

Supplemental Digital Content is available in the text
Keywords: coexpression network, copy-number variation, differentially expressed gene, prostate cancer
Abstract
Background:
This study aimed to identify potential prostate cancer (PC)-related variations in gene expression profiles.
Methods:
Microarray data from the GSE21032 dataset that contained the whole-transcript and exon-level expression profile (GSE21034) and Agilent 244K array-comparative genomic hybridization data (GSE21035) were downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) and copy-number variations (CNVs) were identified between PC and normal tissue samples. Coexpression interactions of DEGs that contained CNVs (CNV–DEGs) were analyzed. Pathway enrichment analysis of CNV–DEGs was performed. Drugs targeting CNV–DEGs were searched using the Drug–Gene Interaction database.
Results:
In total, 679 DEGs were obtained, including 182 upregulated genes and 497 downregulated genes. A total of 48 amplified CNV regions and 45 deleted regions were determined. The number of CNVs at 8q and 8p was relatively higher in PC tissue. Only 16 DEGs, including 4 upregulated and 12 downregulated genes, showed a positive correlation with CNVs. In the coexpression network, 3 downregulated CNV–DEGs, including FAT4 (FAT atypical cadherin 4), PDE5A (phosphodiesterase 5A, cGMP-specific), and PCP4 (Purkinje cell protein 4), had a higher degree, and were enriched in specific pathways such as the calmodulin signaling pathway. Five of the 16 CNV–DEGs (e.g., PDE5A) were identified as drug targets.
Conclusion:
The identified CNV–DEGs could be implicated in the progression of human PC. The findings could lead to a better understanding of PC pathogenesis.
1. Introduction
Prostate cancer (PC) is the most frequently diagnosed malignancy in males, with 220,800 new cases and 27,540 deaths per year in the United States.[1] Despite an improvement in therapeutic schedules, PC-related mortality remains high worldwide.[1] In recent years, remarkable advances have been made in the investigation of molecular mechanisms underlying PC, such as the detection of copy-number variations (CNVs).
CNV is a kind of gene mutation that causes genetic diversity and is defined as a variable copy number of a segment of deoxyribonucleotides (DNA) > 1 kb in size.[2] CNV is universal among humans and is associated with complex diseases like PC by providing phenotypic diversity and conferring variable disease susceptibility.[3,4] The analyses of transcriptomes and CNVs in PC have been reported, and common results include a positive correlation between Transmembrane Protease, Serine 2 (TMPRSS2)-V-Ets avian erythroblastosis virus E26 oncogene homolog (ERG) fusion gene, gene amplifications (e.g., androgen receptor gene, mitogen-activated protein kinase kinase 7 [MAP3K7], and maternal embryonic leucine zipper kinase [MELK]),[5–7] predominant somatic gene mutations (e.g., phosphatase and tensin homolog [PTEN] and olfactomedin 4 gene [OLFM4]), and overexpression of differential display code 3 (DD3PCA3).[8]DD3PCA3 is a novel PC-specific gene, which has been renamed PCA3 to reflect its association with PC.[8] Furthermore, CNVs at 15q21.3 and 12q21.31 are related to the activator protein 1 (AP1) and α-1,3-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase C (MGAT4C) genes, and 8p loss and 8q gain have been found to be associated with PC risk through altered regulation of cell proliferation and migration of PC cells.[9,10] Nevertheless, the relationship between CNVs and gene expression in PC has not been fully elucidated.
Microarrays constructed with genomic clones have been widely used to detect CNVs in vitro.[11,12] Based on array-comparative genomic hybridization (aCGH), whole-transcript expression data, and microRNA expression data, Taylor et al[13] identified NCOA2 (nuclear receptor coactivator 2) as an oncogene in approximately 11% of tumors and revealed that androgen-driven TMPRSS2-ERG fusion was associated with a narrow deletion on 3p14 implicated FOXP1, RYBP, and SHQ1 as potential cooperative tumor suppressors. However, the differentially expressed genes (DEGs) between normal samples and PC samples have not been fully characterized, and numerous CNVs in PC have not been described. The present study focused on focal DNA CNVs and DEGs between PC samples and normal tissue samples to identify differences that could contribute to the progression of human PC.
