

Abstract
The identity and developmental potential of a human cell is specified by its epigenome that is largely defined by patterns of chromatin modifications including histone acetylation. Here we report high-resolution genome-wide mapping of diacetylation of histone H3 at Lys 9 and Lys 14 in resting and activated human T cells by genome-wide mapping technique (GMAT). Our data show that high levels of the H3 acetylation are detected in gene-rich regions. The chromatin accessibility and gene expression of a genetic domain is correlated with hyperacetylation of promoters and other regulatory elements but not with generally elevated acetylation of the entire domain. Islands of acetylation are identified in the intergenic and transcribed regions. The locations of the 46,813 acetylation islands identified in this study are significantly correlated with conserved noncoding sequences (CNSs) and many of them are colocalized with known regulatory elements in T cells. TCR signaling induces 4045 new acetylation loci that may mediate the global chromatin remodeling and gene activation. We propose that the acetylation islands are epigenetic marks that allow prediction of functional regulatory elements.
Keywords: Chromatin, epigenome, transcription
Comparative genomics studies reveal the existence of long conserved noncoding sequences (CNSs) that are thought to play regulatory roles in the expression of the mammalian genomes (Loots et al. 2000; Dermitzakis et al. 2002). However, even though all of the nucleated mammalian cells in one species have the same genome, every cell type has a different epigenome that is mainly defined by post-translational modifications of chromatin and expresses a subset of genes by using only a subset of the regulatory elements. Additional screening strategies are required to identify genome-wide functional regulatory elements.
Histone modifications regulate the accessibility of chromatin and gene activity (for reviews, see Kornberg and Lorch 1999; Strahl and Allis 2000; Turner 2000; Wu and Grunstein 2000; Jenuwein and Allis 2001; Berger 2002; Kurdistani and Grunstein 2003). Many enzymes that regulate histone acetylation and deacetylation are transcriptional cofactors (Kuo and Allis 1998). Histone acetylation is required for gene activation and cell growth (Megee et al. 1990; Durrin et al. 1991). The acetylation of histone H3 at Lys 9 and Lys 14 has been shown to be important for activation of the human interferon-β gene upon viral infection (Agalioti et al. 2002). Genome-wide analyses in yeast and Drosophila melanogaster have correlated acetylation patterns with the transcriptional activity (Kurdistani et al. 2004; Schubeler et al. 2004). While the mechanisms by which histone acetylation regulates chromatin structure and transcription are not fully understood, it is believed that the acetylation status of histones provides complex recognition surfaces or a code for factors that regulate chromatin structure and gene activity (Strahl and Allis 2000). For example, the recruitment of Sir3 to form heterochromatin in yeast requires that H4-K16 is deacetylated (Hecht et al. 1996). The bromodomain, which is found in many chromatin-modifying enzymes, binds specifically to acetylated lysine residues (Dhalluin et al. 1999; Jacobson et al. 2000). Therefore, the acetylated histones may recruit and/or stabilize transcription factors and/or chromatin remodeling enzymes to their target sites in chromatin (Hassan et al. 2001; Agalioti et al. 2002). There is also evidence that histone acetylation regulates gene expression by facilitating transcriptional elongation (Belotserkovskaya et al. 2003; Saunders et al. 2003).
Much in vivo evidence about the function of histone acetylation in regulation of gene expression comes from genetics studies in lower eukaryotic organisms such as yeast (Kurdistani and Grunstein 2003), which has high levels of histone acetylation in the whole genome (Roh et al. 2004). Higher eukaryotic genomes differ from yeast in having much lower acetylation levels and more heterochromatic regions. How does histone acetylation control the chromatin accessibility of active domains in higher eukaryotes? Previous studies suggest that the whole domain becomes generally hyperacetylated when it is activated (Litt et al. 2001). However, analysis of human cells suggests that histone acetylation is localized to the active promoter region (Liang et al. 2004). To clarify the question, an unbiased genome-wide analysis of histone acetylation at high resolution is necessary. Several methods have been reported for the high-throughput evaluation of the chromatin immunoprecipitation (ChIP) DNA sequences (Horak and Snyder 2002; Liang et al. 2004; Roh et al. 2004). One such method, ChIP-on-Chip has been successfully used for the analysis of lower eukaryotes such as Saccharomyces cerevisiae because of the availability of DNA microarrays that contain most of yeast genomic DNA (Bernstein et al. 2002; Kurdistani et al. 2004). However, the currently available human DNA microarrays cover only a small portion of the entire human genome. To detect the true global histone modifications at high resolution, we have recently developed a genome-wide mapping technique (GMAT) that combines ChIP and the serial analysis of gene expression technique (SAGE) (Roh et al. 2003, 2004). GMAT does not depend on preselected DNA sequences. It identifies a tag of 21-bp sequence from each ChIP DNA fragment, which contains sufficient information to be mapped precisely in the human genome. The detection frequency of a tag in the GMAT library reflects directly the level of modification at the locus. Therefore, the level of histone modifications can be compared between different genetic loci.
Using GMAT, we determined the genome-wide distribution of K9/K14 diacetylated histone H3 in resting and activated human T cells. Our data argue that chromatin accessibility of a genetic locus is not caused by generally elevated acetylation; instead, the openness is correlated with hyperacetylation of a limited number of regulatory elements including promoters, locus control regions, and enhancers. We propose a concept “acetylation islands” and show that they are functional regulatory elements in the human genome.
