

Abstract
Background & aims
Iron overload disorders such as hereditary hemochromatosis and iron-loading anemias are a common cause of morbidity from liver diseases and increase risk of hepatic fibrosis and hepatocellular carcinoma (HCC). Once iron-induced damage occurs, treatment options are limited partly because of the lack of animal models recapitulating human disease, which do not exhibit relevant sequelae after chronic iron overload.
Methods
Since liver-specific β-catenin knockout mice (KO) are susceptible to injury, fibrosis and tumorigenesis following chemical carcinogen exposure, iron-overload diet was administered to KO and littermate control (CON) mice for various times. To ameliorate an oxidant-mediated component of tissue injury, N-Acetyl-L-(+)-cysteine (NAC) was added to drinking water of mice on iron overload diet.
Results
KO on iron diet (KO+Fe) exhibited remarkable inflammation, followed by steatosis, oxidative stress, fibrosis, regenerating nodules and occurence of occasional HCC. Increased injury in KO+Fe was associated with activated AKT, ERK, and NFκB, along with reappearance of β-catenin and target gene Cyp2e1, which promoted lipid peroxidation and hepatic damage. Addition of NAC to drinking water protected KO+Fe from hepatic steatosis, injury and fibrosis, and prevented activation of AKT, ERK, NFκB and re-appearance of β-catenin.
Conclusions
We provide evidence that absence of hepatic β-catenin predisposes mice to hepatic injury and fibrosis following iron overload, which was reminiscent of hemochromatosis and associated with enhanced steatohepatitis and fibrosis. Disease progression was notably alleviated by antioxidant therapy, which supports its chemopreventive role in the management of chronic iron overload disorders.
Keywords: Wnt signaling, Hemochromatosis, Oxidative Stress, Lipid Peroxidation
Graphical abstract
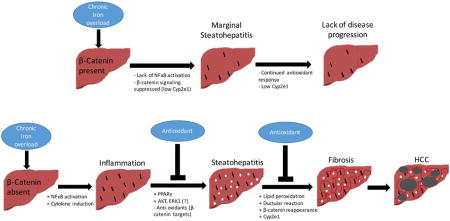
INTRODUCTION
Hereditary hemochromatosis, a prototypical iron overload disorder, is one of the most common genetic disorders in the United States. It occurs in approximately 1 in 200 Caucasians and 40% of healthy population in the US carry a mutation predisposing them to iron overload [1]. There is progressive accumulation of iron in parenchymal cells of various tissues due to mutations in key iron homeostasis genes. Liver is one of the major organs affected and around 75% of clinically affected hemochromatosis patients have evidence of liver disease at presentation. Excess iron deposition in hepatocytes is associated with hepatomegaly, elevated liver enzymes and eventually leads to chronic injury due to oxidative stress, steatosis, inflammation, fibrosis, and cancer [2, 3]. Currently, recurrent phlebotomy is an effective way to prevent chronic iron overload in patients with hereditary hemochromatosis [4] but this option is not available for similarly affected patients with iron-loading anemias, such as β-thalassemia, where iron chelation is used to reduce iron loading.
One major obstacle in studying cellular and molecular mechanisms underlying hemochromatosis disease progression is the lack of relevant animal models. Rodent models fed high iron diet do not display injury or disease progression like patients. Similarly, genetic mouse models targeting iron-metabolism genes like hepcidin and human hemochromatosis protein encoding gene HFE do not recapitulate the severity of fibrosis or liver injury [5]. Thus, there is a significant need for development of relevant animal models that can mimic human disease.
Wnt/β-catenin signaling has been shown to be important in hepatic homeostasis [6]. Wnt ligands inhibit β-catenin degradation allowing it to enter the nucleus where it interacts with T-cell factor (TCF) family of transcription factors to induce target genes involved in proliferation, liver zonation, cell survival, and oncogenic transformation [7]. Wnt/β-catenin signaling can regulate cellular redox states in models such as hepatic ischemia-reperfusion [8]. Liver-specific β-catenin knockout mice (KO) show increased carcinogenesis after diethylnitrosamine treatment, which was associated with increased injury, oxidative stress and fibrosis [9, 10]. Iron has been shown to activate Wnt/β-catenin signaling [11, 12]. Based on these observations, we decided to investigate the effect of iron overload in KO compared to age- and sex-matched littermate controls (CON). We show that KO after high iron diet (KO+Fe) develop inflammation, steatosis, oxidative stress, fibrosis, and occasionally develop HCC. Intriguingly, supplementation with an antioxidant prevents disease pathogenesis. Thus, through its role as a regulator of inflammatory milieu and redox states in hepatocytes, but not through regulation of iron metabolism genes, β-catenin plays a protective role after iron overload.
MATERIALS AND METHODS
Animals
All animal work was performed in accordance with the University of Pittsburgh Institutional Animal Care and Use Committee. Liver-specific β-catenin knockout mice (KO) were generated as previously described [13].
Additional details are available in online supplement and Supplementary CTAT Table.
RESULTS
Mice lacking β-catenin in liver epithelial cells develop steatohepatitis and fibrosis after iron overload
Iron overload diet was fed for 3 months to KO (KO+Fe) and CON (CON+Fe). While both groups tolerated the treatment, KO+Fe livers showed evidence of gross injury showing pale discoloration and rigidity (not shown). Serum ALT levels trended higher in KO+Fe although the differences were not significant (Fig.1A). KO−Fe had significantly lower basal liver weight to body weight ratio (LW/BW) when compared to the CON−Fe group as reported previously (Fig.1B) [13]. Intriguingly, both CON+Fe and KO+Fe showed comparable and significantly higher LW/BW ratio than their respective controls (Fig.1B).
Figure 1. Chronic iron overload causes steatohepatitis in β-catenin knockout mice (KO+Fe).
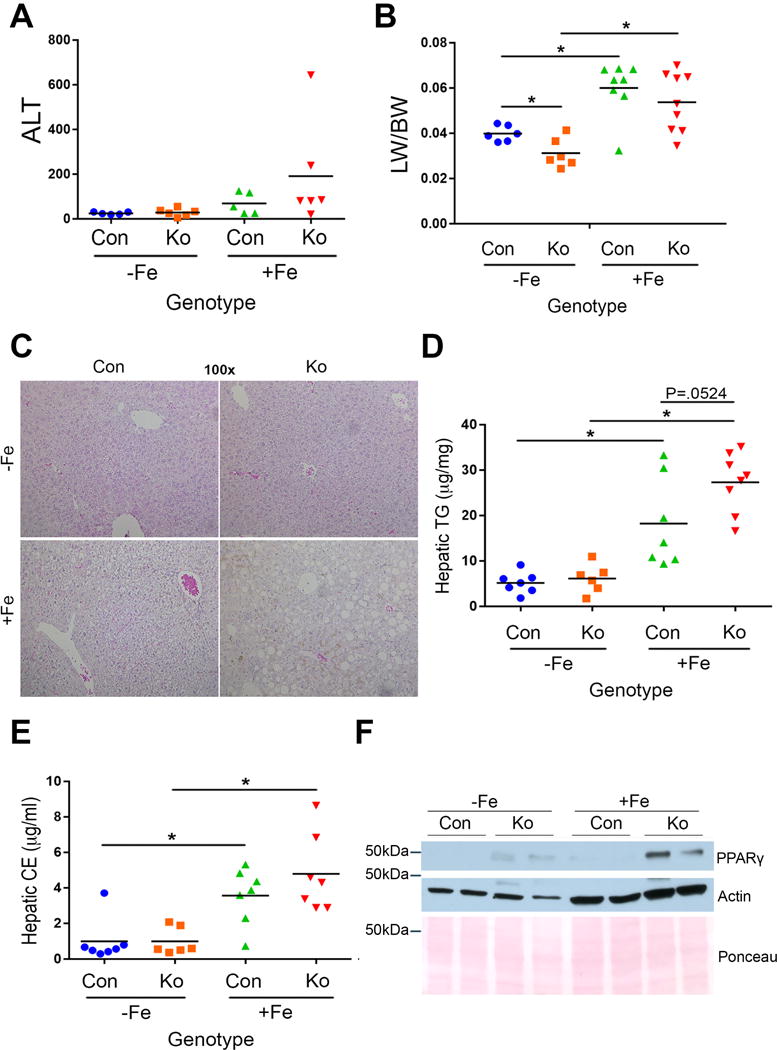
(A) Serum ALT levels reflect a modest but insignificant increase in KO+Fe as compared to other groups. (B) Iron causes increased liver weight to body weight ratio (LW/BW) in CON+Fe and KO+Fe. At baseline, KO show significantly lower LW/BW than CON. (C) Representative H&E staining (100×) shows mild hepatic steatosis in CON+Fe and more profound hepatic steatosis and inflammation in KO+Fe. (D) Increased hepatic triglycerides (TG) (p=0.0524) in KO+Fe as compared to CON+Fe. (E) Cholesterol esters (CE) were comparably increased after iron in KO and CON. (F) Representative WB showing increase in PPARγ in KO+Fe as compared to other groups. *p<0.05 using Student’s T test.