In the present study, using aCGH and whole-transcript expression data deposited by Taylor et al,[13] DEGs and CNVs between PC tissue samples and normal tissue were determined, and coexpression interactions between DEGs were analyzed. Furthermore, functional analysis of DEGs that contained CNVs was performed. Our bioinformatic approaches may be used to analyze other PC datasets (e.g., whole transcriptome expression data, microRNA expression data, and aCGH) or even datasets of any type of cancer to identify novel CNV genes.
2. Material and methods
2.1. Microarray data
The GSE21032 dataset was downloaded from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/),[13] which contained GSE21034 (whole-transcript and exon-level expression data), GSE21035 (Agilent 244K aCGH data), and GSE21036 (microRNA expression data). PC tissue and cell-line microarray data were included, but only those of tissue samples in GSE21034 and GSE21035 were used in the study.
GSE21035 included 218 samples, which consisted of 181 primary PC samples and 37 metastatic PC samples; the associated platform was an Agilent-014693 Human Genome CGH Microarray 244A (GPL4091; Agilent Technologies, Palo Alto, CA). The preprocessed GSE21034 data, which were based on an Affymetrix Human Exon 1.0 ST Array platform (GPL5188; Affymetrix, Santa Clara, CA), consisted of 179 samples, including 29 normal tissue samples, 131 primary PC samples, and 19 metastatic PC samples. This study was conducted with approval from the Memorial Sloan-Kettering Cancer Center Institutional Review Board, and all patients provided informed consent.
2.2. Screening of DEGs
The preprocessed whole-transcript data underwent a log2 transformation. DEGs between the 368 tumor samples and the 29 normal tissue samples were identified using the linear models for microarray data package,[14] which has been previously used to identify DEGs based on microarray data, and the generated P value for each gene was adjusted using the Benjamini–Hochberg (BH) method.[15] Only genes meeting the selection criteria of |logFC (fold change)| ≥0.5 and adjusted P value <.05 were considered DEGs.
2.3. Preprocessing of aCGH microarray data
Raw aCGH data were preprocessed using the package cghMCR in R (version 1.34.0, http://www.bioconductor.org/packages/release/bioc/html/cghMCR.html).[16,17] The method “minimum” in the package was used for background correction, and this method meant that any intensity, which was zero or negative after background subtraction, was set to a value equal to half the minimum of the positively corrected intensities for that array. The approach of locally weighted scatterplot smoothing, a modern modeling method based on classical methods such as linear and nonlinear least squares regression,[18] was employed for data normalization.
2.4. Analysis of CNVs
The circular binary segmentation[19] algorithm was applied for segment analysis of the preprocessed aCGH data to translate noisy intensity measurements into regions of equal copy number. Subsequently, significant CNV regions were predicted using GISTIC 2.0 (genomic identification of significant targets in cancer, http://www.gistic.org/), an online tool to identify genes implicated in somatic copy-number alterations that regulate cancer growth.[20] All sequences were mapped to the reference human genome hg18 (NCBI build 36.1, http://genome.ucsc.edu), and only the sequences meeting the threshold for amplification of 0.3 or deletion of −0.3 were considered significant CNV regions.[21]
2.5. Integrated analysis of CNVs and DEGs
Correlations between DEGs were analyzed using the Pearson correlation coefficient.[22] The raw P value was adjusted by the BH method. Pairs that satisfied an adjusted P value <.05 and |correlation| ≥0.8 were deemed to be significantly correlated. The coexpression network of these DEGs was then visualized using Cytoscape (version 3.2.0, U.S. National Institute of General Medical Sciences, http://cytoscape.org/), an open source software for integrating biomolecular interaction networks.[23] In the network, a “node” represents a gene or protein, and a “line” represents an interaction between 2 nodes. The degree of each node equals the number of nodes that interact with it.
Correlations between the genes that had CNVs and DEGs were performed. Copy number is usually understood to positively correlate with gene expression. Thus, DEGs that had a positive correlation with copy number were chosen for further analysis. Coexpression interactions of CNV–DEGs were extracted from the DEG coexpression network to construct a CNV–DEG coexpression network, which was visualized using Cytoscape. Herein, each CNV–DEG and its connected genes were defined as a network module. With a P value <.05 as the cut-off criterion, a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of genes in the network modules was performed using the Database for Annotation, Visualization, and Integrated Discovery, https://david.ncifcrf.gov/,[24] and the network composed of pathways and CNV–DEGs was visualized using Cytoscape.
