

Abstract
We have developed a new method for determining the DNA-binding specificity of proteins. In DIP-chip (DNA immunoprecipitation with microarray detection), protein·DNA complexes are isolated from an in vitro mixture of purified protein and naked genomic DNA. Whole-genome DNA microarrays are used to identify the protein-bound DNA fragments, and the sequence of the identified fragments is used to derive binding-site descriptions. Using objective criteria for assessing the accuracy of DNA-binding motifs, and using yeast Leu3p as a model, we demonstrate that motifs determined by DIP-chip are as effective at predicting the location of bound proteins in vivo as are motifs determined by conventional low-throughput in vitro methods.
Accurate binding-site descriptions of hundreds, or perhaps thousands of transcription factors and transcription-factor complexes will likely be needed to understand how regulatory factors interact with the genome to generate coordinated transcriptional programs. Well-established methods, like binding-site selection (SELEX) (Oliphant et al. 1989; Tuerk and Gold 1990) and electrophoretic mobility shift assays (EMSA) (Fried and Crothers 1981) can be used to determine binding specificity, but they are labor intensive, not amenable to high-throughput analysis, and do not sample the full range of a protein's natural in vivo DNA substrates. An emerging alternative is to isolate and identify sequences bound in vivo using ChIP-chip experiments, and to infer binding motifs from computational analysis of the ChIP-enriched sequences (Liu et al. 2002). While ChIP-chip is a powerful method, the ability to infer relevant and accurate binding sites is dependent on adequate expression of the protein of interest. Furthermore, the discovery of binding sites from ChIP-chip data is complicated by the effects of protein-protein interactions, and the cooperative and competitive DNA binding of other proteins in vivo. DIP-chip, while similar in concept to ChIP-chip, can overcome these limitations by inferring accurate DNA-binding specificities under well-defined and easily varied in vitro conditions.
To compare DIP-chip with established methods for determining DNA-binding specificity, we also introduce a generally applicable procedure for evaluating and comparing the quality of DNA-binding motifs. Unlike conventional metrics for comparing motifs that are based directly on the motifs themselves (for example, the number of matches to a consensus site or the “distance” between position weight matrices), our procedure is based on how well each motif predicts the results of an actual binding experiment. We use this functional metric to define a set of related motifs obtained from DIP-chip analysis of yeast Leu3p and show that motifs defined by DIP-chip are able to predict Leu3p binding in vivo as well as motifs defined by SELEX and EMSA analysis.
Results
Isolation and identification of protein-bound DNA using the DIP-chip approach
We developed and tested the DIP-chip methodology using the DNA-binding domain of the yeast transcription factor Leu3p. Leu3p was chosen for study because its DNA-binding specificity had been determined previously by EMSA of 50 binding-site variants and by SELEX (Liu and Clarke 2002). Here, the same protein used in those studies, a maltose-binding protein (MBP)-tagged Leu3 DNA-binding domain, was used in DIP-chip assays to allow direct comparison to EMSA and SELEX.
The DIP-chip approach is outlined in Figure 1. Leu3p-MBP protein was mixed with purified yeast genomic DNA (2 μg/mL, 0.3 pM Genome) that had been mechanically sheared to an average size of 600 bp. Two different concentrations of protein, 4 and 40 nM, were tested. After incubation, Leu3p-DNA complexes were enriched by affinity purification on amylose resin. The protein-DNA complexes were then eluted from the resin with maltose. Unlike ChIP, no treatment with formaldehyde or other cross-linking agent is necessary.
Figure 1.

The DIP-ChIP method. A purified DNA-binding protein is incubated with purified, sheared yeast genomic DNA. Protein·DNA complexes are separated from unbound DNA using immunoprecipitation or affinity purification. Purified DNA fragments are amplified, labeled fluorescently, and identified by hybridization to a DNA microarray. Computational methods are then used to define a binding site based on enriched sequences (see text). A detailed description of the DIP-chip experimental methodology is available in the Methods.
To assess the relative abundance of genomic fragments selected by the purification, retained DNA was purified, amplified, and labeled fluorescently. In parallel, total genomic DNA was prepared, amplified, and labeled with a different fluorescent marker (Lieb et al. 2001). The two samples were then analyzed by comparative hybridization to DNA microarrays that cover the entire yeast genome at an average resolution of ∼1 kb (Lieb et al. 2001). DIP experiments were repeated independently at least three times at each concentration. The significance of enrichment for each feature on the array, expressed as a p-value, was estimated using a modified single array error model (Methods) (Ren et al. 2000).
