI] [PubMed] [Google Scholar]
- 8. Pollak M. Overcoming drug development bottlenecks with repurposing: repurposing biguanides to target energy metabolism for cancer treatment. Nat Med 2014;20:591–593. [DOI] [PubMed] [Google Scholar]
- 9. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab 2014;20:953–966. [DOI] [PubMed] [Google Scholar]
- 10. Martinez‐Outschoorn UE, Peiris‐Pagés M, Pestell RG, Sotgia F, Lisanti MP. Nat Rev Clin Oncol. 2017;14:11–31. doi: 10.1038/nrclinonc.2016.60. Review. PMID: 27141887 [DOI] [PubMed] [Google Scholar]
- 11. El‐Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 2000;275:223–228. [DOI] [PubMed] [Google Scholar]
- 12. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti‐diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 13. Wheaton WW, Weinberg SE, Hamanaka RB, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014;3:e02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chandel NS, Avizonis D, Reczek CR, et al. Are Metformin doses used in murine cancer models clinically relevant? Cell Metab 2016;23:569–570. [DOI] [PubMed] [Google Scholar]
- 15. Andrzejewski S, Gravel SP, Pollak M, St‐Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab 2014;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014;510:542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luo Q, Hu D, Hu S, Yan M, Sun Z, Chen F. In vitro and in vivo anti‐tumor effect of metformin as a novel therapeutic agent in human oral squamous cell carcinoma. BMC Cancer 2012;12:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harada K, Ferdous T, Harada T, Ueyama Y. Metformin in combination with 5‐fluorouracil suppresses tumor growth by inhibiting the Warburg effect in human oral squamous cell carcinoma. Int J Oncol 2016;49:276–284. [DOI] [PubMed] [Google Scholar]
- 19. Ueno S, Kimura T, Yamaga T, et al. Metformin enhances anti‐tumor effect of L‐type amino acid transporter 1 (LAT1) inhibitor. J Pharmacol Sci 2016;131:110–117. [DOI] [PubMed] [Google Scholar]
- 20. Martinez‐Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer‐associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol 2014;25:47–60. [DOI] [PubMed] [Google Scholar]
- 21. Romero IL, Mukherjee A, Kenny HA, Litchfield LM, Lengyel E. Molecular pathways: trafficking of metabolic resources in the tumor microenvironment. Clin Cancer Res 2015;21:680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DeNicola GM, Cantley LC. Cancer's fuel choice: new flavors for a picky eater. Mol Cell 2015;60:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012;491:364–373. [DOI] [PubMed] [Google Scholar]
- 24. Martinez‐Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab 2012;15:4–5. [DOI] [PubMed] [Google Scholar]
- 25. Wallace DC. Mitochondria and cancer. Nat Rev Cancer 2012;12:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H symporters. J Mol Med (Berl) 2016;94:155–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Curry JM, Tuluc M, Whitaker‐Menezes D, et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle 2013;12:1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jensen DH, Therkildsen MH, Dabelsteen E. A reverse Warburg metabolism in oral squamous cell carcinoma is not dependent upon myofibroblasts. J Oral Pathol Med 2015;44:714–721. [DOI] [PubMed] [Google Scholar]
- 29. Curry JM, Sprandio J, Cognetti D, et al. Tumor microenvironment in head and neck squamous cell carcinoma. Semin Oncol 2014;41:217–234. [DOI] [PubMed] [Google Scholar]
- 30. Martinez‐Outschoorn UE, Sotgia F, Lisanti MP. Caveolae and signalling in cancer. Nat Rev Cancer 2015;15:225–237. [DOI] [PubMed] [Google Scholar]
- 31. Pavlides S, Whitaker‐Menezes D, Castello‐Cros R, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009;8:3984–4001. [DOI] [PubMed] [Google Scholar]
- 32. Asterholm IW, Mundy DI, Weng J, Anderson RG, Scherer PE. Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin‐1. Cell Metab 2012;15:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwamborn K, Caprioli RM. Molecular imaging by mass spectrometry—looking beyond classical histology. Nat Rev Cancer 2010;10:639–646. [DOI] [PubMed] [Google Scholar]
- 34. Van de Plas R, Yang J, Spraggins J, Caprioli RM. Image fusion of mass spectrometry and microscopy: a multimodality paradigm for molecular tissue mapping. Nat Methods 2015;12:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taverna D, Pollins AC, Sindona G, Caprioli RM, Nanney LB. Imaging mass spectrometry for accessing molecular changes during burn wound healing. Wound Repair Regen 2016;24:775–785. [DOI] [PubMed] [Google Scholar]
- 36. Strohalm M, Hassman M, Kosata B, Kodicek M. mMass data miner: an open source alternative for mass spectrometric data analysis. Rapid Commun Mass Spectrom 2008;22:905–908. [DOI] [PubMed] [Google Scholar]
- 37. Niraula S, Dowling RJ, Ennis M, et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat 2012;135:821–830. [DOI] [PubMed] [Google Scholar]
- 38. Hosono K, Endo H, Takahashi H, et al. Metformin suppresses colorectal aberrant crypt foci in a short‐term clinical trial. Cancer Prev Res (Phila) 2010;3:1077–1083. [DOI] [PubMed] [Google Scholar]
- 39. Cazzaniga M, DeCensi A, Pruneri G, et al. The effect of metformin on apoptosis in a breast cancer presurgical trial. Br J Cancer 2013;109:2792–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 41. Sikka A, Kaur M, Agarwal C, Deep G, Agarwal R. Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle 2012;11:1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skinner HD, Sandulache VC, Ow TJ, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation‐induced senescence. Clin Cancer Res 2012;18:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vitale‐Cross L, Molinolo AA, Martin D, et al. Metformin prevents the development of oral squamous cell carcinomas from carcinogen‐induced premalignant lesions. Cancer Prev Res (Phila) 2012;5:562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Woo SH, Sandulache VC, Yang L, Skinner HD. Evaluating response to metformin/cisplatin combination in cancer cells via metabolic measurement and clonogenic survival. Methods Mol Biol 2014;1165:11–18. [DOI] [PubMed] [Google Scholar]