2.6. Search for drugs targeting CNV–DEGs
Potential drugs targeting CNV–DEGs were searched using the Drug–Gene Interaction database (DGIdb, http://dgidb.genome.wustl.edu/),[25] which contains several common drug–gene interaction databases, including DrugBank,[26] therapeutic target database,[27] and pharmacogenomics knowledge database.[28]
3. Results
3.1. Identification of DEGs
In total, 679 DEGs were screened from the 368 PC samples compared with the 29 normal samples. Among these, 182 genes were upregulated and 497 were downregulated in PC tumor cells (supplementary Tumor vs Normal, DEGs).
3.2. Analysis of CNV regions
In total, 48 amplified CNV regions and 45 deleted regions were obtained (Fig. 1A and B). The amplified regions were located on chromosomes 1 to 12, 14 to 17, and 22. The peak density at 8q13.2 to 8q24.3 was relatively higher, and the peak height at 1q21.3 was clearly larger compared with normal samples (Fig. 1A). Meanwhile, the deleted regions were located on chromosomes 1 to 18, 21, and 22. Compared with the normal tissue samples, the peak density at 5q11.2 to 5q21.3, 6q15 to 6q21, 8p23.1 to 8p21.3, 13q14.2 to 13q22.1, and 16q22.1 to 16q24.2 was relatively higher (Fig. 1B). All identified CNVs were located on euchromosomes, not idiochromosomes. Amplified regions contained 226 genes and deleted regions contained 372 genes (supplementary files-amplified and deleted regions).
Figure 1.
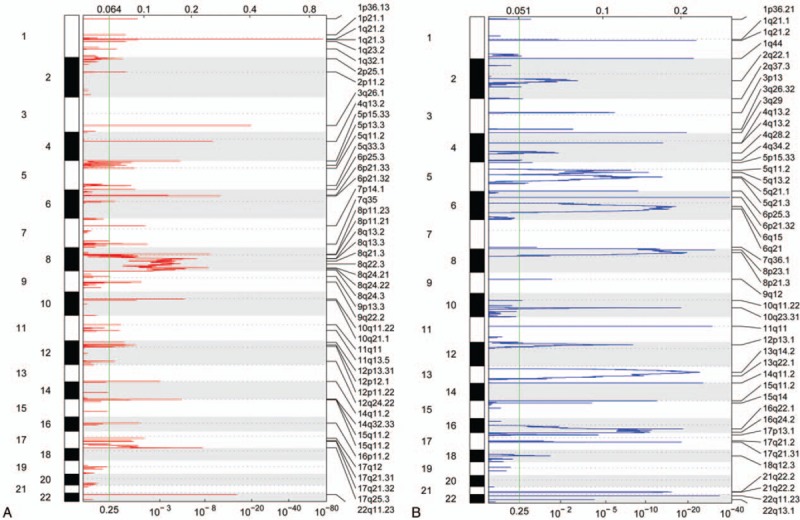
Amplified and deleted regions of copy-number variations (CNVs) on chromosomes. (A) Amplified regions of CNVs. (B) Deleted regions of CNVs. The numbers on the left vertical axis represent chromosomes, and the labels on the right represent varied regions of CNVs. The values on the top horizontal axis represent G-scores, and values on the horizontal axis at the bottom represent q values. The larger G-score and smaller q value indicate that the CNVs were more significant.