Results
Sequencing the GMAT library
To generate GMAT libraries for sequencing analysis, resting CD3+ T cells were isolated from human blood using negative selections. The cells were either used immediately or activated with anti-CD3 and anti-CD28 for 24 h before ChIP with anti-K9/K14 diacetylated histone H3 antibodies. The GMAT libraries were prepared from the ChIP DNA as described previously (Roh et al. 2003, 2004). As shown in Table 1A, the human genome contains a total of 24,577,210 tags, which represent 18,559,414 kinds of tags (or different 21-bp sequences). By sequencing 32,544 GMAT clones, we obtained a total of 803,439 tags, which represent 414,655 kinds of tags. Therefore, 2.2% of the tag kinds in the human genome were detected in the GMAT library. To determine whether we have covered the genome sufficiently, we quantified the level of H3-K9/K14 acetylation in resting T cells using ChIP as described (Litt et al. 2001). The results showed that ∼1.2% of nucleosomes in total chromatin is associated with K9/K14 acetylated H3 (data not shown). Since the repetitive sequences, which make up ∼40% of the human genome, were associated with hypoacetylated histone H3 (Table 1B), we estimate that ∼2% of the unique sequences in the genome are associated with the acetylated histone. This is consistent with the data that 2.1% of unique tags were detected in the GMAT library (Table 1B). We conclude that our sequencing has covered most of the acetylated regions.
Table 1.
GMAT analysis of the human genome
| (A) Tag distribution in the human genome
|
|
(C) The detection frequency of unique tags varies widely
|
||||
|---|---|---|---|---|---|---|
| Detected in GMAT library
|
||||||
| Calculated in the genome | Resting T cell | Activated T cell | Detection frequency | Number of tags | Percentage in library | |
| Total tags | 24,577,210 | 803,439 | 761,624 | 1 | 272,477 | 40.7% |
| Tag kinds | 18,559,414 | 414,655 | 425,762 | 2 | 81,334 | 12.1% |
| Unique tags | 17,688,797 | 670,073 | 626,692 | 3 | 49,404 | 7.4% |
| (72%) | (83%) | (82%) | 4 | 38,864 | 5.8% | |
| Repetitive tags | 6,890,413 | 133,366 | 134,932 | 5 | 31,675 | 4.7% |
| (2-91,831) | (28%) | (17%) | (18%) | 6-10 | 99,963 | 14.9% |
| 11-65 | 96,356 | 14.4% | ||||
| (B) Repetitive sequences are associated with lower levels of histone H3 acetylation in resting T cells
|
(D) Most tags are truly positive tags
|
|
|
||||
|---|---|---|---|---|---|---|---|
| Repeat number in genome | Calculated tag kinds | Detected tag kinds | Percentage of detection | Distance to the nearest tag (kb) | Percentage of detected tags | Number of tested tags | Positive (%) |
| 1 | 17,686,797 | 365,250 | 2.1% | Multiple-copy tags | |||
| 2 | 517,461 | 3497 | 0.7% | na | 59.3 | 15 (15) | 100.0 |
| 3 | 133,541 | 517 | 0.4% | Single-copy tags | |||
| 4 | 64,570 | 169 | 0.3% | 0-5 | 32.5 | 42 (57) | 73.7 |
| 5-100 | 151,854 | 182 | 0.1% | ||||
| 101-91,831 | 5191 | 0 | 0 | >5 | 7.9 | 9 (25) | 36.0 |
(A) The 21-bp tag distribution in the human genome and in GMAT libraries. The calculated column shows the calculated number of the 21-bp tags derived from NlaIII cleavage of human genome sequence downloaded from http://genome.ucsc.edu/downloads.html#human. The detected column indicates the number of the 21-bp tags detected in the GMAT library from the resting and activated T cells. Total tags represent the total number of the 21-bp tags existing in the human genome or detected in the libraries. Unique tags indicate the number of the 21-bp tags whose sequence is found only once in the human genome. Repetitive tags indicate the number of the 21-bp tags whose sequence is found two or more times in the human genome. Tag kinds represent the number of the 21-bp tags including both the unique tags and repetitive tags, all of which are counted once even if they appear more than once in the human genome.
(B) Repetitive sequences are associated with lower levels of the H3 acetylation. Repeat number in genome indicates the number of times a 21-bp tag appears in the human genome. Repeat number “1” indicates a unique sequence. Repeat number “2” indicates a sequence that is found two times in the genome. Calculated tag kinds indicate the total number of tag kinds with the same copy number in the human genome. Detected tag kinds indicate the total number of tag kinds with the same copy number detected in the GMAT library. The percentage of detection (%) was calculated by dividing the detected tag kinds in the GMAT library by the calculated tag kinds in the human genome.
(C) The tag detection frequency varies widely. Only the tags from unique sequences in the resting T cell GMAT library were analyzed. Detection frequency indicates the number of times a tag was detected in the GMAT library. Single-copy tags are those with a detection frequency of 1.
(D) Most tags are truly positive tags. Single-copy or multiple-copy tags were randomly selected for analysis using PCR on ChIP DNA. A 200-300-bp fragment of DNA containing the selected tag was amplified from the ChIP DNA in the presence of α-32P(dCTP) for PhosphorImager analysis. The band intensity was normalized to the band intensities of two control regions from the β-globin domain. A value of three-fold or more is considered positive. Distance to the nearest tag indicates how far a neighboring detected tag is located. The percentage of detected tags indicates the percentage of the categorized tags in the GMAT library. Number of tested tags indicates the number of positive tags and total number of tested tags (in parentheses), respectively, in the PCR analysis.
Table 1B shows that 2.1% of the unique sequences (1 repeat) in the genome were detected in the GMAT library, while the percentage of detection decreased as the repetitiveness (repeat number) increased, indicating that higher repetitive sequences are associated with lower levels of the histone H3 acetylation. Since the repetitive sequences were not associated with significant levels of the H3 acetylation and the repetitive tags that are associated with acetylated H3 could not be mapped precisely in the human genome, we considered only the unique tags for further analysis.