The DIP-chip method was validated initially by the strong enrichment of DNA upstream of genes known to be regulated by Leu3p in vivo (Kohlhaw 2003). Of seven well-characterized Leu3p-regulated genes, promoter regions of six were among the top 30 sequences enriched by binding at 40 nM protein (p-value for enrichment of each sequence ≤ 2e-6). The seventh is bound more weakly (p-value = 0.01), but still ranks among the top 4% among all 12,000 array features. The enrichment of these sequences provided encouragement for the analyses described below, which demonstrate that DNA-binding motifs relevant to the location of bound protein in vivo can be determined using genomic sequences bound in vitro, as detected by the DIP-chip method.
Systematic determination of DNA-binding specificity from DIP-chip data
To further validate DIP-chip, we sought to determine the DNA-binding specificity of Leu3p from the DIP-chip data alone, and compare it with binding-site descriptions obtained by EMSA and SELEX. To that end, we devised a systematic and objective procedure to define position weight matrices (PWMs) (Stormo and Fields 1998) that maximizes the distinction between protein-bound and unbound sequences. The procedure consists of two steps. The first step uses the motif discovery programs BioProspector and MDScan (Liu et al. 2001, 2002) to define PWMs in a way that ensures that the number of input sequences has a minimal effect on the results. The second step evaluates the discovered motifs using the entire experimental data set.
In the motif discovery step (Fig. 2A), we used fixed numbers of the most highly enriched DNA sequences identified by DIP-chip. Specifically, BioProspector and MDScan were run using the sequences of the top 10, 20,..., 100 arrayed features, as ranked by enrichment p-value (Methods). For each set of input sequences, the first PWM reported by each program was recorded, resulting in a total of 20 motifs for each of the two experiments (4 and 40 nM). To simplify comparison to the 10-bp binding sites defined by SELEX and EMSA Kd, motif searches were restricted to widths of 10 bp, although searches unrestricted by width find similar sites (data not shown). Most, but not all of the PWMs identified by this procedure resemble the Leu3p consensus DNA-binding site (33/40 have the consensus base as the most favored at each position).
Figure 2.

Motif discovery procedure. (A) For each of the two protein concentrations used, array features were ranked according to enrichment p-value. The sequences corresponding to the top 10, 20, 30,..., 100 features were used as input to BioProspector and MDscan. For each set of features, indicated by arrows, a single position weight matrix (PWM) was obtained from each of the two programs. For illustrative purposes, we show the two motifs discovered using the top 70 features from the 4-nM experiment (black arrow). This set is interesting because it provides a contrast between an excellent PWM (MDscan) and a poor one (Bioprospector, see B and C). Motifs are represented as sequence logos with the height of each column representing the information content of that position in the binding site (Schneider and Stephens 1990). (B) Computationally defined occupancy scores for the top 75 enriched array features and for every 200th feature thereafter (4-nM experiment; note the break in the y-axis and the change in scale). Occupancy scores were calculated using the two PWMs shown in A (Methods). Filled circles represent the 23 features that meet the 1% false discovery rate criterion for significance; all other features are shown as open circles. Only the PWM defined by MDscan (consensus sequence CCGGTACCGG) shows a marked tendency for the DIP-enriched sequences to have higher occupancy scores than the nonenriched sequences. (C) A Receiver Operator Characteristic (ROC) curve (Hanley and McNeil 1982) showing the power of a PWM to distinguish DIP-enriched sequences from nonenriched. The heavy line with the shaded area below is for the PWM defined by MDscan in A, while the light line is for the PWM defined by BioProspector. The curves are equivalent to a plot of the true positives vs. false positives for all possible values of the occupancy scores that, for a given PWM, would be used to predict enrichment (see text). Each of the 20 PWMs discovered at each protein concentration was judged based on the area under the ROC curve (ROC AUC) obtained using occupancy scores calculated with that PWM. A ROC AUC value of 0.5, corresponding to a diagonal ROC curve, is expected by chance, while a value of 1.0 indicates perfect predictive value for the motif. In this case, the BioProspector-defined motif shows no predictive power (ROC AUC = 0.49), while the MDscan motif does (ROC AUC = 0.91). Note that the ability of MDscan to outperform Bioprospector is specific to this example and does not occur in every case.