3.3. Correlations between DEGs and CNVs
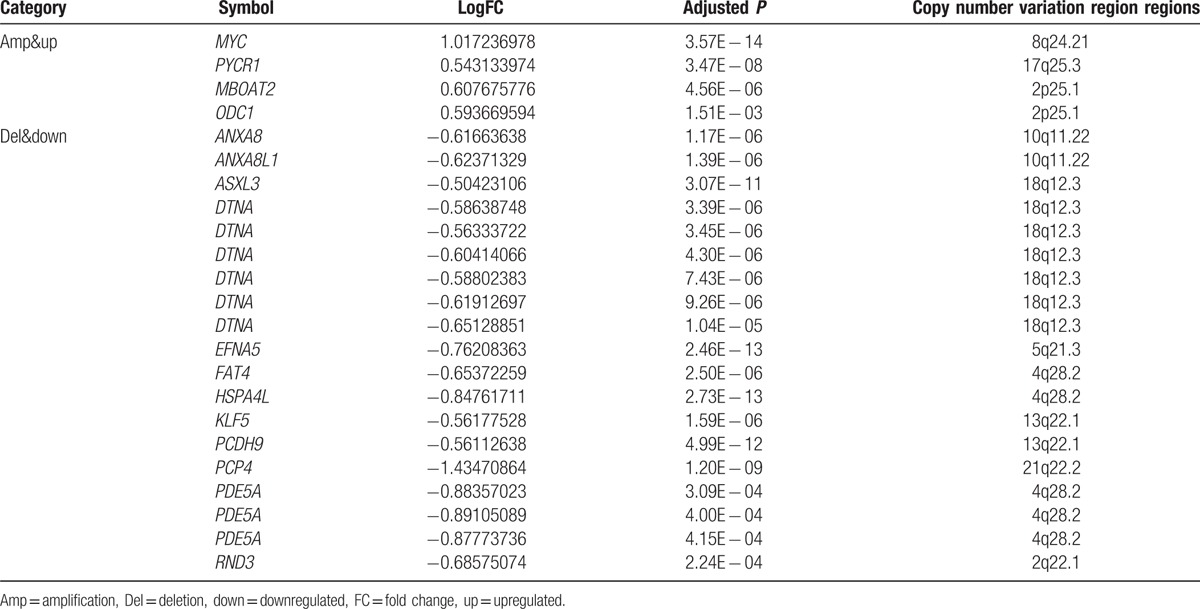
To investigate whether DEGs had CNVs, DEGs were compared with CNV genes. Notably, expression changes of most DEGs showed no positive correlation with CNVs. Only 16 DEGs had a positive correlation with CNVs (CNV–DEGs), including 4 upregulated ones that had amplified CNV regions and 12 downregulated ones that had deleted regions (Table 1).
Table 1.
DEGs mapped to CNV regions.
3.4. Analysis of CNV–DEG coexpression network and module functions
To reveal interactions between CNVs and DEGs, coexpression interactions were extracted from the DEG coexpression network to construct the CNV–DEG coexpression network. There were 181 interactions and 113 nodes in the CNV–DEG coexpression network (Fig. 2). Eight nodes were CNV–DEGs, including PCP4 (Purkinje cell protein 4), PDE5A (phosphodiesterase [PDE] 5A, cGMP-specific), FAT4 (FAT atypical cadherin 4), RND3 (Rho family GTPase 3), ASXL3 (additional sex combs-like 3, Drosophila), HSPA4L (heat shock 70 kDa protein 4-like), ANXA8L1 (annexin A8-like 1), and ANXA8 (annexin A8); all of these genes were downregulated. Among the 8 CNV–DEGs, PCP4, PDE5A, and FAT4 had a higher degree. PDE5A interacted with both PCP4 and FAT4. Here, each CNV–DEG and its interacting genes were considered as a network module.
Figure 2.
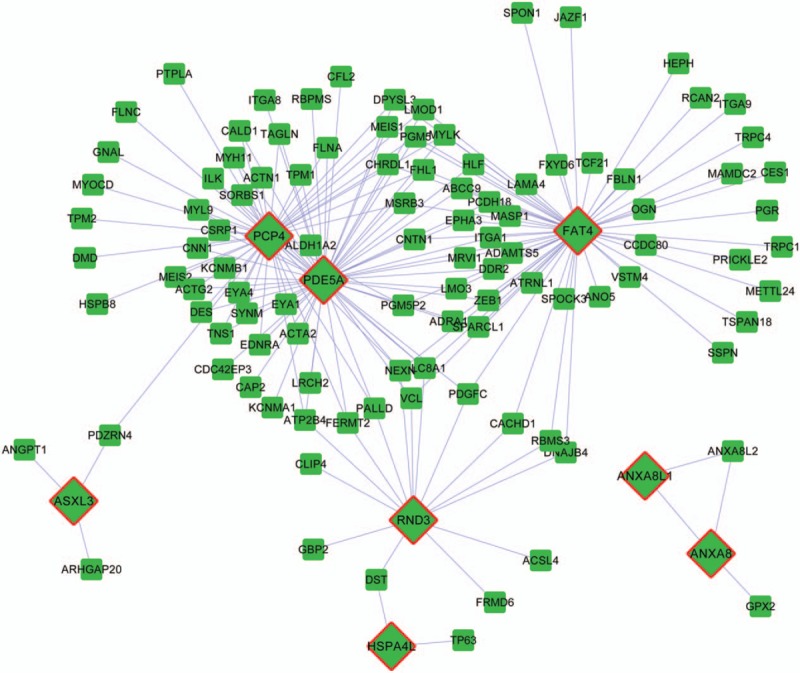
Coexpression network of differentially expressed genes (DEGs) with copy-number variations (CNVs) and DEGs without CNVs. Squares represent DEGs without copy-number variations. Rhombuses represent DEGs with copy-number variations. Lines represent the coexpression correlations between genes.