The GMAT tags are truly associated with H3 acetylation
We detected 670,073 tags (Table 1A) derived from unique sequences. The detection frequency of the tags in the GMAT library ranges from 1 to 65 times (Table 1C). Of these tags, 40.7% were detected only once (single-copy tag) and 59.3% were detected multiple times in the library (Table 1C). Since the specific antibody against the diacetylated (K9/K14) histone H3 pulled down 100-fold more DNA than nonspecific rabbit IgGs in the ChIP experiments, the majority of the ChIP DNA and therefore the tags detected in the GMAT library were associated with the acetylated histone. However, we could not completely avoid of low levels of nonspecific pull-down of DNA in the procedure. One nonspecific fragment may generate a maximum of two tags irrespective of the fragment size. To confirm that the majority of the tags detected in the library truly represent the levels of histone modification and are not derived from contamination during ChIP experiments, we randomly picked ∼100 tags and analyzed them by quantitative PCR using specific primers. Our analysis of 15 loci with a two-copy or higher-copy GMAT tag indicates that all of them were enriched in the ChIP DNA. The enrichment was >10-fold over the control β-globin sequence (data not shown). The results were reproduced with four different ChIP samples obtained from chromatin prepared from four independent cultures. Therefore, we can confidently conclude that all of the tags that were detected two or more times are truly positive tags. Approximately 59.3% of all the unique tags detected in the GMAT library belonged to this category (Table 1C).
For the single-copy tags, 74% of them were enriched more than threefold over the control sequences when the distance to the nearest detected tag was within 5 kb. The results were confirmed in four different experiments using different ChIP DNA samples (Table 2). When the nearest detected tag was found at a distance of >5 kb, only 36% of the tags were enriched more than threefold (Table 1D). However, only 7.9% of the tags fell into this category. Therefore, we conclude that all of the multiple-copy tags and most of the single-copy tags detected in the GMAT library were associated with the acetylated histone H3.
Table 2.
Most single-copy tags are reproducibly enriched in independent experiments
| Enrichment fold
|
||||
|---|---|---|---|---|
| No. | 1 | 2 | 3 | 4 |
| 1 | 11.03 | 27.64 | 0.320 | 11.60 |
| 2 | 13.0 | 15.34 | 10.62 | 13.3 |
| 3 | 3.12 | 6.17 | 3.88 | 0.09 |
| 4 | 64 | 12.2 | 3.67 | 17.2 |
| 5 | 21.8 | 6.97 | 4.30 | 3.63 |
| 6 | 21.7 | 10.3 | 3.95 | 13.5 |
| 7 | 19.5 | 5.14 | 0.36 | 4.96 |
| 8 | 11.84 | 2.48 | 3.58 | 4.03 |
| 9 | 10.73 | 13.75 | 11.47 | 8.83 |
| 10 | 49.3 | 17.42 | 7.62 | 11.52 |
| 11 | NA | 10.7 | 5.51 | 0.58 |
| 12 | NA | 0.17 | 1.67 | 19 |
| 13 | 7.17 | 8.84 | 4.91 | 1.67 |
| 14 | 5.9 | 10.86 | 4.73 | 3.81 |
| 15 | 8.46 | 4.43 | 5.12 | 2.27 |
| 16 | 70.3 | 30.1 | 18.9 | 8.18 |
| 17 | 17.5 | 24.3 | 12.8 | 3.43 |
| 18 | 14.6 | 4.28 | 7.1 | 0.09 |
| 19 | 6.09 | 1.66 | 2.25 | 0.39 |
| 20 | 15.35 | 2.69 | 2.04 | 2.05 |
Single-copy tags were randomly selected for analysis using PCR on ChIP DNA. A 200-300-bp fragment of DNA containing the selected tag was amplified from the input and four ChIP DNA samples in the presence of α-32P(dCTP). The products were resolved by PAGE and quantified by PhosphorImager analysis. A pair of primers specific for the β-globin domain (chromosome 11: 5,250,645-5,250,913) was used as a control in column 1 and a pair of primers amplifying a fragment on chromosome 4 (57,858,032-57,858,304) was used as a control for columns 2, 3, and 4. The fold of enrichment is calculated by normalizing the tag signals from each sample to the control tag signals. Then, the normalized signals of each tag site from the ChIP samples were normalized to the input signals to obtain the fold of enrichment. The values that are less than threefold are shown in bold. (NA) not available.
To reconfirm that the GMAT tags were derived from the acetylated but not from nonacetylated regions by contamination, we examined the β-globin domain that is not expressed and exists as condensed chromatin in resting T cells. The 120-kb region contained 1016 predicted tags. Only three tags from this region were detected in the GMAT library. We cannot conclusively determine if these three tags represent true low levels of acetylation or if they represent background. However, these data indicate that the background level was very low and most of the tags detected in the library were derived from the specific association with the acetylated H3.
Promoter regions are highly acetylated in the human genome
To determine if the H3 acetylation is biased toward any functional regions in the human genome, we arbitrarily defined the genome as consisting of three parts: 2-kb promoter regions including 1 kb upstream and 1 kb downstream of the transcription initiation site, gene body regions including introns and exons, and intergenic regions (Fig. 1A). The 2-kb promoter region makes up 0.8% of the genome (Fig. 1B). The gene body and intergenic regions make up 27.3% and 71.9% of the genome, respectively. Interestingly, 23.5% of the tags detected in the GMAT library were derived from the promoter region, and 30.0% and 46.4% were from the gene body and intergenic regions, respectively. These results indicate that the distribution of the acetylated H3 is biased toward the promoter region in the genome, which is consistent with the results obtained from the analysis of 57 human genes (Liang et al. 2004).
Figure 1.