Liver histology revealed notable macrovesicular steatosis and inflammation in KO+Fe only (Fig.1C). To confirm hepatic steatosis, we examined levels of hepatic lipids. CON+Fe and KO+Fe had significantly greater hepatic triglycerides and cholesterol esters than respective controls although KO+Fe showed highest and most significant increase (Fig.1D,E). PPARγ, a known adipogenic factor negatively regulated by β-catenin [14], was marginally increased in KO basally but was further induced in KO+Fe (Fig.1F). KO−Fe livers show a small increase in CD45-positive inflammatory cells as reported previously [15]. Iron diet led to more pronounced inflammation in KO along with F480-positive macrophage infiltration (Fig.2A,B). Enhanced inflammation in KO+Fe was further verified by the presence of increased inflammatory markers IL-6 and CCL2 by Western blots (WB), both of which migrate slower likely due to glycosylation [16, 17] (Fig.2C). Presence of steatohepatitis was also associated with increased fibrosis as revealed by Sirius Red staining showing extensive collagen accumulation in KO+Fe only (Fig.2D and Supplementary Fig.1A). This was further validated by the presence of activated myofibroblasts seen by increased α-smooth muscle actin (α-SMA) by both WB and IHC in KO+Fe only (Fig.2E,F). Presence of fibrosis was further supported by elevated PDGFRβ levels (Fig.2F).
Figure 2. KO+Fe have increased hepatic inflammation and fibrosis after chronic iron overload.
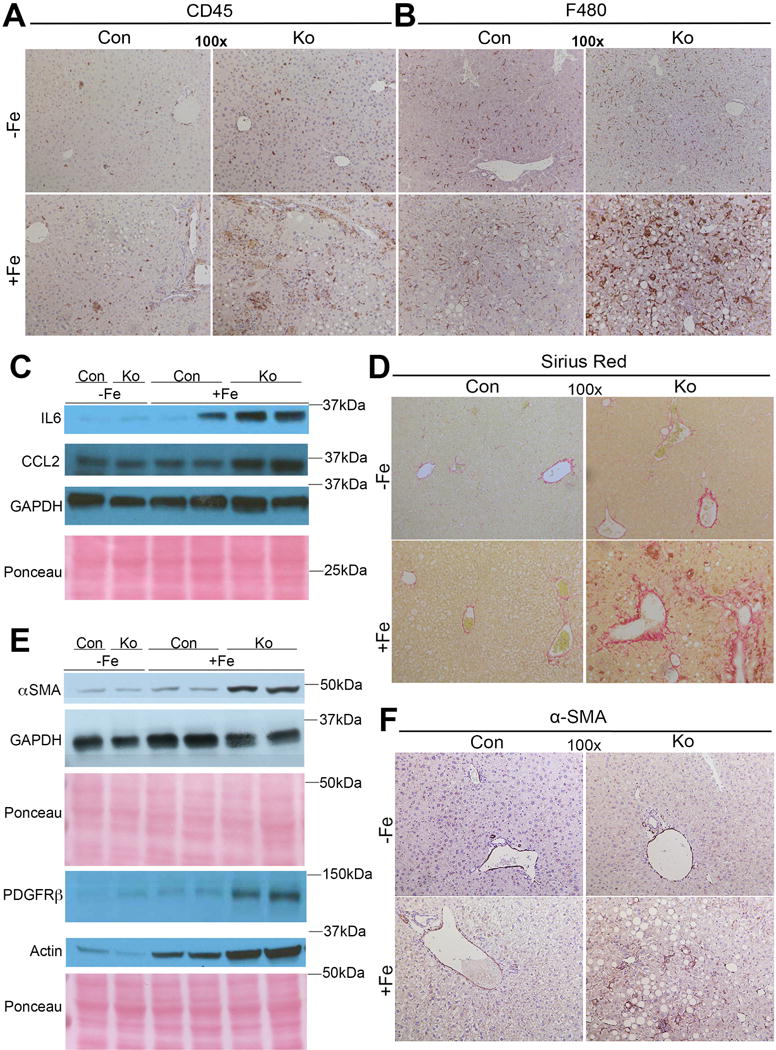
(A) Representative IHC images demonstrate increased CD45-positive inflammatory cells in KO+Fe as compared to other groups. (B) Representative IHC images demonstrate increased F480-positive macrophages in KO+Fe only. (C) Representative WB shows KO+Fe have increased inflammatory markers including IL-6 and CCl2. IL-6 and CCl2 appear to be migrating slower due to glycosylation. (D) KO+Fe have increased Sirius Red staining suggesting increased collagen deposition. (E). WB show increased levels of α-SMA and PDGFRβ substantiating presence of fibrosis. (F) IHC for α-SMA also confirms presence of activated myofibroblasts in KO+Fe only. (Image magnification: 100×)
KO display enhanced ductular reaction in response to injury due to iron overload
We next investigated cellular injury and repair in KO+Fe. A marginal increase in cell death as seen by TUNEL staining was evident in CON+Fe, which was exacerbated in KO+Fe (Fig.3A). This was associated with modest but comparable increase in S-phase marker Ki-67, in hepatocytes in CON+Fe and KO+Fe (Fig.3B). Likewise, a mid-zonal increase in Cyclin-D1 expression was evident in CON+Fe, which was reduced in KO+Fe and instead observed in periportal hepatocytes (Fig.3B). Chronic hepatic injury can lead to progenitor cell response in the form of ductular reaction, especially if hepatocyte proliferation is disproportionally lower than ongoing death [18]. Indeed, KO+Fe uniquely showed increased sox-9, EpCAM and CK19 positive ductular reaction (Fig.3D–F).
Figure 3. Analysis of cell death, proliferation and ductular reaction in CON and KO on normal and high iron diet.