In the second step of our analysis (Fig. 2B,C), each of the PWMs found in the first step was evaluated for its ability to distinguish significantly DIP-enriched features from all other microarray features. Significantly DIP-enriched features were defined using a 1% expected false discovery rate (FDR) (Benjamini and Hochberg 1995). By this criterion, 23 features were enriched at 4 nM protein (0.23 features expected by chance; enrichment p-value ≤ 1.8e-5) and 60 were enriched at 40 nM protein (0.60 features expected by chance; enrichment p-value ≤ 4.6e-5). We then assessed the ability of each DIP-derived PWM to distinguish these significantly DIP-enriched sequences from all other sequences on the array.
The scoring function and statistical metrics used in evaluating the binding-site descriptions have been described previously (Liu and Clarke 2002; Clarke and Granek 2003). Briefly, each PWM was used to predict the relative Leu3p binding affinity of all possible binding sites in the genome. We then used these relative affinities (which are unit-less) to predict the probability of Leu3p binding at each site. This calculation requires a unit-less parameter analogous to protein concentration, and we set the value of this parameter such that the consensus binding site is predicted to be half-occupied. Having estimated the occupancy of each binding site, we then determined an “occupancy score” for each microarray feature by calculating the probability that at least one site in each feature would be occupied by Leu3p. The degree to which DIP-enriched features exhibit high Leu3p occupancy scores, compared with nonenriched features, was defined by the area under a receiver operator characteristic curve (ROC AUC) (Fig. 2C). The PWM with the highest ROC AUC value in each experiment was considered the best motif that could be defined from that experiment (Fig. 3). At both protein concentrations, the motifs defined by DIP-chip analysis are similar to those defined by SELEX and EMSA (Fig. 3).
Figure 3.

Four representations of Leu3p-binding specificity, derived from the indicated in vitro binding experiments.
Many motif descriptions derived from DIP-chip describe the data equally well
The procedure described above yields the single best motif from each experiment, but many of the other motifs are similar in appearance, and indeed, are functionally similar by the objective criterion of ROC AUC value. We sought to determine whether these motifs were significantly worse than the best motifs, or were really members of a family of motifs that cannot be meaningfully distinguished from one another. This issue arises frequently in the field of motif analysis whenever a variety of binding-site descriptions are generated by experimental or computational methods. To resolve this issue, we estimated the 95% confidence interval for the ROC AUC values of the best motifs using bootstrap resampling of the occupancy scores and DIP-enrichment values (Efron and Gong 1983). We found that half of the 40 discovered motifs fall within the 95% confidence intervals (four from the 4 nM experiment and 16 from the 40 nM). We conclude that multiple variants of the motif can explain the DIP-chip data indistinguishably well. This procedure is generally applicable, and could be used widely to determine whether motif representations can be distinguished with confidence. Indeed, we use the same procedure below to compare DIP-chip-defined motifs with motifs defined by other methods, but, in this case, using a completely independent in vivo experimental data set as our standard for comparison.
DIP-chip predicts in vivo targets as well as EMSA and SELEX
As described above, the DIP-chip experimental protocol, combined with computational procedures for motif discovery, yields binding sites that resemble superficially those defined by SELEX or EMSA (Fig. 3). To establish the relative utility of these motifs, we determined how well each of the PWMs derived from SELEX, EMSA, or DIP-chip could predict the location of Leu3p binding in vivo. We performed ChIP-chip experiments to determine the in vivo binding location of the same epitope-tagged Leu3p fragment used in the DIP-chips. ChIP-chips were repeated independently five times, and 22 array features were identified as being bound in vivo (1% FDR). For each DIP-derived PWM, occupancy scores were calculated for all arrayed sequences. The scores of the ChIP-enriched sequences were then compared with those of unenriched sequences by ROC analysis (Fig. 4). All Leu3p PWMs tested, whether defined by classical assays or by DIP-chip, were found to explain in vivo binding similarly well (Fig. 4B,C). Specifically, all of the PWMs fall well within the 95% confidence interval of ROC AUC values for the PWM derived from EMSA Kd data. The PWMs defined by 40-nM DIP-chip appear to perform slightly better than both the classically defined PWMs and the PWMs defined by the 4-nM DIP-chip experiment (Fig. 4C). This result highlights a key aspect of DIP-chip, which is the ability to easily control the protein concentration. This allows one to find the concentration at which the derived in vitro PWM best explains the in vivo binding distribution.