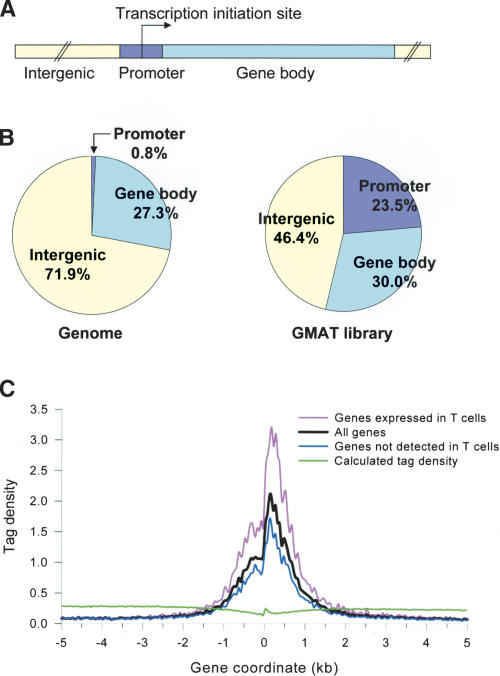
High levels of the H3 acetylation are detected in the promoter regions. (A) A schematic showing the human genome arbitrarily separated into three parts: 2-kb promoter regions, intergenic regions, and gene body regions. (B, left) Calculated percentage of each region in the genome. (Right) The percentage of the tags detected in the GMAT library from each region. (C) A 10-kb region of 21,355 genes was aligned relative to their transcription initiation sites (X-axis). The Y-axis shows the tag density that was obtained by normalizing the total number of detected tags with the number of expected NlaIII sites in a 50-bp window. The green line represents the calculated value, provided the tags were detected randomly. The black line represents the tag density of all of the 21,355 genes. The pink line represents the tag density of the 8000 highly active genes. The blue line represents the rest of the genes that are either silent or expressed at lower levels.
We aligned 21,355 annotated genes relative to their transcription initiation sites and plotted the tag density, which is derived by normalizing the detected number of tags in the GMAT library to the number of expected NlaIII sites in a 50-bp window across a 10-kb region (Fig. 1C). The green line represents the calculated value, which shows that the calculated tags are distributed evenly across the whole region. Interestingly, a significant peak of tags was detected in the GMAT library in the 2-kb promoter region, as revealed by the black line for all the genes. A chi-squared statistical test indicates the enrichment of the H3 acetylation in the promoter region is significant (p < 0.0001). To rule out the possibility that the enrichment of the GMAT tags in the promoter region was caused by the exclusion of repetitive tags from the data, we analyzed the distribution of the repetitive tags detected in the GMAT library. As shown in Supplementary Figure 1B, the repetitive tags were distributed in the genome with a slight enrichment in the promoter region. Therefore, removal of these tags from the analysis should not generate a bias in favor of detecting the unique tags in the promoter region. The pink line represents the acetylation level of the 8000 highly expressed genes (van der Kuyl et al. 2002) and the blue line indicates the acetylation level of 14,000 genes whose expression was not detected. The data showed that more active promoters have higher levels of histone H3 acetylation than less active promoters. A similar observation has been made from the genome-wide analysis of Drosophila cells (Schubeler et al. 2004). It is noteworthy that the 1-kb region 3′ of the transcription initiation site has the highest acetylation level. Since the genome-wide expression data in literature did not allow us to distinguish true silent genes from the genes expressed at lower levels, we examined the acetylation of several genes including β-globin, BAF53b (Olave et al. 2002), NEUROD1 (Naya et al. 1995), and PITX2 (Semina et al. 1996), which are not expressed in T cells and are true silent genes. The analysis indicates that no acetylation was detected in their promoter regions (data not shown).
CpG islands are highly acetylated
Many human promoters contain CpG islands, which are important transcription-controlling elements and are unmethylated under normal circumstances (Bird et al. 1985; Gardiner-Garden and Frommer 1987). The mechanisms preventing CpG islands from being methylated remain elusive. We decided to examine the acetylation status of CpG islands. The human genome project has identified 27,058 CpG islands in the human genome (Karolchik et al. 2003). As suggested by previous studies (Larsen et al. 1992), we found that the CpG islands were highly concentrated in a 1-kb region surrounding the transcription initiation site of human genes (Supplementary Fig. 2), which is consistent with the overrepresentation of the CpG dinucleotides in human promoter sequences (Marino-Ramirez et al. 2004). Our data indicate that 64.2% of the CpG islands were acetylated (Fig. 2A), which is much higher than the 1.2% average acetylation level in the whole genome. In active genes and the genes whose expression was not detected (from -10 kb to the gene end), 78.2% and 67.8% of CpG islands, respectively, were acetylated. Interestingly, ∼33% of CpG islands were located in the intergenic regions and were also acetylated at a level of 49.1%. A chi-squared test indicates that the association of H3 acetylation with CpG islands is significant (p = 0.001). To study the relationship between CpG islands and associated chromatin acetylation status, we analyzed acetylation boundaries of 4973 CpG islands that have a size ranging from 1 to 2 kb (Fig. 2B). The analysis revealed that the acetylation was quite evenly distributed within the CpG islands. However, sequences 200 bp away from CpG islands showed a significant decrease in the acetylation. Furthermore, some of the GMAT tags neighboring CpG islands may have been brought down by the acetylation within the CpG islands because of the heterogeneous size of the chromatin fragments used for ChIP experiments. The acetylation level decreased rapidly as the distance to CpG islands increased. It reached background level when the distance was ∼1 kb. These data suggest that the H3 hyperacetylation may serve as a mechanism to prevent the CpG islands from being methylated.
Figure 2.
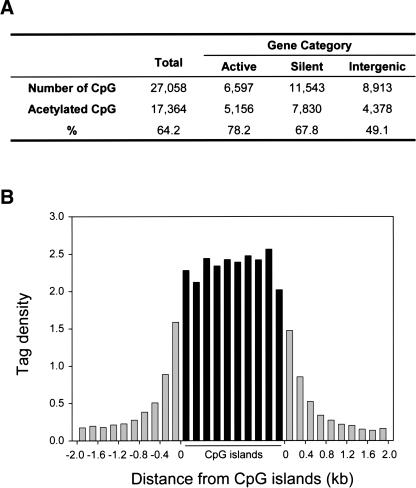
CpG islands are highly acetylated. (A) CpG islands are highly acetylated. “Active” and “silent” indicate the CpG islands identified between the -10- and +10-kb regions in active genes and the genes whose expression was not detected, respectively. “Intergenic” indicates the CpG islands identified in the intergenic region beyond -10 kb upstream of the transcription initiation site. (B) Sharp boundaries of histone H3 acetylation are detected surrounding CpG islands. The detected GMAT tags within the 4973 CpG islands that have a size ranging from 1 to 2 kb were counted in a window of 10% of the CpG island length and normalized to the number of NlaIII sites in the window to get the tag density. The GMAT tags outside of the CpG islands were counted in a 0.2-kb window and normalized to the number of NlaIII sites in the window to get the tag density. Each bar represents 10% of the sequence length within the CpG islands and 0.2 kb outside of the CpG islands. Black bars indicate the tag density within the CpG islands and the gray bars indicate the tag density outside of the CpG islands.