To further explore functions of genes in the network modules, a KEGG pathway enrichment analysis was conducted. Three modules, including FAT4, PDE5A, and PCP4, as well as their interacting genes, were significantly enriched in pathways (Fig. 3). Common pathways included arrhythmogenic right ventricular cardiomyopathy, focal adhesion, hypertrophic cardiomyopathy, dilated cardiomyopathy, and calcium signaling pathway.
Figure 3.
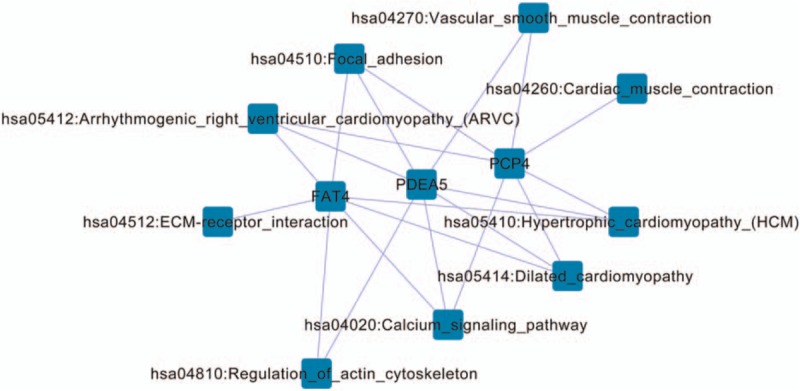
Network of differentially expressed genes with copy-number variations and their enriched pathways.
3.5. Drugs targeting CNV–DEGs
Drugs targeting the CNV–DEGs were searched using the DGIdb database. Five gene products of identified CNV–DEGs are drug targets, including MYC (v-myc avian myelocytomatosis viral oncogene homolog), PYCR1 (pyrroline-5-carboxylate reductase 1), ODC1 (ornithine decarboxylase 1), PDE5A, and RND3.
4. Discussion
In PC, deletions and increases in DNA copy-number contribute to alterations in the expression of tumor-related genes and are involved in PC pathogenesis.[29] In the present study, amplified CNV regions containing 226 genes and deleted regions containing 372 genes were identified through genomic analyses of PC samples. The peak density of amplified regions at 8q13.2 to 8q24.3 in PC samples was higher than that in normal samples, which was consistent with the results reported by Sun et al.[30] Compared with normal groups, the peak density of deleted regions at 5q11.2 to 5q21.3, 6q15 to 6q21, 8p23.1 to 8p21.3, 13q14.2 to 13q22.1, and 16q22.1 to 16q24.2 was higher in PC samples. In total, 182 upregulated and 497 downregulated genes were screened from PC samples compared with normal samples. Among the identified DEGs, 2 upregulated genes and 14 downregulated genes contained CNVs. In the CNV–DEG coexpression network, there were 8 CNV–DEGs, including FAT4, PDE5A, and PCP4, which had a higher degree and were significantly enriched in several pathways.