Figure 4.

A comparison of the ability of DNA-binding motifs derived from different in vitro experiments to explain in vivo binding patterns. (A) Receiver Operator Characteristic (ROC) curve for quantitating how well a DIP-chip derived PWM can predict the results of a ChIP-chip experiment. The best PWM defined by the 4-nM DIP-chip data was used to calculate this plot (Methods; Fig. 2). Identical analyses were performed on PWMs derived from SELEX, EMSA, and all DIP-chip PWMs. (B) Areas under the ROC curve for PWMs evaluated against the ChIP data. The 95% confidence interval for the EMSA Kd ROC AUC value was estimated by bootstrap resampling of the occupancy scores and enrichment values for the 22 ChIP-enriched features. For the DIP-chip defined PWMs, the PWM that scored best when evaluated against the DIP data itself is shown as a filled circle. Other PWMs that are within the confidence interval of the best when evaluated against the DIP data are shown as open circles. (C) Same as B, but with a zoomed-in ROC AUC scale.
Discussion
We have developed DIP-chip as a new method for determining DNA-binding specificity and have demonstrated its utility using the DNA-binding domain of yeast Leu3p. The method is simple, and with the widespread availability of microarrays, it should be possible to quickly determine the in vitro specificity of any epitope-tagged transcription factor. Using genomic DNA and microarrays derived from the same organism as the tested protein facilitates comparisons to ChIP-chip data (see below) and ensures that all of the naturally occurring binding sites are present in the reaction mix. However, the same yeast genomic DNA and yeast microarrays used here could be used to determine the specificity of a DNA-binding protein from any organism, not just yeast. Since, in theory, genomic DNA and microarrays from any organism could be used to determine the specificity of a protein from any other organism, a genome sequence with low base-composition bias and broad representation of all possible binding sequences might be best for general use in DIP-chip.
DIP-chip has several advantages over established, nonarray-based methods for defining specificity like SELEX or EMSA. First, it requires only a fraction of the time and effort of either of those methods. Second, bound sequences are determined in a single binding reaction, avoiding the problem of overselection that is associated with iterative selection in SELEX experiments (Roulet et al. 2002). Third, long genomic sequences are used rather than short oligonucleotides, ensuring that binding sites are found within a relevant sequence context. These advantages also apply to microarray methods that directly detect the binding of proteins to spotted DNA microarrays (Bulyk et al. 2001). Direct detection avoids the need to enrich bound sequences by affinity purification, but there are likely to be greater challenges in obtaining adequate sensitivity in direct detection.
PWMs have also been defined using ChIP-chip data, but if the goal is to define intrinsic binding specificities, DIP-chip has substantial advantages. First, because of the confounding effects of other proteins binding cooperatively or in competition with the protein of interest, the sequences enriched in a ChIP-chip experiment are not a simple function of binding specificity. Second, in a DIP-chip experiment, the protein concentration can be adjusted easily to ensure that a sufficiently large number of sequences are significantly enriched. Without the ability to adjust protein concentration, the number of DNA sequences sampled by the motif-finding software may be too small for the binding-site description to be accurate. Concentrations can be varied in vivo, but not easily and not always reproducibly. Third, DIP-chip uses no chemical cross-linking step, avoiding the potential for protein-specific or DNA sequence-based biases in cross-linking efficiency that are inherent in ChIP-chip experiments. It might be necessary to use cross-linking in a DIP-chip experiment as well, if an exceptionally weak binding protein were being studied, but this should not generally be necessary.