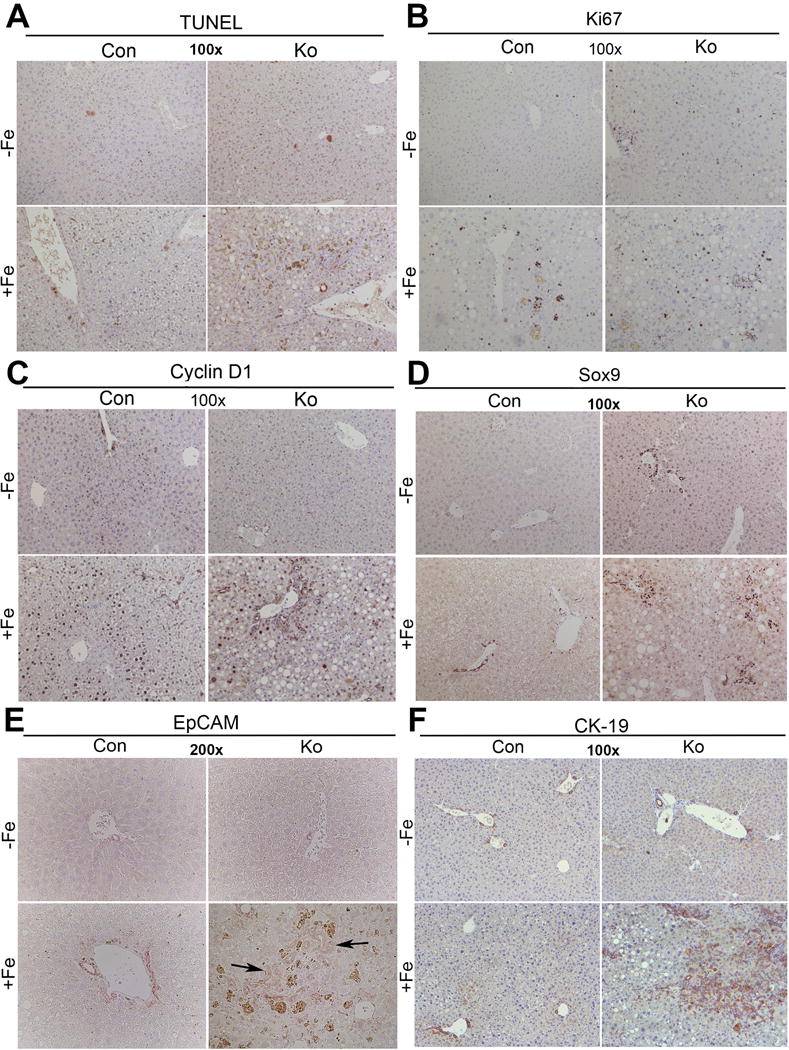
(A) Representative images of TUNEL staining shows marginal increase in cell death in CON+Fe, but a greater increase in KO+Fe. (B) Ki-67 IHC demonstrates comparable and increased number of cells in S-phase after iron overload in both CON+Fe and KO+Fe. (C) Cyclin-D1 staining is increased in midzonal hepatocytes in CON+Fe compared to CON−Fe. KO+Fe exhibit less Cyclin-D1 staining in periportal hepatocytes and in ductular cells. (D) KO+Fe have increased ductular reaction as evident by IHC for Sox9. (E) Increased ductular reaction in KO+Fe was confirmed by EpCAM staining. (F) Increased ductular reaction in KO+Fe was also confirmed by IHC for CK19. (Image magnification:100×, except EpCAM:200×).
Continued injury in KO+Fe led to development of bridging fibrosis outlining regenerating nodules (Supplemental Fig.1A). We also observed occurrence of occasional HCC nodule in KO+Fe (Supplementary Fig.1B). Intriguingly, tumors were β-catenin and GS-positive (Supplementary Fig.1B).
Inflammation precedes steatosis, which precedes fibrosis in β-catenin KO after iron overload
To address the disease development in KO+Fe, we subjected CON and KO mice to 4-week iron-overload diet. Notable steatosis was evident in KO+Fe, but not in CON+Fe (Fig.4A). An associated increase in CD45- and F480-positive cells was also observed in KO+Fe only along with mild fibrosis (Fig.4A). These observations suggest that fibrosis follows inflammation and steatosis in KO+Fe.
Figure 4. Increased levels of inflammatory cytokines precede steatohepatitis and fibrosis in KO+Fe as shown by time course after iron-feeding.
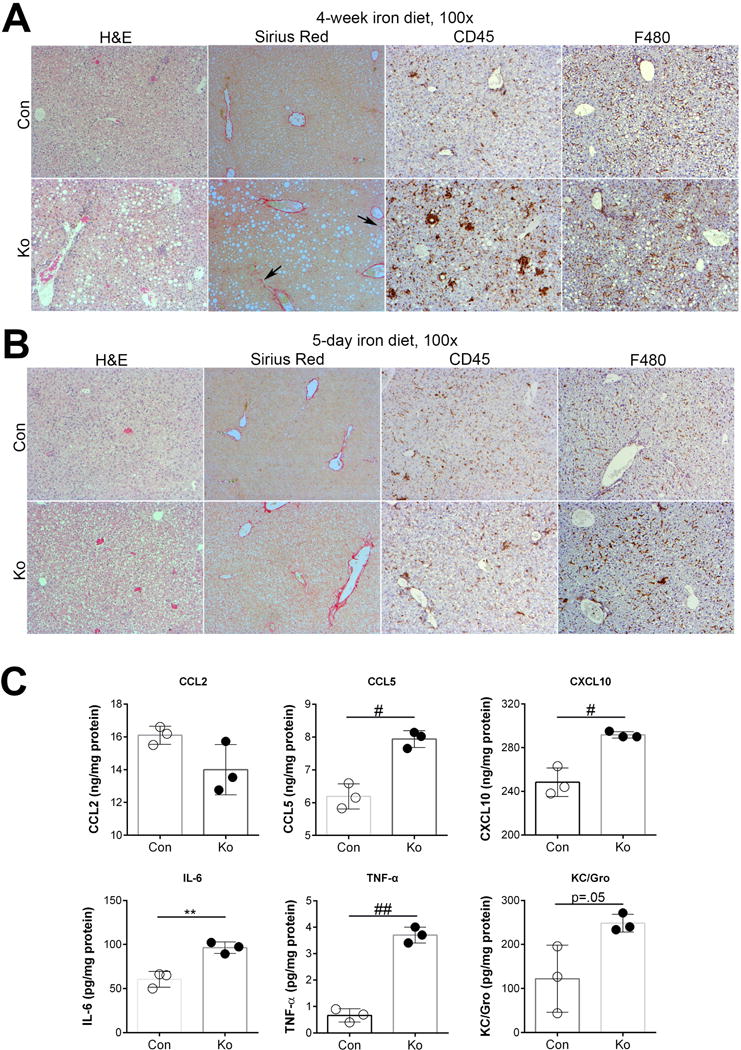
(A) H&E, Sirius Red and IHC for CD45 and F480 in CON and KO livers after 4-week iron diet shows presence of steatohepatitis in KO+Fe and initiation of fibrosis. (B) H&E, Sirius Red and IHC for CD45 and F480 in CON and KO livers after 5-day iron diet. Con+FE and KO+Fe lack steatosis and fibrosis while CD45-positive inflammatory cells and F480-macrophages appear to be comparably increased in both groups. (C) Cytokine assay using liver lysates from 5-day CON+Fe and KO+Fe reveals comparable increased protein levels of CCL5, CXCL10, IL-6, TNFα, and KC/Gro in KO+Fe but no change in CCl2. *p<0.05, **p<0.01, #p<0.005, #p<0.001 using Student’s T test. (Image magnification: 100×)
We next challenged KO and CON with high iron diet for 5 days. No steatosis or fibrosis was evident in either group (Fig.4B). While no discernible differences in CD45-positive cells were evident, a small increase in F480-positive infiltrate was evident in KO+Fe (Fig.4B). To more conclusively address differences, we assessed these livers for pro-inflammatory cytokines. A significant upregulation of CCL5, CXCL10, IL-6, and TNF-α was seen in KO+Fe as compared to CON+Fe (Fig.4C).
Thus, iron-overload in absence of β-catenin in liver leads to sequential increase in inflammation followed by steatosis and fibrosis.
Comparable hepatic iron accumulation in β-catenin KO and CON following iron overload
Next, we address if iron metabolism is altered in KO. Prussian blue staining revealed iron accumulation in CON predominantly in the periportal region (Fig.5A). Iron accumulation showed a more pan-zonal distribution in KO+Fe (Fig.5B). An increase in aggregates of iron pigment in KO+Fe reminiscent of iron-laden macrophages was also observed (Fig.5B). However, both CON+Fe and KO+Fe had an increase in total liver iron content compared to respective controls, and differences in the hepatic iron content between CON+Fe and KO+Fe were insignificant (Fig.5C). Interestingly, KO+Fe showed increased serum iron when compared to KO−Fe, whereas CON+Fe and CON−Fe showed no differences (Fig.5D).