The highest H3 acetylation is detected in gene-rich regions
Based on the sequence information, we mapped the GMAT tags onto the 24 chromosomes in the human genome (Supplementary Fig. 3). The X-axis in the upper panel indicates the chromosomal coordinates of the tags and the Y-axis indicates the detection frequency or number of times detected for a tag in the GMAT library. The data show that the acetylated H3 was not evenly distributed on chromosomes. Instead, there appear to be clusters of the tags and large chromosomal regions with low levels of tags, indicating that there are chromatin domains that are highly or poorly acetylated. For example, chromosome 12 contains several large gaps due to low or no histone acetylation (Supplementary Fig. 4). Analysis of gene distribution suggests that the highly acetylated regions on the chromosomes were correlated with generich regions (r = 0.80), as shown in Supplementary Figure 4. This observation indicates that gene-rich regions tend to exist in an open chromatin structure, since the H3 acetylation is generally associated with active chromatin. This is consistent with the observation that open chromatin fibers correlate with regions of highest gene density, obtained from sucrose sedimentation analysis (Gilbert et al. 2004).
An interactive high-resolution acetylation map linked to the University of California, Santa Cruz (UCSC), human genome database can be found at http://dir.nhlbi.nih.gov/labs/lmi/zhao/epigenome/G&D2005.htm.
Active chromatin domains are not uniformly hyperacetylated
Supplementary Figure 4 indicates that active chromatin domains correlate with high levels of H3 acetylation. Is an open chromatin domain generally highly acetylated? To answer this question, we examined the high-resolution map of the H3 acetylation of the STAT2 locus on chromosome 12 (Fig. 3A). The 160-kb region (chromosome 12: 54,920,000-55,080,000) harbors five expressed genes as indicated by the arrows in Figure 3A. Interestingly, there were no sustained high levels of the H3 acetylation throughout the entire domain. Instead, high peaks of acetylation were detected within promoter regions. To determine whether this is a general phenomenon, we examined the high-resolution maps of more active loci. BAF53A (Zhao et al. 1998) and Sp1 are constitutively expressed in almost every cell type, and STAT5B (Liu et al. 1995) are constitutively expressed in T cells. As shown in Figure 3B, C, and D, all of these loci have high levels of acetylation in their promoter regions. However, no general hyperacetylation was detected in their transcribed regions, except for a few isolated clusters of tags in some genes. The same is also true for the CD4 locus (Fig. 4A; data not shown). These results indicate that active domains are not uniformly hyperacetylated but correlated with hyperacetylation of critical regulatory elements, suggesting that localized acetylation may contribute to maintain the openness of the entire domain.
Figure 3.
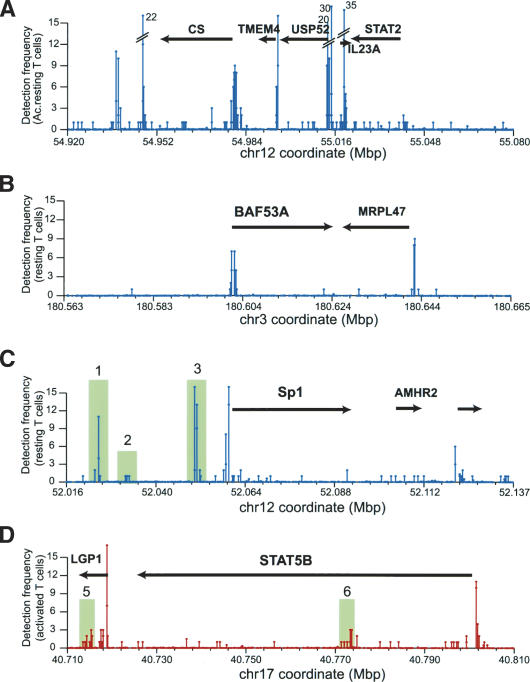
Active domains are not uniformly acetylated. (A) A gene-rich region on chromosome 12. The transcribed regions and gene orientations are indicated by the arrows. The acetylation levels of the locus in resting T cells are shown. The Y-axis indicates the detection frequency and the X-axis indicates the chromosome coordinate. The numbers above the broken lines indicate the detection frequency. (B) BAF53A locus. (C) The Sp1 locus. Acetylation islands are highlighted and numbered. (D) The STAT5B locus.
Figure 4.
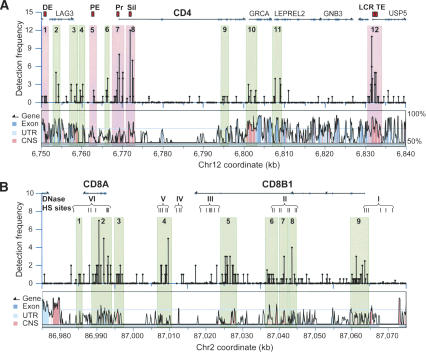
Colocalization of acetylation islands with known regulatory elements. (A) CD4 locus. The upper panel shows the acetylation data, above which gene positions and known functional regulatory elements are indicated. The lower panel shows the VISTA human and mouse sequence comparison. (DE) Distal enhancer; (PE) proximal enhancer; (Pr) promoter; (Sil) silencer; (LCR) locus control region; (TE) thymocyte enhancer. The acetylation islands colocalized with known regulatory elements are highlighted in pink. The acetylation islands with no known functions are highlighted in green. (B) CD8 locus. The acetylation data and VISTA sequence analysis are shown as in A. The positions of the six clusters of DNase hypersensitive sites (HS) are indicated below the genes.