FAT4 encodes a gene family member belonging to the protocadherin family, and this protein is involved in the regulation of planar cell polarity.[31] Loss of Fat4 expression in tumors is related to human FAT4 promoter methylation.[32] To date, there is no evidence to demonstrate CNVs in FAT4 in PC. However, it has been demonstrated that loss of FAT4 expression is present in a large fraction of human breast tumor cell lines and primary tumor tissues, and Fat4 has been identified as a potential tumor suppressor gene in breast cancer.[32] Moreover, single-nucleotide polymorphisms in FAT4 have been discovered in spontaneous pulmonary adenoma,[33] and mutations in FAT4 have been detected in colorectal cancer.[34] Therefore, FAT4 could play a role in PC progression through deleted CNVs.
PDE5A encodes a cGMP-specific PDE, which belongs to the cyclic nucleotide PDE family. This PDE is correlated with the regulation of intracellular concentrations of cyclic nucleotides.[35] PDE5 is mainly observed in glandular structures of the prostate.[36] A previous study found that PDE5 inhibition reverses hypoxia-induced shedding of the immune stimulatory molecule MHC class I-related chain A and attenuates the growth of human prostate tumors.[37]PDE5A was found to be targeted by a set of drugs in this study. As reported in previous studies, PDE5 inhibitors can mediate smooth muscle tone in the prostate.[36,38] However, there is no evidence in previous studies to demonstrate CNVs in PDE5A in PC. In this study, PDE5A was coexpressed with FAT4 and PCP4. PCP4 encodes Purkinje cell protein 4, also known as PEP19.[39] The hemizygous deletion of PCP4 has been discovered in the PC cell line LuCap35,[40] and downregulation of PCP4 has also been observed in PC.[41] In this study, PCP4 was significantly enriched in the calmodulin signaling pathway, along with FAT4 and PDE5A. The association between PCP4 and calmodulin signaling has been previously reported.[42] Ca2+ is a ubiquitous intracellular messenger responsible for regulating multiple biological processes, including mitosis, cell death, gene transcription, contraction, and muscle relaxation.[43] Calmodulin kinase II (CaMKII), a major target of the Ca2+/calmodulin second messenger system, promotes PC cell survival.[44] Furthermore, Ca2+/calmodulin-dependent protein kinase kinase beta regulates PC cell growth via 5′-adenine monophosphate-activated protein kinase.[45] Collectively, PCP4, as well as FAT4 and PDE5A, could exert pivotal functions related to PC progression via deleted CNVs and the calmodulin signaling pathway.
In addition, as reported in the article of Taylor et al[13] who are the contributors of the GSE21032 dataset used in this study, NCOA2 is amplified in primary and metastatic PC, and androgen-driven TMPRSS2-ERG fusion is correlated with a deletion at 3p14, which is related to FOXP1 (forkhead box P1), RYBP (RING1 and YY1 binding protein), and SHQ1 (H/ACA ribonucleoprotein assembly factor). The amplifications of chromosome 8 (spanning MYC) and chromosome 11 (CCND1) were increased, while the amplifications of 17p13.1 (TP53), 12p13.1 (CDKN1B), and others were reduced, and SPINK1 messenger ribonucleic acid was found to be frequently overexpressed in PC.[46] These CNVs and genes were not identified in our study. Instead, we screened out the FAT4, PDE5A, and PCP4 genes that contained CNVs. We speculated that the different findings in our study compared with those in the Taylor et al study could be due to the different analytical methods used to evaluate CNVs. The analytical methods used in this study were applicable to identifying genes in somatic CNVs that regulate cancer growth and help identify genes targeted by CNVs that drive cancer growth. Conversely, Taylor methods were applicable to high-resolution genomic data, which are used to assess the extent and function of copy-number alterations in cancer. Moreover, in the Taylor et al[13] article and The Cancer Genome Atlas (TCGA) paper,[46] the most frequent molecular abnormalities were chromosomal arm-level copy-number alterations (gains and losses), while our study focused on focal DNA copy-number alterations, which enabled the discovery of 16 CNV–DEGs that could contribute to human PC progression. Most of the samples in the Taylor et al[13] study were derived from non-Hispanic white subjects in a clinical T1c stage with a lower Gleason score. However, the majority of samples in the TCGA paper were from Caucasian subjects in a pathological T2c/T3a stage with a higher Gleason score.[46] These differences could have contributed to the different results between this study and the TCGA paper.