Figure 5. Comparable hepatic iron levels in CON+Fe and KO+Fe, despite increased serum iron and altered iron zonation in KO+Fe.
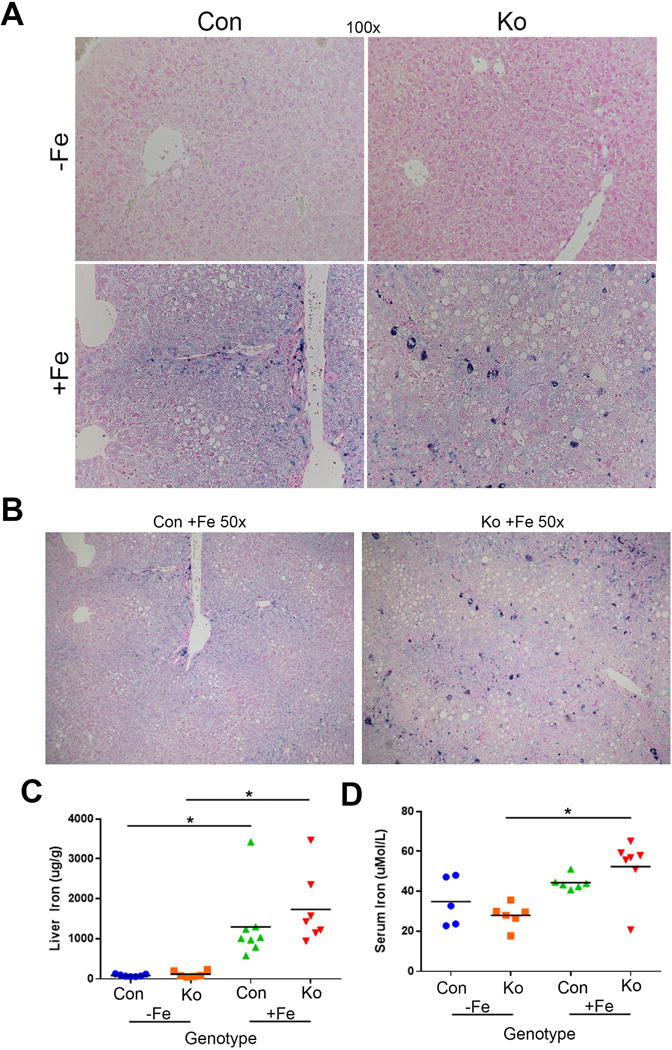
(A) Prussian blue staining (100×) shows iron accumulation is periportal in CON+Fe. In KO+Fe, iron deposition is pan-zonal. Aggregates of iron pigment resembling iron-laden macrophages were observed in KO+Fe (B) A Periportal Prussian blue staining in periportial region in CON+Fe versus more pan-zonal staining in KO+Fe can be appreciated in lower magnification (50×). (C) Liver iron levels increase comparably in CON+Fe and KO+Fe compared to CON−Fe and KO−Fe. (D) Serum iron levels are increased in KO+Fe compared to KO−Fe, but there is no difference between KO+Fe and CON+Fe. *p<0.05 using Student’s T test.
Next, we determined expression levels of genes commonly mutated in hemochromatosis or otherwise involved in iron metabolism by RT-PCR. Hepcidin, a gene that inhibits iron export from a cell [19] was comparably induced in CON and KO after iron overload as reported previously [20] (Supplementary Fig.2A). Human hemochromatosis protein and hemojuvelin, two genes frequently mutated in different hemochromatosis subtypes [5], demonstrated no significant differences in expression (Supplementary Fig.2B,C). Expression of ceruloplasmin, which converts toxic ferrous form to a nontoxic ferric form [21], was increased in KO+Fe as compared to CON+Fe, with an insignificant increase compared to KO−Fe (Supplemental Fig.2D). Transferrin receptors 1 and 2 are cell surface receptors required for iron entry into cells [22, 23]. Although TRF1 expression is low in the liver [23], we saw a further reduction in CON+Fe and KO+Fe compared to CON−Fe and KO−Fe (Supplemental Fig.2E). TRF2 was not significantly altered in CON+Fe compared to CON−Fe, as also reported elsewhere [23]. Interestingly, a modest but statistically significant decrease in TRF2 was observed in KO+Fe when compared to KO−Fe (Supplemental Fig.2F). Ferroportin, the iron export protein negatively regulated by hepcidin [24], is increased in CON and KO after iron although increase is significantly greater in KO+Fe (Supplemental Fig.2G). Overall, our data suggests that major iron regulatory genes are unaffected by β-catenin signaling basally, but KO livers show altered expression of some iron regulatory genes likely in an attempt to compensate for increased hepatic injury after high iron diet.
Iron overload decreases β-catenin signaling in CON but induces other cell survival and mitogenic signaling in KO
Next, we asked if chronic iron overload impacts β-catenin signaling in the liver. Livers from mice fed high iron diet or basal diet were assessed for liver-specific β-catenin targets. Intriguingly, we observed decrease in Glutamine synthetase (GS), Cyp1a2 and Cyp2e1 (Fig.6A.B). In the absence of any evidence of β-catenin activation by iron overload, the mechanism of worse injury in high iron-fed KO mice required additional investigation.
Figure 6. Chronic iron overload affects β-catenin signaling in CON+Fe, but induces AKT, ERK, and NFκB signaling and lipid peroxidation in KO+Fe.
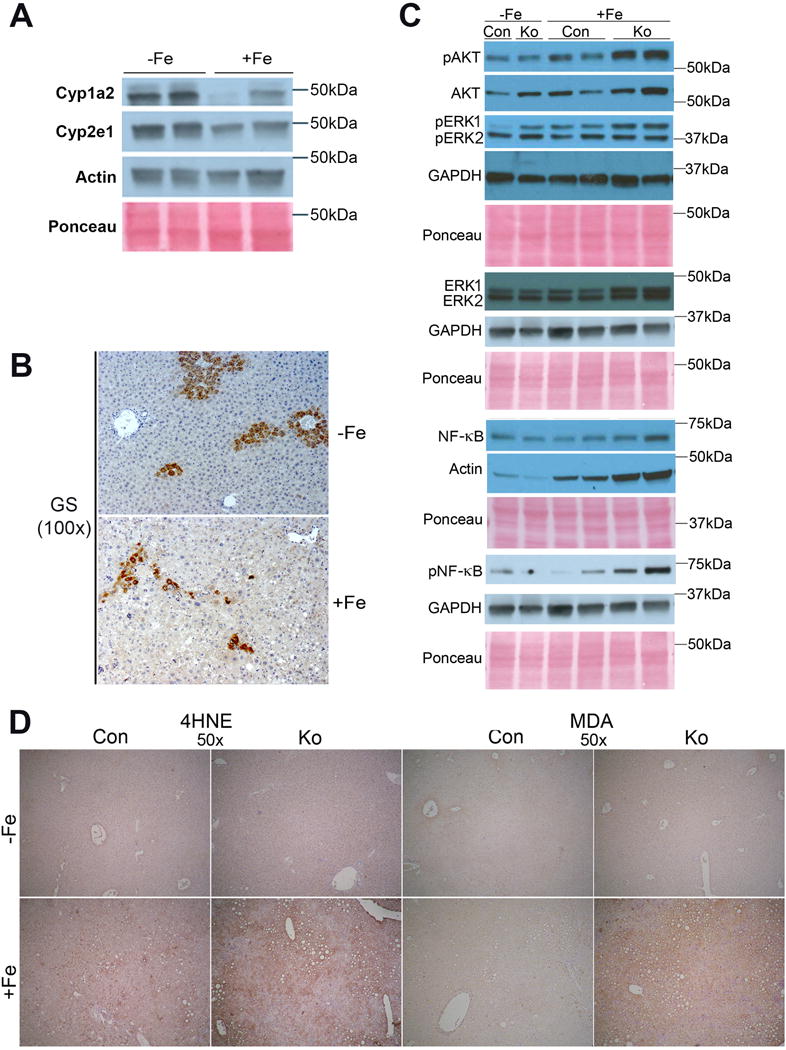
(A) Representative WB show a decrease in hepatic levels of Cyp1a2 and Cyp2e1 levels after iron overload. (B) Representative IHC for GS shows decreased uniformity of GS staining in pericentral hepatocytes in CON after iron overload (100×). (C) Representative WB show greater increases in pAKT (Thr308) and pERK1 (Thr202/Thr204) in KO+Fe as compared to CON+Fe. pNFкB (Ser536) is induced only in KO+Fe. (D) Representative IHC images show increased staining for lipid peroxidation markers 4-hydroxynonenal (4HNE) and malondialdehyde (MDA) in KO+Fe. (50×).