Identification of acetylation islands
Examination of the high-resolution maps revealed that there were clusters of two or more acetylation tags along the chromatin fiber, which we have named “acetylation islands” (highlighted and numbered in Figs. 3, 4). The acetylation islands were detected both in the intergenic and transcribed regions. We identified a total of 21,481 and 25,332 acetylation islands in the intergenic and transcribed regions, respectively. Our discovery of the acetylation islands suggests that besides promoters, other regulatory elements may also be marked by histone acetylation.
Most acetylation islands colocalize with CNSs
Comparative genomics studies have identified ∼240,000 CNSs in the human and mouse genomes (for review, see Hardison 2000), which are believed to be regulatory elements in the mammalian genomes. Because of the general role that histone acetylation plays in the regulation of transcription and chromatin structure, we investigated whether there is a correlation between the acetylation islands and CNSs. As shown in Figure 4, most of the acetylation islands are associated with CNSs that are revealed by the VISTA analysis (http://pipeline.lbl.gov/cgi-bin/gateway2) at the lower part of the figure.
Comparative analysis of human chromosome 21 with syntenic regions of the mouse genome has revealed 2262 CNSs in the intergenic regions (Dermitzakis et al. 2002). Comparison between the CNSs and H3 acetylation indicates that there are 187 acetylated CNS, which accounts for 8.3% of CNSs. Genome-wide analysis of the acetylation status of CNSs revealed that 15.7% of the 241,222 CNSs identified in the VISTA database (http://pipeline.lbl.gov/cgi-bin/gateway2) were associated with the acetylated H3. Chi-squared statistical tests suggest that the correlation between CNSs and the H3 acetylation is significant (p = 0.001).
Colocalization of acetylation islands with known regulatory elements in T cells
The significant colocalization of acetylation islands with CNSs suggests that the acetylation islands may represent functional regulatory elements in T cells. Therefore, we examined whether they are correlated with known regulatory elements in T cells.
CD4 is a critical T-cell coreceptor that assists antibody production. DNase hypersensitive sites (HS) mapping combined with transgenic studies has identified several regulatory elements that collectively mediate the specific expression of the CD4 gene (for reviews, see Ellmeier et al. 1999; Siu 2002). As expected, strong acetylation was detected in the promoter (Fig. 4A, highlight 7). Interestingly, the proximal enhancer, which is conserved between human and mouse and is located 6.5 kb upstream of its transcription initiation site in human, is colocalized with an acetylation island (Fig. 4A, highlight 5). The distal enhancer located upstream of the LAG3 gene was also acetylated (Fig. 4A, highlight 1). The CD4 gene is also regulated by a locus control region (LCR) and a thymocyte enhancer (TE) that are located 30 kb downstream of the CD4 gene with several intervening genes. A significant acetylation island was detected in the LCR/TE region (Fig. 4A, highlight 12). Besides the known regulatory elements, we detected several other significant acetylation islands within the locus (Fig. 4A, highlights 2-4, 6, and 9-11). It will be interesting to determine if these also represent important regulatory elements for the CD4 gene.
The CD8α and CD8β coreceptors play critical roles in mediating cell killing. The minimal functional elements of the human CD8 genes, which render their specific expression, are contained in a 95-kb region (Kieffer et al. 1997). Six clusters of DNase HSs are present in the locus (Kieffer et al. 2002), as summarized in Figure 4B. Interestingly, most of these HS sites coincided well with significant acetylation islands. However, HS cluster III was not colocalized with any significant acetylation islands. Instead, a significant acetylation island (Fig. 4A, highlight 5) was detected ∼5 kb away from HS III. Since the expression level of the CD8 genes contained in the 95-kb region varies depending on integration site, it does not contain a LCR, suggesting a LCR may be located outside of the region. Examining the acetylation map revealed a highly acetylated region and a TCR signaling-induced island, 100 and 40 kb downstream of the CD8α gene, respectively (data not shown). It will be interesting to test whether these regions function as LCRs for the CD8 locus.
Examination of the IL2Rα (Lin and Leonard 1997) and BCL3 (Ohno et al. 1990) loci (Supplementary Fig. 5) also revealed that known transcription and chromatin regulatory elements are well correlated with the acetylation islands, suggesting that the acetylation islands may represent a functional regulatory network of gene expression programs in T cells.
Genome-wide change of histone H3 acetylation induced by TCR signaling
TCR signaling induces thousands of genes required for cellular differentiation and immunologic functions, accompanied by massive chromatin decondensation (Crabtree 1989). To identify cis regulatory elements that initiate the global chromatin remodeling upon T-cell activation, we analyzed the histone H3 acetylation of T cells after 24 h of TCR signaling (Table 1A). We determined the changes in acetylation levels between the resting and activated T cells by comparing the average tag density, which is obtained by dividing the total number of detected tags by the number of NlaIII sites in the region, within a 3-kb window. Changes of three times or more were plotted along chromosome coordinates (Fig. 5; Supplementary Fig. 6). The analysis revealed that increased acetylation was detected at 4045 loci and decreased acetylation was detected at 4178 loci (Supplementary Table 1), indicating that the TCR signaling induced a genome-wide acetylation change.
Figure 5.
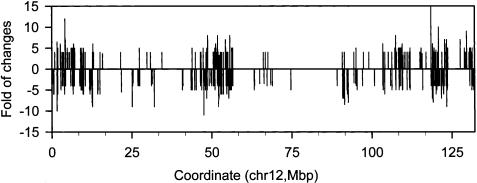
Genome-wide changes of acetylation induced by TCR signaling. The average tag density was derived by normalizing the total number of detected tags to the number of NlaIII site in a 3-kb window. The average tag densities from resting and activated T cells were compared directly to obtain the fold change. Changes of threefold or more between activated and resting T cells were plotted along the chromosome coordinates. The data for chromosome 12 are shown. The other chromosomes are shown in Supplementary Figure 6.