In this study, we have focused on the use of DIP-chip to define DNA-binding specificities and have described computational procedures for defining and evaluating position weight matrices obtained in this way. However, the experimental protocol for DIP-chip can also be used for a rather different purpose, which is comparing the sites of binding in vitro with the sites of binding in vivo, as defined by ChIP-chip. As expected, we find a significant overlap among the microarray features that are enriched by binding of the Leu3p DNA binding domain in vitro and in vivo (P ≈ 10-130 for features enriched at a 1% false discovery rate; X. Liu, N.D. Clarke, and J.D. Lieb, in prep.). Comparisons of DIP-chip and ChIP-chip experiments will be useful in determining how much of the specificity of in vivo interactions depends on chromatin and other nuclear factors, and how much is inherent to the protein and DNA itself. We also envision DIP-chip experiments being performed with additional proteins in the reaction mixture to observe how they affect DNA-binding specificity. The ultimate goal of this line of experimentation would be to reconstruct in vitro the precise binding distribution observed in vivo. We conclude that DIP-chip is a powerful adjunct to ChIP-chip experiments, and as described here, is an efficient and accurate method for determining in vitro DNA-binding specificity.
Methods
Protein purification
Plasmid pMAL-c2-Leu3(1-147) (Liu and Clarke 2002) was used as follows to express an MBP-tagged Leu3p DNA-binding domain (MBP-leu3pDB) in Escherichia coli BL21 cells. Bacteria were grown in rich medium (yeast extract 5 g/L, tryptone 10 g/L, NaCl 5 g/L, and glucose 2 g/L supplemented with 100 μg/mL of ampicillin) with shaking at 37°C to an A600 of 0.5 and induced by addition of IPTG (0.3 mM). After 2 h, cells were collected and frozen in column buffer (20 mM TriṡCl at pH 7.4, 200 mM NaCl, 1 mM EDTA, and 1 mM DTT) overnight at -20°C. Cells were thawed in a cold water bath and disrupted by sonication. Cell debris was removed by centrifugation at 9000g, and the supernatant was passed through an amylose resin column (NEB). MBP-Leu3pDB was eluted with column buffer + 10 mM Maltose. Maltose was removed from the eluted protein by successive concentration and resuspension using a 30-kD molecular weight cutoff spin column (Microcon-30, Amicon). Protein purity was determined by SDS-PAGE, and concentration was determined by the Bio-Rad protein assay (Bio-Rad).
Genomic DNA purification
Strain BY4720-leu3Δneo carrying pRS416-TEF1-MBPLeu3pDB was cultured in YPD medium (yeast extract 10 g/L, peptone 20 g/L, supplemented with 2% glucose) to an A600 of 2. Collected cells were suspended in 200 μL solution A (2% Triton X-100, 1% SDS, 0.1 M NaCl, 10 mM Tris at pH 8, and 1 mM EDTA), 200 μL phenol/chloroform and 0.3 g acid-washed glass beads (Sigma G8772), and vortexed for 5 min. We added 200 μL TE (pH 8) to the extract and centrifuged for 5 min to collect the upper aqueous layer. The supernatant was then sonicated to fragment DNA to an average size of ∼0.6 kb. DNA was purified by phenol and phenol/chloroform extractions and ethanol precipitation. Genomic DNA was resuspended in TE (pH 8) + 30 μg/mL RNaseA at 37°C for 15 min. It was then re-extracted with phenol/chloroform, ethanol precipitated, and resuspended in 10 mM TriṡCl (pH 8). DNA concentration and purity were determined by absorption spectroscopy.
DNA immunoprecipitation (DIP) reactions
Purified MBP-leu3pDB and genomic DNA were mixed in 100 μL of binding/washing buffer (10 μM ZnSO4, 2 mM MgCl2, 2 mM TriṡCl at pH 7.4, 100 mM KCl, and 10% glycerol) and incubated at 30°C for 30 min. The final protein concentration was either 4 or 40 nM, and the DNA concentration was 2 μg/mL (equal to 0.3 pM genome). The solution was then mixed with 10 μL buffer-washed amylose resin, incubated at 30°C for 15 min with mixing by repeated pipetting, and then washed with binding/washing buffer four times. Protein-DNA complexes were eluted with column buffer supplemented with 10 mM maltose. The 4-nM MBP-Leu3pDB DIP experiment was repeated independently three times (independent mixtures of protein and DNA, and independent DNA microarrays), while the 40-nM DIP experiment was repeated independently four times. Control experiments using the MBP protein itself were performed six times.