We first assessed factos like Protein Kinase B (Akt), extracellular signal-regulated kinase 1/2 (Erk1/2), and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB). Akt, which is involved in cell survival, migration, and proliferation [25], showed increased phosphorylation in KO+Fe compared to other groups (Fig.6C). Erk1 phosphorylation, which is associated with hepatic fibrosis [26], was increased at baseline in KO, was induced by iron in both CON and KO, and was highest in KO+Fe (Fig.6C). Erk2 phosphorylation showed only a modest increase in KO−Fe with no notable differences between CON+Fe or KO+Fe (Fig.6C). NFκB, which regulates genes involved in inflammation [27], proliferation and survival [28], showed increased phosphorylation in KO+Fe compared to other groups (Fig.6C). Taken together, these data suggest that iron overload leads to decreased β-catenin signaling but in its absence, leads to activation of Akt, Erk and NFκB, which may contribute to greater hepatic injury and repair response in KO+Fe.
Evidence of increased hepatic oxidative stress in β-catenin KO after iron overload
Since β-catenin has been shown to regulate redox homeostasis in hepatic tissue through multiple mechanisms [8, 10, 29], we next investigated if β-catenin-deficient livers are inept at counteracting oxidative stress generated normally by iron accumulation and inflammation, and known to contribute to chronic injury, fibrosis and cancer [30, 31]. Indeed, increased oxidative stress in the form of lipid peroxidation was seen in KO+Fe by IHC for both 4-hydroxynonenal (4HNE) and malondialdehyde (MDA) (Fig.6D). Predictably, lipid peroxidation products were visible in and around areas of steatosis in the KO+Fe.
Since Cyp2e1 has been implicated in contributing to harmful reactive oxygen species (ROS) [32] and regulated by β-catenin signaling in liver [13, 33], we assessed it’s expression in CON and KO after iron. Decrease in Cyp2e1 staining and its localization to sparse areas of steatosis was observed in CON+Fe as compared to CON−Fe (Fig.7A). Intriguingly, while no Cyp2e1 was detected in KO−Fe, iron overload led to an unexpected increase, which was localized to steatotic areas in KO+Fe (Fig.7A). The reappearance of Cyp2e1 was also confirmed by WB (Fig.7B). Likewise, while total β-catenin levels decreased in CON after iron overload, its levels increased in KO+Fe (Fig.7B–D). Similarly, GS staining decreased and was irregular in CON+Fe as compared to uniform pericentral staining in hepatocytes in CON−Fe (Fig. 7E), However, while GS was absent in KO−Fe, we observed occasional areas of GS-positive hepatocytes especially in steatotic and even periportal areas in KO+Fe (Fig.6B,7E). Thus, presence of chronic injury in KO after iron overload led to some repopulation by β-catenin-positive hepatocytes, which led to reappearance of Cyp2e1.
Figure 7. Reappearance of β-catenin and its targets following chronic iron overload in KO.
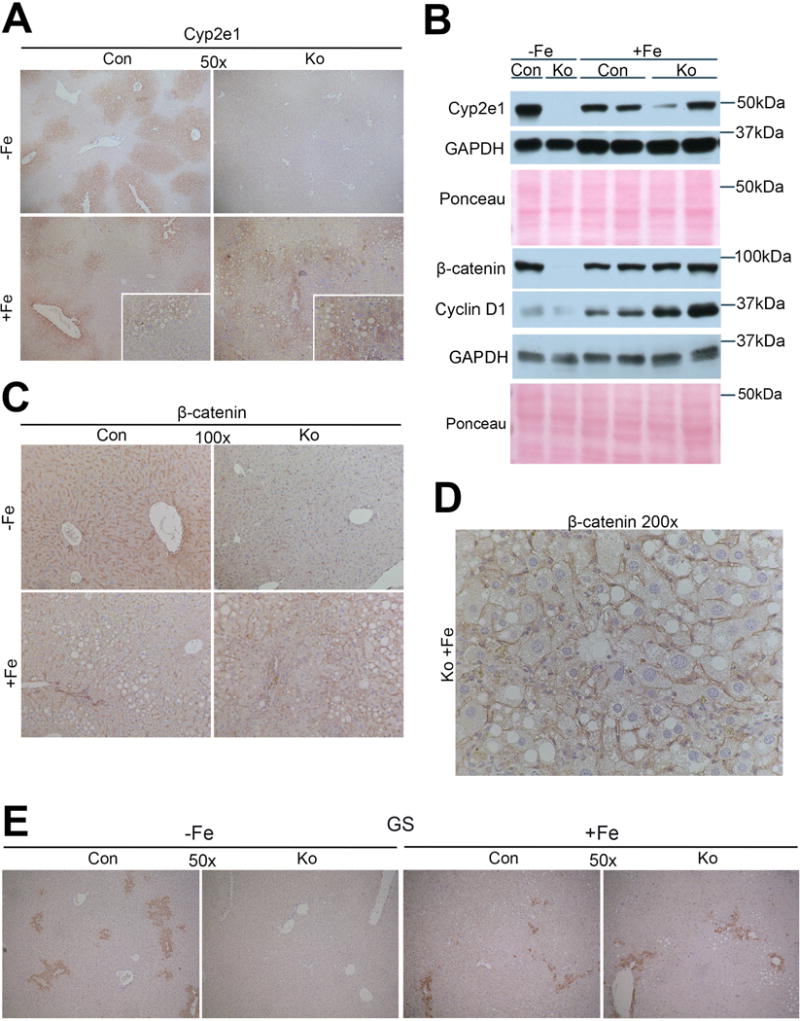
(A) IHC for Cyp2e1 shows decrease in pericentral staining for Cyp2e1 in CON after iron diet. KO lack Cyp2e1 basally, however KO+Fe show its re-appearance particularly in areas of steatosis. (B) WB confirm increases in levels of Cyp2e1, β-catenin, and Cyclin-D1 in KO+Fe compared to KO−Fe. (C) Representative IHC images of β-catenin also confirm re-expression of β-catenin especially in steatotic areas in KO+Fe (100×). (D) Higher magnification image of β-catenin IHC from KO+Fe liver shows positive staining (200×). (E) Representative IHC of GS shows decreased staining in CON livers after iron diet, however in KO which lack GS basally, show re-appearance of GS (50×).
N-Acetyl-L-(+)-cysteine (NAC) protects β-catenin KO after iron overload against injury and fibrosis
Since increased lipid peroxidation appeared to be associated with steatohepatitis and fibrosis in KO+Fe, we next administered NAC, an antioxidant, to CON+Fe and KO+Fe via drinking water for 3 months. NAC alleviated lipid peroxidation as observed by IHC for MDA and 4-HNE in KO+Fe (Fig.8). Simultaneously, we observed a modest decrease in steatosis, which was also reflected by a partial reduction in PPARγ levels (Fig.8, Supplementary Fig.3A). NAC also led to a pronounced decrease in α-SMA IHC along with a dramatic decrease in fibrosis by Sirius Red staining (Fig.8). This was verified by decreased PDGFRβ and α-SMA protein levels (Supplemental Fig.3A). Although numbers of CD45-positive cells remained unaffected by NAC in KO, inflammation markers IL-6 and MCP1 lowered to match CON+Fe+NAC levels (Supplemental Fig.3A). Zonal iron distribution was also partially restored in KO+Fe+NAC group (Fig.8).