To determine whether the acetylation islands induced by TCR signaling are involved in T-cell activation, we specifically examined the Th2 cytokine locus that harbors three TCR signaling-induced cytokine genes, IL5, IL13, and IL4 (Abbas et al. 1996). Our analysis of resting T cells indicates that the IL13 and IL4 promoters were acetylated even in silent state (Fig. 6A, upper panel). Interestingly, two acetylation islands (Fig. 6A, highlights 1 and 2) were induced downstream of the IL13 gene by TCR signaling (Fig. 6A, middle panel), even though there were no significant changes of histone H3 acetylation in the promoter regions. Acetylation Island 2 colocalized with a conserved sequence that is known to be required for the coordinated expression of the cytokine genes (Loots et al. 2000). These data indicate that the TCR signaling-induced Acetylation Islands 1 and 2 may have an important function in initiating the chromatin opening and regulating the expression of the cytokine genes.
Figure 6.
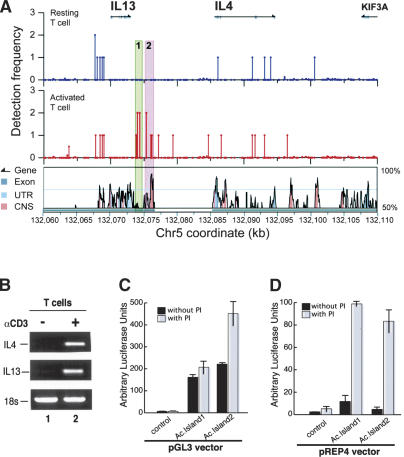
TCR signaling-induced acetylation islands activate transcription in a chromatin-dependent manner. (A) IL13/IL4 locus. The upper and middle panels show the acetylation data from resting and activated T cells, respectively. The lower panel shows the VISTA human and mouse sequence comparison. The gene locations are indicated above the acetylation data. Also indicated is the CNS-1 identified by Loots et al. (2000). (B) TCR signaling induces the expression of the IL13 and IL4 genes in T cells. Total RNA was isolated from resting T cells treated with anti-CD3 and anti-CD28 for 24 h. The expression of the IL13 and IL4 genes were analyzed by RT-PCR with specific primers. The 18s RNA was used as a control. (C) Acetylation Islands 1 and 2 have constitutive enhancer activity in a nonchromatin vector. The 1.5-kb DNA containing Acetylation Islands 1 and 2 in A or a control sequence from the neighboring unacetylated region (chromosome 5: 132,080,058-132,081,677) were inserted upstream of a minimal GM-CSF1 promoter in the pGL3 luciferase reporter vector. The constructs were transfected into Jurkat cells for 48 h, followed by stimulation with 1 μg/mL ionomycin and 10 ng/mL PMA for 15 h. The luciferase activity was analyzed with the dual luciferase system from Promega as described (Liu et al. 2001). (PI) PMA and Ionomycin. (D) The enhancer activity of Acetylation Islands 1 and 2 is dependent on TCR signaling in a chromatin-forming vector. The 1.5-kb DNA containing Acetylation Islands 1 and 2 in A or the control sequence from the neighboring unacetylated region were inserted upstream of a minimal GM-CSF1 promoter in the pREP4 luciferase reporter vector. The constructs were similarly transfected into Jurkat cells and analyzed as above.
The TCR-induced acetylation islands act as transcription enhancers
Accompanying the induction of Acetylation Islands 1 and 2, the IL13 and IL4 genes were induced by the TCR signaling (Fig. 6B, cf. lanes 1 and 2). To demonstrate whether the TCR signaling-induced acetylation islands are functional regulatory elements, we tested the activities of Acetylation Islands 1 and 2 in a luciferase reporter assay. As shown in Figure 6C, both Acetylation Islands 1 and 2 activated the GM-CSF1 promoter in pGL3 vector in Jurkat cells even without stimulation, while the control insert from the unacetylated neighboring region (chromosome 5: 132,080,058-132,081,677) did not have any detectible activity. Upon stimulation with ionomycin and PMA, which mimicked TCR signaling, Acetylation Island 2 further activated the promoter twofold. Next, we cloned these sequences into the episomal pREP4 vector, which replicates and forms a regular chromatin structure in cells. Both Acetylation Islands 1 and 2 only modestly increased the promoter activity without stimulation (Fig. 6D). Interestingly, they dramatically activated the promoter upon ionomycin and PMA stimulation (Fig. 6D). These results suggest that TCR signaling induced an activity that can overcome the inhibitory effect of chromatin, possibly by modification of the chromatin structure, which is consistent with the observation that the acetylation of Acetylation Islands 1 and 2 was induced by TCR signaling in T cells. It is noteworthy that even though Acetylation Island 1 was not conserved between human and mouse, it had strong activity in activating transcription, indicating that comparative genomics studies may miss important regulatory elements. These results indicate that Acetylation Islands 1 and 2 activate transcription in a chromatin-dependent manner and the TCR signaling-induced acetylation islands are indeed epigenetic marks for functional regulatory elements.
Discussion
The human genome contains ∼35,000 genes (Baltimore 2001; Lander et al. 2001). At least 10,000-15,000 genes are expressed in any tissues, which occupy ∼15% of the human genome. We found that only 1.2% of the chromatin is associated with the K9/K14 diacetylated histone H3 in human T cells. Therefore, this level of acetylation does not allow all of the active chromatin domains to be uniformly highly acetylated, as suggested by the data from studies in yeast. Indeed, we found that most of intergenic and transcribed regions of active chromatin do not have generally elevated levels of histone acetylation. Our analysis of the 21,355 annotated human genes indicates that the promoter region is highly acetylated, as suggested by a previous smaller scale study (Liang et al. 2004). A higher acetylation level in the promoter region is correlated with more active genes, consistent with a previous study in Drosophila (Schubeler et al. 2004). Furthermore, we find that other functional regulatory elements such as enhancers, LCRs, and insulators are also marked by the histone acetylation. Our data argue for a model in which the chromatin accessibility and expression of a gene are controlled by histone modifications of a number of regulatory elements, which act to actively limit the spreading of neighboring heterochromatin and allow binding of transcription factors and assembly of the transcription machinery.