Chromatin immunoprecipitation (ChIP) reactions
Yeast strain BY4720-leu3Δneo, carrying a plasmid that expresses MBP-leu3pDB (pRS416-TEF1-MBPLeu3pDB) was used for the ChIP-chip experiments. The strain was constructed, in this work, by replacement of the LEU3 coding region by the NEO gene in Saccharomyces cerevisiae strain BY4720 (MATalpha lys2Δ0 trp1Δ63 ura3Δ0) (Brachmann et al. 1998). pRS416-TEF1-MBPLeu3pDB was constructed by insertion of the MBPLeu3p-DB coding sequence into plasmid pRS416-TEF1 (URA3+ ampr), which contains the TDH3 promoter and CYC1 terminator (Sewing et al. 1994). Yeast were grown in uracil dropout medium (YNB-AA (Sigma) 6.7 g/L, 0.77 g/L Ura DO Supp. (Clonetech), and 2% glucose supplemented with G418 at 200 mg/L) with shaking at 30°C to an A600 of ∼1.0, at which point formaldehyde was added to a final concentration of 1%. The culture was maintained with shaking at 30°C for 15 min. Cells were collected and lysed by Beadbeater with glass beads (Sigma G8772) in bead-beater lysis buffer (50 mM Hepes-KOH at pH 7.5, 10 mM MgCl2, 150 mM KCl, 0.1 mM EDTA, 10% glycerol, 0.1% NP-40, 1 mM DTT, 1mM sodium metabisulfate and protease inhibitors). The supernatant was then sonicated to fragment DNA to an average size of ∼0.5 kb.
We carried out ChIP assays as described (Lieb et al. 2001) with anti-MBP (Abcam ab65) and protein G agarose (SIGMA 83219), except that after the ChIP, we washed protein G-agarose beads twice with lysis buffer (0.1% SDS, 0.5% Triton X-100, 20 mM TriṡCl at pH 8.0, 150 mM NaCl, and protease inhibitors), twice with lysis buffer + 2mM EDTA (pH 8.0), once with LiCl Buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA at pH 8.0 and 10 mM TriṡCl at pH 8.0) and twice with TE. Both the input protein-DNA mixture and the IP-enriched DNA were purified by QIAquick PCR purification kit (QIAGEN) after reversing cross-links at 65°C for 6 h. ChIP of the MBP-Leu3pDB was performed in replicate five times. A control experiment using an MBP-tagged fragment of Leu3p incapable of DNA binding (deletion of residues 13-601) was performed independently seven times.
DNA amplification, labeling, and microarray hybridization
IP-enriched DNA and input DNA (used as reference) was amplified as described, with a random-primed, PCR-based method (Lieb et al. 2001). Amplified DNA was labeled by either Cy5 or Cy3 monofunctional ester. In half of the experiments, IP-enriched DNA was labeled with Cy5 and reference DNA by Cy3, while in the other half, the fluorophores were swapped. Cy5- and Cy3-labeled DNA samples were mixed and hybridized to a genomic DNA microarray. Detailed protocols are available at http://www.bio.unc.edu/faculty/lieb/labpages/Protocols.shtml.
Array images were acquired with a GenePix 4000B scanner (Axon Instruments), and data extracted using GenePix Pro 4.0 software. These data were uploaded to the University of North Carolina (UNC) Microarray Database (https://genome.unc.edu/), from which were retrieved the normalized median intensity values for each channel and the standard deviation of the background intensity for each channel.
Data analysis
The intensities of each channel at each spot were analyzed using a single-array error model (Ren et al. 2000; Roberts et al. 2000) with minor modifications. The significance attached to differences in intensity at a particular spot, i, is given by a statistic X,
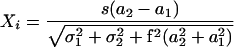 |
where a1 and a2 are the intensities measured in the two channels at that spot, σ1 and σ2 are estimated errors for the two intensities due to background subtraction, and f and s are terms that are adjusted to achieve the desired distribution of X values. f is adjusted so that the distribution of X values is close to normal for an experiment in which no significant enrichment is expected at all. This is the case, for example, with a control experiment in which the protein used has no DNA-binding activity. Even in a real experiment, if relatively few features are bound, then the distribution of X values is expected to be close to normal. s is adjusted so that the variance of X values is equal to 1; this allows comparisons among replicate experiments.