Figure 8. Inclusion of N-Acetyl-L-(+)-cysteine (NAC) in drinking water normalizes histology and affects oxidative stress, steatosis, fibrosis and iron staining in KO+Fe, while inflammation remains unaltered.
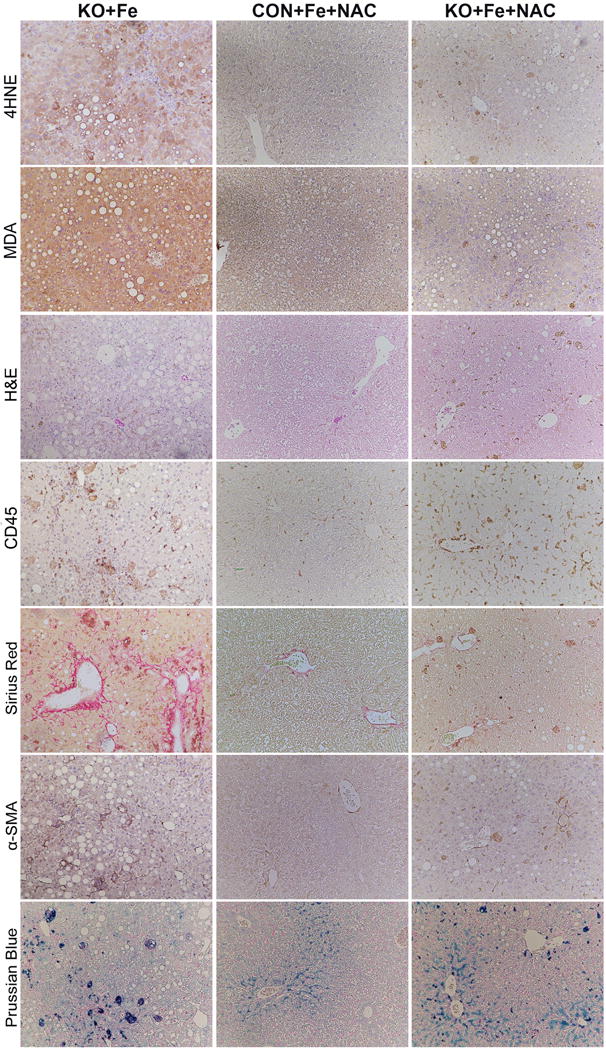
Representative images from livers of KO+Fe, CON+Fe+NAC, and KO+Fe+NAC mice showing a profound effect of NAC in reducing overall hepatic injury in KO+Fe, which is depicted by decrease in staining for 4HNE, MDA, Sirius Red and αSMA. H&E shows normalization of histology along with an overall decrease in steatosis. Prussian blue staining shows shift of pan-zonal staining to periportal staining after NAC treatment of KO+Fe. No quantitative differences in the numbers of CD45 cells were seen. (100×)
Since we considered resurgence of β-catenin and its targets Cyp2e1 and GS as injury responses, we next posited that NAC may prevent such increase if injury is successfully alleviated. Indeed, levels of total β-catenin, Cyclin-D1 and Cyp2e1 were diminished to non-existent levels as basal KO, following NAC treatment as also verified by IHC (Supplementary Fig.3B,C). NAC also prevented activation of AKT and NF-κB although pERK1 remained higher in KO+Fe+NAC (Supplementary Fig.3D). Intriguingly, while pERK2 levels were markedly increased in both CON and KO after iron and NAC treatment (Supplementary Fig.3E). Lastly, normalization of LW/BW ratio was evident in KO+Fe+NAC group which was significantly lower than CON+Fe+NAC (Supplementary Fig.3F). Thus, alleviating oxidative stress by NAC prevented several relevant complications of chronic iron overload in KO.
DISCUSSION
Wnt/β-catenin signaling plays diverse roles in liver pathobiology [6], although its role in iron metabolism in the liver remains unknown. A previous study reported increased Wnt signaling after addition of cellular iron to APC-mutant cell lines [11]. We used previously characterized liver-specific β-catenin KO [13, 33] to determine any changes in the expression of key iron-metabolism genes. None of the major genes were differentially expressed between CON and KO. To determine if iron had an impact on β-catenin signaling, control mice were fed a diet with excessive iron content. Two groups had individually identified iron chelators that inhibited Wnt signaling in different cancer models [12, 34]. Interestingly, we observed decreased β-signaling after high iron diet as indicated by decreased expression of target genes Cyp2e1, Cyp1a2 and GS. Intriguingly, despite decreased signaling, a complete lack of β-catenin was a greater detriment to mice after hepatic iron overload, which exhibited activation of AKT, ERK, and NFκB, steatohepatitis, fibrosis, HCC and progenitor response, which led to reappearance of β-catenin and its targets inadvertently making the injury worse. Thus overall, KO on high iron diet recapitulated hemochromatosis-like disease. Addition of antioxidant NAC abrogated hepatic damage and normalized histology and cell signaling in KO+Fe.
There was differential regulation of a few iron metabolism genes between KO and CON after high iron diet. We noted decreased transferrin receptor 2 mRNA levels while ferroportin and ceruloplasmin mRNA levels were increased in KO+Fe. Since transferrin receptor 2 allows entry of iron into hepatocytes and ceruloplasmin converts toxic ferrous form to a nontoxic ferric form, the changes in KO are likely compensatory to limit injury in KO. This could also explain increased serum iron levels in KO+Fe, which however could simply reflect an increase in overall hepatic injury in KO. Ferroportin mRNA is increased in KO+Fe and CON+Fe. Ferroportin is negatively regulated by hepcidin at a posttranslational level [24]. Hepcidin binds to ferroportin to induce its internalization and degradation [19]. Thus, we believe that increase in ferroportin mRNA in KO+Fe and CON+Fe is in response to its enhanced degradation by increased hepcidin. However, a significantly greater increase in ferroportin in KO+Fe versus CON+Fe may be due to additional mechanisms like inflammation and IL-6, which occur in KO+Fe and have been shown to induce hepcidin expression [35].
Progression of injury in KO after iron overload lends insights into pathogenesis of disease like hemochromatosis. Increase in inflammatory cytokines like CCL5, CXCL10, IL-6, and TNFα appear to be the first molecular event following iron overload in the absence of β-catenin in hepatocytes, which was evident at 5 days after iron diet. We believe that NFκB activation is responsible for inflammation in KO+Fe [36]. We have previously shown that β-catenin associates with NFκB to prevent its nuclear translocation, and β-catenin loss accelerates NFкB activation [15].
Steatosis follows inflammation as seen by the presence of steatohepatitis at 4 weeks. Steatosis in KO is likely due to inflammation and also due to increased hepatic PPARγ, master regulator of adipogenesis [37]. β-Catenin signaling suppress adipogenesis while its inhibition promotes pre-adipocyte differentiation to adipocyte through activation of lipogenic factors PPARγ and CEBPβ [38]. How iron overload induces PPARγ uniquely in KO remains under investigation. We also observe activation of AKT, ERK, and NFκB after long term iron diet administration to KO. AKT activation impacts proteins involved in survival, cell cycle, autophagy, and protein synthesis [39, 40], including FoxO, which normally inhibits cell growth and/or induces apoptosis [40]. AKT phosphorylates and inhibits FoxO [41]. Intriguingly, β-catenin can also act as a transcriptional cofactor for FoxO [42]. While further studies are required to precisely address the role of AKT in KO+Fe, we posit that excess iron enhances β-catenin and FoxO to induce apoptotic genes to clear damaged cells, thereby promoting proliferation of healthy cells. However, when β-catenin is absent, AKT phosphorylates and inactivates FoxO, allows persistence of damaged cells, eventually leading to HCC. We also observe increased ERK1 in KO+Fe compared to CON+Fe. Whether ERK1 and ERK2 have distinct roles is controversial [26]. A rat study showed ERK1 upregulation augmented fibrosis and its suppression decreased fibrosis and increased hepatocyte proliferation [43]. Therefore, it is likely that ERK1 activation in KO+Fe contributes to fibrosis. Phospho-ERK2 levels increased in CON and KO after iron and NAC treatment. Since silencing ERK2, but not ERK1, affected hepatocyte proliferation [44], we believe that NAC-induced ERK2 activation contributes to hepatic regenerative response after iron overload. NFκB can also be activated by inflammation and lipid peroxidation [45]. As a transcription factor, it regulates expression of a number of inflammatory cytokines and chemokines and also Cyclin-D1 [46]. This may explain in part the Cyclin-D1 induction seen in KO+Fe. Eventually steatohepatitis leads to cell death, stellate cell activation, fibrosis, and occasionally to development HCC.