Despite the aforementioned results, there were several limitations in this study. The predicted results should be confirmed by laboratory data. In our future studies, the CNVs and DEGs will be validated by multiple ligation-dependent probe amplification and real-time polymerase chain reaction analyses. Coexpression relationships between PDE5A and PCP4/FAT4 will be confirmed by pull-down assays.
In conclusion, amplified CNV regions containing 226 genes and deleted regions containing 372 genes were identified in PC tissue. The number of CNVs at 8q and 8p in PC samples was relatively higher than that in normal tissue samples. Furthermore, a total of 182 upregulated and 497 downregulated genes were identified between PC and normal tissues. Among the 16 CNV–DEGs, differences in FAT4 and PDE5A in PC were not previously reported. These 2 genes and PCP4 were coexpressed with multiple DEGs, and all of these were enriched in pathways, including the calmodulin signaling pathway. These CNV–DEGs could play important roles in the progression of human PC. The findings could lead to a greater understanding of PC pathogenesis.
Supplementary Material
Supplementary Material
Footnotes
Abbreviations: aCGH = array-comparative genomic hybridization, BH = Benjamini–Hochberg, CNV = copy-number variation, DEG = differentially expressed gene, FC = fold change, KEGG = Kyoto Encyclopedia of Genes and Genomes, PC = prostate cancer.
YH and XJ have contributed equally to the article.
The authors have no funding and conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29. [DOI] [PubMed] [Google Scholar]
- [2].Almal SH, Padh H. Implications of gene copy-number variation in health and diseases. J Hum Genet 2011;57:6–13. [DOI] [PubMed] [Google Scholar]
- [3].Perry GH, Tchinda J, McGrath SD, et al. Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci U S A 2006;103:8006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ledet EM, Hu X, Sartor O, et al. Characterization of germline copy number variation in high-risk African American families with prostate cancer. Prostate 2013;73:614–23. [DOI] [PubMed] [Google Scholar]
- [5].Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997;275:1943–7. [DOI] [PubMed] [Google Scholar]
- [6].Ross-Adams H, Lamb A, Dunning M, et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: a discovery and validation cohort study. EBioMedicine 2015;2:1133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li H, Chen Y, Liu W, et al. Olfactomedin 4 plays a tumor-suppressor role and is a novel candidate biomarker in the prostate cancer progression and independent of PSA. Cancer Res 2015;75(15 suppl):4344–14344. [Google Scholar]
- [8].Schalken JA, Hessels D, Verhaegh G. New targets for therapy in prostate cancer: differential display code 3 (DD3 PCA3), a highly prostate cancer-specific gene. Urology 2003;62(5 suppl 1):34–43. [DOI] [PubMed] [Google Scholar]
- [9].Demichelis F, Setlur SR, Banerjee S, et al. Identification of functionally active, low frequency copy number variants at 15q21.3 and 12q21.31 associated with prostate cancer risk. Proc Natl Acad Sci U S A 2012;109:6686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644–8. [DOI] [PubMed] [Google Scholar]
- [11].Pollack JR, Perou CM, Alizadeh AA, et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet 1999;23:41–6. [DOI] [PubMed] [Google Scholar]
- [12].Carter NP. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat Genet 2007;39:S16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gentleman R, Carey VJ, Huber W, et al. Bioinformatics and computational biology solutions using R and Bioconductor, vol. 746718470. New York: Springer; 2005. [Google Scholar]
- [15].Benjamini Y, Hochberg Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J Educ Behav Stat 2000;25:60–83. [Google Scholar]
- [16].R package version 1.24.0, Zhang J, Feng B, Zhang MJ. biocViews Microarray C. Package ‘cghMCR’. 2013. [Google Scholar]
- [17].R package version 1.24.0, Zhang J, Feng B. How to Use cghMCR. 2013. [Google Scholar]
- [18].Cleveland WS, Devlin SJ. Locally weighted regression: an approach to regression analysis by local fitting. J Am Stat Assoc 1988;83:596–610. [Google Scholar]
- [19].Olshen AB, Venkatraman E, Lucito R, et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004;5:557–72. [DOI] [PubMed] [Google Scholar]