The X statistic, calculated for each spot on an array, is used to weight measurements obtained from replicate experiments. For each replicate, the enrichment, Ri, of feature i is defined as the log of the ratio of intensities [log(a2/a1)]. The weighted mean enrichment is then:
 |
where wi is the weight for the enrichment of spot i from a particular array and the summations are over all arrays. The weights are calculated from the significance statistic Xi by first defining the uncertainty (σi) in the enrichment (Ri) as σi = Ri/Xi. The weight attached to each value of Ri is then wi = 1/σi2. For the results reported here, the calculation of the weighted mean, as defined above and by Ren et al. (2000), was modified to
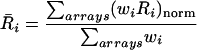 |
where the term (wiRi)norm means that the values of wiRi are first normalized so that the standard deviation of the values was the same in each array. This was done because of overall differences in the intensities of hybridization from replicate to replicate, but in practice, this normalization had only a small effect on the identification of enriched features.
In order to now estimate the p-value for enrichment, we first estimate the standard deviation, σm, in the mean enrichment for each spot by propagating the uncertainty in the enrichment of that spot in each array:
 |
The more the mean ratio of intensities exceeds this estimate of the standard deviation, the more likely it is that there is true enrichment. Thus,
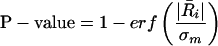 |
where the erf() function is the standard cumulative distribution function for the area under a normal curve area. The error model was implemented in R (version 1.8.0).
Sequence manipulation and computational analysis
The yeast genome sequence and its annotations were downloaded from the Saccharomyces Genome Database http://www.yeastgenome.org. The sequences of microarray features, and their genomic coordinates, were derived by J. Granek (Johns Hopkins) based on the primer sequences used to amplify the array features. Primer sequences are available from the UNC Microarray Database https://genome.unc.edu. Genomic features enriched in control experiments (P < 0.001) and mitochondrial features were not used in the analysis.
Occupancy scores of microarray features were calculated as described for gene regulatory regions (Liu and Clarke 2002), but with modifications. Briefly, an occupancy score for a feature is based on the probability of binding to each subsequence that is either within the feature, or within 1000 bp on either side of the feature. The probability of binding to a particular subsequence, pi, is based on the equation for a simple binding isotherm:
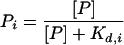 |
where [P] is the free protein concentration and Kd,i is the equilibrium dissociation constant of site i. Since the free concentration of protein in vivo is unknown, we used a value for [P] equal to the estimated Kd for the optimal binding site (Kd,optimal), which gives a binding probability of 0.5 for that site. The optimal binding site is defined as the variant of the binding site in which the most favored base is found at each position. Dividing both the numerator and denominator of the previous equation by Kd,optimal, and remembering that [P] is defined as being equal to Kd,optimal, the probability of binding to site i becomes:
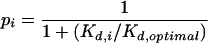 |
Equilibrium constants are estimated from a position weight matrix. The simplest such calculation can be performed when the elements of the PWM represent the contribution of each base to the free energy of binding. PWMs of this type can be obtained from a fit to experimental data for the binding affinity of a large number of binding sites (Liu and Clarke 2002). In this case, the binding free energy, ΔGi, can be estimated from summation of the relevant PWM terms, and Kd,i = exp(ΔGi,/RT). Fortunately, similar PWMs can be derived using the base frequency at each position in a set of known or suspected binding sites, which is a more common source of information on DNA-binding specificity. The ratio of the observed frequency of a base at a particular position to the expected frequency (base composition) can be thought of as an equilibrium constant, which can then be converted to a PWM element representing the contribution to the free energy of binding (Stormo and Fields 1998).
Having calculated the probability of binding, pi, to all possible sites, the occupancy score, S, for the entire feature is defined as
 |
where the summation is over all possible sites, i, within the feature or within 1000 bp of the ends. wi is the weight assigned to site i. Sites within the feature are given a weight of 1, while those flanking the feature are given weights between 1 and 0, declining as a linear function of distance from the feature boundary. The calculation of occupancy scores, and the assessment of the correlation between predicted occupancy and observed enrichment in the ChIP or DIP experiments, were performed using a computer program called GOMER (Generalizable Objective Model of Expression Regulation) (J. Granek and N.D. Clarke, in prep.). GOMER is available upon request from N.D. Clarke at nclarke@jhmi.edu.