The presence of increased oxidative stress appears to be chief perpetrator in disease progression in KO+FE. We observe both increased MDA and 4HNE, especially in the areas of steatotic hepatocytes. Indeed, iron overload in genetically obese mice is associated with oxidative stress, injury and a NASH-like phenotype [47]. Since mice are notoriously resistant to injury after high iron diet, we posit that lack of notable oxidative stress after iron feeding may explain the absence of a phenotype in control mice. Cyp2e1 is typically associated with excessive lipid peroxidation and hepatic injury following iron overload, alcohol and high fat diet [32, 48–50]. Cyp2e1 is a bona fide β-catenin target in pericentral hepatocytes [13, 33, 51]. Our study shows that feeding high iron diet to control mice leads to decreased Wnt/β-catenin signaling and as a result, lowers Cyp2e1 levels. This may be the protective mechanism, which limits generation of oxidative stress to make mice resistant to iron overload injury.
Despite lack of Cyp2e1 in KO, these mice showed worse injury to iron overload. This is likely a consequence of enhanced inflammation due to NFκB activation and dearth of β-catenin-dependent anti-oxidant mechanisms. Loss of β-catenin in liver is associated with enhanced oxidative stress following ischemia-reperfusion injury and chemical carcinogenesis [8–10]. This is due to the lack of adequate glutathione content resulting from altered expression of several glutathione S-transferases, regucalcin (required for ascorbic acid biosynthesis) and interactions with HIF1α [8, 29, 52, 53]. However, another unexpected and paradoxical event in KO+Fe further underscores the importance of Cyp2e1 and oxidative stress as a mediator of iron-induced hepatic injury. Due to enhanced injury in KO+Fe, we observed progenitor response characterized by Sox9, EpCAM, and CK19-positive ductular reaction [18]. As a consequence, there is reappearance of β-catenin-positive hepatocytes in KO+Fe, along with targets like Cyp2e1 especially in the steatotic areas. A caveat is that as progenitor cells would differentiate into hepatocytes, they will re-express albumin which would lead to expression of cre-recombinase and β-catenin deletion. It is likely hepatocyte differentiation is suboptimal and hence these “hepatocyte-like” cells may not have optimum albumin-cre expression. Another possibility is repopulation by β-catenin-positive hepatocytes due to suboptimal deletion by ‘leaky’ albumin-cre, which has been reported during chronic injuries like 3,5-diethoxycarbonyl-1, 4-dihydrocollidine (DDC) diet [54]. In the current study however, the survival advantage attributed to the β-catenin deletion-escaped hepatocytes comes at an unexpected cost of reappearance of Cyp2e1, which is normally downregulated following iron overload in controls. This pivots the balance towards greater lipid peroxidation, injury and disease progression.
Alleviation of oxidative stress and lipid peroxidation in β-catenin KO with NAC following iron overload had a dramatic effect on the disease process. While inflammation was unaffected quantitatively, NAC treatment led to notable decreases in pro-inflammatory cytokines, steatosis, stellate cell activation and fibrosis, thus normalizing histology and phenotype and making KO+Fe indistinguishable in phenotype from CON+Fe. It may be beneficial to examine any role of antioxidants for chemopreventive use in iron overload disorders.
Supplementary Material
Lay Summary.
Lack of models for iron overload disorders makes it hard to study the disease process for improving therapies. Feeding high iron diet to mice that lack β-catenin in liver cells led to increased inflammation followed by fat accumulation, cell death and wound healing that mimicked human disease. Administration of an antioxidant prevented hepatic injury in this model.
Acknowledgments
We thank Dr. Murugesan Velayutham, Amalea Misse and Greg Logan for help with experimentation.
Financial Support Statement: This study was funded by NIH grants 1R01DK62277, 1R01DK100287 and Endowed Chair for Experimental Pathology to SPSM.
Abbreviations
- CON
control
- KO
knockout
- +Fe
iron overload diet
- −Fe
basal diet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: No conflicts of any authors pertinent to the current study.
Author contributions:
M.E.P: acquisition of data, data analysis, data interpretation, drafting of article; S.S: acquisition of data; E.V.V and C.J: acquisition of data; B.P: acquisition of data; M.P: acquisition of data; S.N: concept design and interpretation of data; T.G: concept design and interpretation of data; S.M: concept design, analysis and interpretation of data, drafting of article.
References
- 1.Burt MJ, George PM, Upton JD, Collett JA, Frampton CM, Chapman TM, et al. The significance of haemochromatosis gene mutations in the general population: implications for screening. Gut. 1998;43:830–836. doi: 10.1136/gut.43.6.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang X. Iron overload and its association with cancer risk in humans: evidence for iron as a carcinogenic metal. Mutat Res. 2003;533:153–171. doi: 10.1016/j.mrfmmm.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 3.Valenti L, Fracanzani AL, Dongiovanni P, Bugianesi E, Marchesini G, Manzini P, et al. Iron depletion by phlebotomy improves insulin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: evidence from a case-control study. Am J Gastroenterol. 2007;102:1251–1258. doi: 10.1111/j.1572-0241.2007.01192.x. [DOI] [PubMed] [Google Scholar]
- 4.Camaschella C. Treating iron overload. N Engl J Med. 2013;368:2325–2327. doi: 10.1056/NEJMcibr1304338. [DOI] [PubMed] [Google Scholar]
- 5.Delima RD, Chua AC, Tirnitz-Parker JE, Gan EK, Croft KD, Graham RM, et al. Disruption of hemochromatosis protein and transferrin receptor 2 causes iron-induced liver injury in mice. Hepatology. 2012;56:585–593. doi: 10.1002/hep.25689. [DOI] [PubMed] [Google Scholar]
- 6.Monga SP. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology. 2015;148:1294–1310. doi: 10.1053/j.gastro.2015.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monga SP. Role of Wnt/beta-catenin signaling in liver metabolism and cancer. Int J Biochem Cell Biol. 2011;43:1021–1029. doi: 10.1016/j.biocel.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehwald N, Tao GZ, Jang KY, Sorkin M, Knoefel WT, Sylvester KG. Wnt-beta-catenin signaling protects against hepatic ischemia and reperfusion injury in mice. Gastroenterology. 2011;141:707–718. 718 e701–705. doi: 10.1053/j.gastro.2011.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Awuah PK, Rhieu BH, Singh S, Misse A, Monga SP. beta-Catenin loss in hepatocytes promotes hepatocellular cancer after diethylnitrosamine and phenobarbital administration to mice. PLoS One. 2012;7:e39771. doi: 10.1371/journal.pone.0039771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang XF, Tan X, Zeng G, Misse A, Singh S, Kim Y, et al. Conditional beta-catenin loss in mice promotes chemical hepatocarcinogenesis: role of oxidative stress and platelet-derived growth factor receptor alpha/phosphoinositide 3-kinase signaling. Hepatology. 2010;52:954–965. doi: 10.1002/hep.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brookes MJ, Boult J, Roberts K, Cooper BT, Hotchin NA, Matthews G, et al. A role for iron in Wnt signalling. Oncogene. 2008;27:966–975. doi: 10.1038/sj.onc.1210711. [DOI] [PubMed] [Google Scholar]