- [20].Mermel CH, Schumacher SE, Hill B, et al. GISTIC2. 0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sia D, Hoshida Y, Villanueva A, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013;144:829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry 2010;77:733–42. [DOI] [PubMed] [Google Scholar]
- [23].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Da Wei Huang BTS, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2008;4:44–57. [DOI] [PubMed] [Google Scholar]
- [25].Griffith M, Griffith OL, Coffman AC, et al. DGIdb: mining the druggable genome. Nat Methods 2013;10:1209–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 2006;34(suppl 1):D668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhu F, Shi Z, Qin C, et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res 2011;40(Database issue):D1128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hewett M, Oliver DE, Rubin DL, et al. PharmGKB: the pharmacogenetics knowledge base. Nucleic Acids Res 2002;30:163–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hieronymus H, Schultz N, Gopalan A, et al. Copy number alteration burden predicts prostate cancer relapse. PNAS 2014;111:11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sun J, Liu W, Adams TS, et al. DNA copy number alterations in prostate cancers: a combined analysis of published CGH studies. Prostate 2007;67:692–700. [DOI] [PubMed] [Google Scholar]
- [31].Alders M, Al-Gazali L, Cordeiro I, et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome. Hum Genet 2014;133:1161–7. [DOI] [PubMed] [Google Scholar]
- [32].Qi C, Zhu YT, Hu L, et al. Identification of Fat4 as a candidate tumor suppressor gene in breast cancers. Int J Cancer 2009;124:793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Berndt A, Cario CL, Silva KA, et al. Identification of Fat4 and Tsc22d1 as novel candidate genes for spontaneous pulmonary adenomas. Cancer Res 2011;71:5779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yu J, Wu WK, Li X, et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut 2015;64:636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Loughney K, Hill TR, Florio VA, et al. Isolation and characterization of cDNAs encoding PDE5A, a human cGMP-binding, cGMP-specific 3′,5′-cyclic nucleotide phosphodiesterase. Gene 1998;216:139–47. [DOI] [PubMed] [Google Scholar]
- [36].Ückert S, Oelke M, Stief CG, et al. Immunohistochemical distribution of cAMP- and cGMP-phosphodiesterase (PDE) isoenzymes in the human prostate. Eur Urol 2006;49:740–5. [DOI] [PubMed] [Google Scholar]
- [37].Hamilton TK, Hu N, Kolomitro K, et al. Potential therapeutic applications of phosphodiesterase inhibition in prostate cancer. World J Urol 2013;31:325–30. [DOI] [PubMed] [Google Scholar]
- [38].Kang KK, Kim JM, Yu JY, et al. Effects of phosphodiesterase type 5 inhibitor on the contractility of prostate tissues and urethral pressure responses in a rat model of benign prostate hyperplasia. Int J Urol 2007;14:946–51. [DOI] [PubMed] [Google Scholar]
- [39].Cabin DE, Gardiner K, Reeves RH. Molecular genetic characterization and comparative mapping of the human PCP4 gene. Somat Cell Mol Genet 1996;22:167–75. [DOI] [PubMed] [Google Scholar]
- [40].Demichelis F, Setlur SR, Beroukhim R, et al. Distinct genomic aberrations associated with ERG rearranged prostate cancer. Genes Chromosomes Cancer 2009;48:366–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ross AE, Marchionni L, Vuica-Ross M, et al. Gene expression pathways of high grade localized prostate cancer. Prostate 2011;71:1568–77. [DOI] [PubMed] [Google Scholar]
- [42].Mouton-Liger F, Thomas S, Rattenbach R, et al. PCP4 (PEP19) overexpression induces premature neuronal differentiation associated with Ca(2+)/calmodulin-dependent kinase II-delta activation in mouse models of Down syndrome. J Comp Neurol 2011;519:2779–802. [DOI] [PubMed] [Google Scholar]
- [43].Zayzafoon M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem 2006;97:56–70. [DOI] [PubMed] [Google Scholar]
- [44].Rokhlin O, Taghiyev AF, Bayer KU, et al. Calcium/calmodulin-dependent kinase II plays an important role in prostate cancer cell survival. Cancer Biol Ther 2007;6:732–42. [DOI] [PubMed] [Google Scholar]
- [45].Frigo DE, Shi Y, Han JJ, et al. CaMKK2-AMPK signaling facilitates androgen-mediated prostate cancer cell metabolism. Cancer Res 2014;74(19 suppl):2450–12450. [Google Scholar]
- [46].Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.