Acknowledgments
We thank Josh Granek for help with GOMER and the preparation of sequence files and Giovanni Parmigiani for helpful discussions on the statistics of ROC analysis. This work was supported by NIH grants to N.D.C. (GM065179) and J.D.L. (HG002577).
Footnotes
[Raw data, array images, and compiled tabular data are publicly available as Supplemental material online at www.genome.org and from the UNC Microarray database at https://genome.unc.edu.]
Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.3256505. Article published online before print in February 2005.
References
- Benjamini, Y. and Hochberg, Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289-300. [Google Scholar]
- Bulyk, M.L., Huang, X., Choo, Y., and Church, G.M. 2001. Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc. Natl. Acad. Sci. 98: 7158-7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann, C.B., Davies, A., Cost, G.J., Caputo, E., Li, J., Hieter, P., and Boeke, J.D. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115-132. [DOI] [PubMed] [Google Scholar]
- Clarke, N.D. and Granek, J.A. 2003. Rank order metrics for quantifying the association of sequence features with gene regulation. Bioinformatics 19: 212-218. [DOI] [PubMed] [Google Scholar]
- Efron, B. and Gong, G. 1983. A leisurely look at the bootstrap, the jackknife and cross-validation. J. Amer. Stat. Soc. 37: 36-48. [Google Scholar]
- Fried, M. and Crothers, D.M. 1981. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 9: 6505-6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley, J.A. and McNeil, B.J. 1982. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143: 29-36. [DOI] [PubMed] [Google Scholar]
- Kohlhaw, G.B. 2003. Leucine biosynthesis in fungi: Entering metabolism through the back door. Microbiol. Mol. Biol. Rev. 67: 1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb, J.D., Liu, D., Botstein, X., and Brown, P.O. 2001. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 28: 327-334. [DOI] [PubMed] [Google Scholar]
- Liu, X. and Clarke, N.D. 2002. Rationalization of gene regulation by a eukaryotic transcription factor: Calculation of regulatory region occupancy from predicted binding affinities. J. Mol. Biol. 323: 1-8. [DOI] [PubMed] [Google Scholar]
- Liu, X., Brutlag, D.L., and Liu, J.S. 2001. BioProspector: Discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac. Symp. Biocomput. : 127-138. [PubMed]
- ____. 2002. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 20: 835-839. [DOI] [PubMed] [Google Scholar]
- Oliphant, A.R., Brandl, C.J., and Struhl, K. 1989. Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: Analysis of yeast GCN4 protein. Mol. Cell. Biol. 9: 2944-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, B., Robert, F., Wyrick, J.J., Aparicio, O., Jennings, E.G., Simon, I., Zeitlinger, J., Schreiber, J., Hannett, N., Kanin, E., et al. 2000. Genome-wide location and function of DNA binding proteins. Science 290: 2306-2309. [DOI] [PubMed] [Google Scholar]
- Roberts, C.J., Nelson, B., Marton, M.J., Stoughton, R., Meyer, M.R., Bennett, H.A., He, Y.D., Dai, H., Walker, W.L., Hughes, T.R., et al. 2000. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science 287: 873-880. [DOI] [PubMed] [Google Scholar]
- Roulet, E., Busso, S., Camargo, A.A., Simpson, A.J., Mermod, N., and Bucher, P. 2002. High-throughput SELEX SAGE method for quantitative modeling of transcription-factor binding sites. Nat. Biotechnol. 20: 831-835. [DOI] [PubMed] [Google Scholar]
- Schneider, T.D. and Stephens, R.M. 1990. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 18: 6097-6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewing, A., Ronicke, V., Burger, C., Funk, M., and Muller, R. 1994. Alternative splicing of human cyclin E. J. Cell Sci. 107(Pt 2): 581-588. [DOI] [PubMed] [Google Scholar]
- Stormo, G.D. and Fields, D.S. 1998. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem. Sci. 23: 109-113. [DOI] [PubMed] [Google Scholar]
- Tuerk, C. and Gold, L. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249: 505-510. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.bio.unc.edu/faculty/lieb/labpages/Protocols.shtml; Common microarray protocols.
- http://www.yeastgenome.org; Saccharomyces Genome Database.
- https://genome.unc.edu; UNC Microarray Database.