- 12.Coombs GS, Schmitt AA, Canning CA, Alok A, Low IC, Banerjee N, et al. Modulation of Wnt/beta-catenin signaling and proliferation by a ferrous iron chelator with therapeutic efficacy in genetically engineered mouse models of cancer. Oncogene. 2012;31:213–225. doi: 10.1038/onc.2011.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology. 2006;131:1561–1572. doi: 10.1053/j.gastro.2006.08.042. [DOI] [PubMed] [Google Scholar]
- 14.Christodoulides C, Lagathu C, Sethi JK, Vidal-Puig A. Adipogenesis and WNT signalling. Trends Endocrinol Metab. 2009;20:16–24. doi: 10.1016/j.tem.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nejak-Bowen K, Kikuchi A, Monga SP. Beta-catenin-NF-kappaB interactions in murine hepatocytes: a complex to die for. Hepatology. 2013;57:763–774. doi: 10.1002/hep.26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu ZG, Haelens A, Wuyts A, Struyf S, Pang XW, Proost P, et al. Isolation of a lymphocyte chemotactic factor produced by the murine thymic epithelial cell line MTEC1: identification as a 30 kDa glycosylated form of MCP-1. Eur Cytokine Netw. 1996;7:381–388. [PubMed] [Google Scholar]
- 17.May LT, Shaw JE, Khanna AK, Zabriskie JB, Sehgal PB. Marked cell-type-specific differences in glycosylation of human interleukin-6. Cytokine. 1991;3:204–211. doi: 10.1016/1043-4666(91)90018-9. [DOI] [PubMed] [Google Scholar]
- 18.Williams MJ, Clouston AD, Forbes SJ. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology. 2014;146:349–356. doi: 10.1053/j.gastro.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 19.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 20.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 21.Patel BN, Dunn RJ, Jeong SY, Zhu Q, Julien JP, David S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J Neurosci. 2002;22:6578–6586. doi: 10.1523/JNEUROSCI.22-15-06578.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aisen P. Transferrin receptor 1. Int J Biochem Cell Biol. 2004;36:2137–2143. doi: 10.1016/j.biocel.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Fleming RE, Migas MC, Holden CC, Waheed A, Britton RS, Tomatsu S, et al. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc Natl Acad Sci U S A. 2000;97:2214–2219. doi: 10.1073/pnas.040548097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossi E. Hepcidin—the iron regulatory hormone. Clin Biochem Rev. 2005;26:47–49. [PMC free article] [PubMed] [Google Scholar]
- 25.Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul. 2014;55:28–38. doi: 10.1016/j.jbior.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Busca R, Pouyssegur J, Lenormand P. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front Cell Dev Biol. 2016;4:53. doi: 10.3389/fcell.2016.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradford JW, Baldwin AS. IKK/nuclear factor-kappaB and oncogenesis: roles in tumor-initiating cells and in the tumor microenvironment. Adv Cancer Res. 2014;121:125–145. doi: 10.1016/B978-0-12-800249-0.00003-2. [DOI] [PubMed] [Google Scholar]
- 29.Tan X, Yuan Y, Zeng G, Apte U, Thompson MD, Cieply B, et al. Beta-catenin deletion in hepatoblasts disrupts hepatic morphogenesis and survival during mouse development. Hepatology. 2008;47:1667–1679. doi: 10.1002/hep.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung FL, Pan J, Choudhury S, Roy R, Hu W, Tang MS. Formation of trans-4-hydroxy-2-nonenal-and other enal-derived cyclic DNA adducts from omega-3 and omega-6 polyunsaturated fatty acids and their roles in DNA repair and human p53 gene mutation. Mutat Res. 2003;531:25–36. doi: 10.1016/j.mrfmmm.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Wang XW, Hussain SP, Huo TI, Wu CG, Forgues M, Hofseth LJ, et al. Molecular pathogenesis of human hepatocellular carcinoma. Toxicology. 2002;181–182:43–47. doi: 10.1016/s0300-483x(02)00253-6. [DOI] [PubMed] [Google Scholar]
- 32.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sekine S, Lan BY, Bedolli M, Feng S, Hebrok M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology. 2006;43:817–825. doi: 10.1002/hep.21131. [DOI] [PubMed] [Google Scholar]
- 34.Song S, Christova T, Perusini S, Alizadeh S, Bao RY, Miller BW, et al. Wnt inhibitor screen reveals iron dependence of beta-catenin signaling in cancers. Cancer Res. 2011;71:7628–7639. doi: 10.1158/0008-5472.CAN-11-2745. [DOI] [PubMed] [Google Scholar]
- 35.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, et al. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl Acad Sci U S A. 1993;90:10193–10197. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 38.Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 39.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 40.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813:1978–1986. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 42.Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007;9:210–217. doi: 10.1038/ncb1534. [DOI] [PubMed] [Google Scholar]
- 43.Zhong W, Shen WF, Ning BF, Hu PF, Lin Y, Yue HY, et al. Inhibition of extracellular signal-regulated kinase 1 by adenovirus mediated small interfering RNA attenuates hepatic fibrosis in rats. Hepatology. 2009;50:1524–1536. doi: 10.1002/hep.23189. [DOI] [PubMed] [Google Scholar]
- 44.Fremin C, Ezan F, Boisselier P, Bessard A, Pages G, Pouyssegur J, et al. ERK2 but not ERK1 plays a key role in hepatocyte replication: an RNAi-mediated ERK2 knockdown approach in wild-type and ERK1 null hepatocytes. Hepatology. 2007;45:1035–1045. doi: 10.1002/hep.21551. [DOI] [PubMed] [Google Scholar]
- 45.Faux SP, Howden PJ. Possible role of lipid peroxidation in the induction of NF-kappa B and AP-1 in RFL-6 cells by crocidolite asbestos: evidence following protection by vitamin E. Environ Health Perspect. 1997;105(Suppl 5):1127–1130. doi: 10.1289/ehp.97105s51127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446:475–482. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 47.Handa P, Morgan-Stevenson V, Maliken BD, Nelson JE, Washington S, Westerman M, et al. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am J Physiol Gastrointest Liver Physiol. 2016;310:G117–127. doi: 10.1152/ajpgi.00246.2015. [DOI] [PubMed] [Google Scholar]
- 48.Cederbaum AI. Iron and CYP2E1-dependent oxidative stress and toxicity. Alcohol. 2003;30:115–120. doi: 10.1016/s0741-8329(03)00104-6. [DOI] [PubMed] [Google Scholar]
- 49.Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radic Biol Med. 2010;49:1406–1416. doi: 10.1016/j.freeradbiomed.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zong H, Armoni M, Harel C, Karnieli E, Pessin JE. Cytochrome P-450 CYP2E1 knockout mice are protected against high-fat diet-induced obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2012;302:E532–539. doi: 10.1152/ajpendo.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loeppen S, Koehle C, Buchmann A, Schwarz M. A beta-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis. 2005;26:239–248. doi: 10.1093/carcin/bgh298. [DOI] [PubMed] [Google Scholar]
- 52.Liu B, Zhang R, Tao G, Lehwald NC, Liu B, Koh Y, et al. Augmented Wnt signaling as a therapeutic tool to prevent ischemia/reperfusion injury in liver: Preclinical studies in a mouse model. Liver Transpl. 2015;21:1533–1542. doi: 10.1002/lt.24331. [DOI] [PubMed] [Google Scholar]
- 53.Nejak-Bowen KN, Zeng G, Tan X, Cieply B, Monga SP. Beta-catenin regulates vitamin C biosynthesis and cell survival in murine liver. J Biol Chem. 2009;284:28115–28127. doi: 10.1074/jbc.M109.047258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thompson MD, Wickline ED, Bowen WB, Lu A, Singh S, Misse A, et al. Spontaneous repopulation of beta-catenin null livers with beta-catenin-positive hepatocytes after chronic murine liver injury. Hepatology. 2011;54:1333–1343. doi: 10.1002/hep.24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.