- 45. Ben Sahra I, Laurent K, Giuliano S, et al. Targeting cancer cell metabolism: the combination of metformin and 2‐deoxyglucose induces p53‐dependent apoptosis in prostate cancer cells. Cancer Res 2010;70:2465–2475. [DOI] [PubMed] [Google Scholar]
- 46. Tomic T, Botton T, Cerezo M, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis 2011;2:e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pollak M. Potential applications for biguanides in oncology. J Clin Invest 2013;123:3693–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gronroos TJ, Lehtio K, Soderstrom KO, et al. Hypoxia, blood flow and metabolism in squamous‐cell carcinoma of the head and neck: correlations between multiple immunohistochemical parameters and PET. BMC Cancer 2014;14:876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Javeshghani S, Zakikhani M, Austin S, et al. Carbon source and myc expression influence the antiproliferative actions of metformin. Cancer Res 2012;72:6257–6267. [DOI] [PubMed] [Google Scholar]
- 50. Birsoy K, Possemato R, Lorbeer FK, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014;508:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Labi V, Erlacher M. How cell death shapes cancer. Cell Death Dis 2015;6:e1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sotgia F, Martinez‐Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin‐1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol 2012;7:423–467. [DOI] [PubMed] [Google Scholar]
- 53. Harris AL. Metabolic compartments in tumor tissue: implications for therapy. Cell Cycle 2012;11:13–14. [DOI] [PubMed] [Google Scholar]
- 54. Martinez‐Outschoorn UE, Pavlides S, Whitaker‐Menezes D, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin‐1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010;9:2423–2433. [DOI] [PubMed] [Google Scholar]
- 55. Bartholomew JN, Volonte D, Galbiati F. Caveolin‐1 regulates the antagonistic pleiotropic properties of cellular senescence through a novel Mdm2/p53‐mediated pathway. Cancer Res 2009;69:2878–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995;92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim YM, Seo YH, Park CB, Yoon SH, Yoon G. Roles of GSK3 in metabolic shift toward abnormal anabolism in cell senescence. Ann N Y Acad Sci 2010;1201:65–71. [DOI] [PubMed] [Google Scholar]
- 59. Ros S, Schulze A. Linking glycogen and senescence in cancer cells. Cell Metab 2012;16:687–688. [DOI] [PubMed] [Google Scholar]
- 60. Favaro E, Bensaad K, Chong MG, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab 2012;16:751–764. [DOI] [PubMed] [Google Scholar]
- 61. Kaplon J, Zheng L, Meissl K, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene‐induced senescence. Nature 2013;498:109–112. [DOI] [PubMed] [Google Scholar]
- 62. Li M, Durbin KR, Sweet SM, Tipton JD, Zheng Y, Kelleher NL. Oncogene‐induced cellular senescence elicits an anti‐Warburg effect. Proteomics 2013;13:2585–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Salmenpera P, Karhemo PR, Rasanen K, Laakkonen P, Vaheri A. Fibroblast spheroids as a model to study sustained fibroblast quiescence and their crosstalk with tumor cells. Exp Cell Res 2016;345:17–24. [DOI] [PubMed] [Google Scholar]
- 64. Bovenzi CD, Hamilton J, Tassone P, et al. Prognostic indications of elevated MCT4 and CD147 across cancer types: a meta‐analysis. Biomed Res Int 2015;2015:242437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hadad S, Iwamoto T, Jordan L, et al. Evidence for biological effects of metformin in operable breast cancer: a pre‐operative, window‐of‐opportunity, randomized trial. Breast Cancer Res Treat 2011;128:783–794. [DOI] [PubMed] [Google Scholar]
- 66. Dowling RJ, Lam S, Bassi C, et al. Metformin pharmacokinetics in mouse tumors: implications for human therapy. Cell Metab 2016;23:567–568. [DOI] [PubMed] [Google Scholar]
- 67. Sandulache VC, Skinner HD, Ow TJ, et al. Individualizing antimetabolic treatment strategies for head and neck squamous cell carcinoma based on TP53 mutational status. Cancer 2012;118:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jinhua X, Ji W, Shouliang C, et al. Expression of immune checkpoints in T cells of esophageal cancer patients. Oncotarget 2016;7:63669–63678. doi: 10.18632/oncotarget.11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Quan H, Fang L, Pan H, et al. An adaptive immune response driven by mature, antigen‐experienced T and B cells within the microenvironment of oral squamous cell carcinoma. Int J Cancer 2016;138:2952–2962. [DOI] [PubMed] [Google Scholar]
- 70. Economopoulou P, Kotsantis I, Psyrri A. Checkpoint inhibitors in head and neck cancer: rationale, clinical activity, and potential biomarkers. Curr Treat Options Oncol 2016;17:40. [DOI] [PubMed] [Google Scholar]
- 71. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune‐mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A 2015;112:1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tian Y, Tang B, Wang C, et al. Metformin mediates resensitivity to 5‐fluorouracil in hepatocellular carcinoma via the suppression of YAP. Oncotarget 2016;7:46230–46241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Peng M, Huang Y, Tao T, et al. Metformin and gefitinib cooperate to inhibit bladder cancer growth via both AMPK and EGFR pathways joining at Akt and Erk. Sci Rep 2016;6:28611. [DOI] [PMC free article] [PubMed] [Google Scholar]