Comparison of the human and mouse genomic sequences reveals the existence of 240,000 conserved noncoding sequences that may function to direct the expression programs of the genomes (Hardison 2000). However, even though all of the nucleated human cells have the same genome, each cell type has a different “epigenome.” Each cell type expresses a different set of genes, which requires a different set of regulatory elements. We show that the acetylation islands identified in this study are well correlated with known DNase HSs and functional regulatory elements, suggesting that the 46,813 acetylation islands may represent the functional regulatory network required for the expression programs in T cells. Even though many of the acetylation islands are not colocalized with any significant CNSs, they may have important regulatory functions, as revealed by the transcription enhancer activity of Acetylation Island 1 of the IL13 locus. It would be interesting to test whether the nonconserved syntenic locus in mouse is also acetylated and required for the coordinated expression of the cytokine locus. In summary, our data in this study provide valuable information for further identification of cis regulatory elements and for elucidation of regulatory mechanisms of transcription of almost every gene expressed in T cells.
Materials and methods
Isolation and stimulation of human T cell
Human resting T cells were purified sequentially using the lymphocyte separation medium (Mediatech) and Pan T-cell isolation kit II (Miltenyi Biotech). The final purity of T cells was >98%. T cells were activated with 1 μg/mL of anti-CD3 and anti-CD28 monoclonal antibodies (BD Pharmingen) for 24 h.
ChIP and preparation of the GMAT libraries
ChIP using anti-diacetylated K9/K14 histone H3 antibody (Upstate) and generation of GMAT library were performed as described (Roh et al. 2004). The method was refined based on our initial report (Roh et al. 2003).
Verification of H3 acetylation at randomly selected tag sites
An approximately 250-bp region encompassing a single or multicopy acetylation tag site was amplified. One region of β-globin domain (chromosome 11: 5,250,645-5,250,913) and one region on chromosome 4 (57,858,032-57,858,304) were used as controls. The PCR products from five DNA samples (input and four ChIP samples) were labeled by incorporating α-32P(dCTP) in the reaction and were separated on a denaturing polyacrylamide gel for quantification using the PhosphorImager (Molecular Dynamics). To calculate the fold of enrichment for each tag site, the signals from each sample were first normalized to the control signals. Then, the normalized signals of each tag site from the ChIP samples were normalized to the input signals to obtain the fold of enrichment.
Data analysis
A theoretical reference library of 21-bp sequence tags was derived from the UCSC July 2003 human sequence (hg16) using SAGE2000 version 4.5 software (Johns Hopkins Oncology Center). The GMAT library was generated by extracting 21-bp tags from raw sequencing data files using the SAGE2000 software. All other calculations and analyses were performed using in-house PERL programs. Detection frequency was determined by normalizing tag count to the genomic copy number. Tag density was calculated by dividing the detection frequency by the number of expected NlaIII sites in a 50-bp window.
An acetylation island was defined by the following criteria: (1) It is composed of tags from more than two adjacent NlaIII sites; (2) the detection frequency of all the tags is more than or equal to one; and (3) neighboring acetylation islands are separated by >500 bp.
The information on CpG islands and nonredundant RefSeq genes was obtained from the UCSC Genome Browser (Karolchik et al. 2003). The list of highly transcribed active genes in CD4+ T cell was downloaded from the NCBI Genome Expression Omnibus (GEO) database. Gene names were synchronized using UniGene ClusterID and LocusLink RefSeq ID.
For comparative analysis of human and mouse genomes, graphic alignments were adopted from the VISTA browser (Couronne et al. 2003) that shows gene information and sequences of >50% homology with October 2003 mouse genome assembly.
To compare the acetylation level between resting and activated T cells, the average detection frequency was calculated by normalizing the total number of tags to the number of NlaIII sites in the 3-kb window. The fold of change was calculated by dividing the average detection frequency of activated T cell by that of resting T cell.
Acknowledgments
We thank Drs. Tian Chi, David Clark, Warren J. Leonard, Niveen Mulholland, and Carl Wu for critical reading of the manuscript, and members of Leonard and Zhao laboratories for discussion. We thank Dr. Zheng Wu for help with the purification of T cells. This work was supported by intramural grants to NHLBI, NIH.
Supplemental material is available at http://www.genesdev.org.
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1272505.
References
- Abbas A.K., Murphy, K.M., and Sher, A. 1996. Functional diversity of helper T lymphocytes. Nature 383: 787-793. [DOI] [PubMed] [Google Scholar]
- Agalioti T., Chen, G., and Thanos, D. 2002. Deciphering the transcriptional histone acetylation code for a human gene. Cell 111: 381-392. [DOI] [PubMed] [Google Scholar]
- Baltimore D. 2001. Our genome unveiled. Nature 409: 814-816. [DOI] [PubMed] [Google Scholar]
- Belotserkovskaya R., Oh, S., Bondarenko, V.A., Orphanides, G., Studitsky, V.M., and Reinberg, D. 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301: 1090-1093. [DOI] [PubMed] [Google Scholar]
- Berger S.L. 2002. Histone modifications in transcriptional regulation. Curr. Opin. Gen. Dev. 12: 142-148. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Humphrey, E.L., Erlich, R.L., Schneider, R., Bouman, P., Liu, J.S., Kouzarides, T., and Schreiber, S.L. 2002. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. 99: 8695-8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A., Taggart, M., Frommer, M., Miller, O.J., and Macleod, D. 1985. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 40: 91-99. [DOI] [PubMed] [Google Scholar]
- Couronne O., Poliakov, A., Bray, N., Ishkhanov, T., Ryaboy, D., Rubin, E., Pachter, L., and Dubchak, I. 2003. Strategies and tools for whole-genome alignments. Genome Res. 13: 73-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G.R. 1989. Contingent genetic regulatory events in T lymphocyte activation. Science 243: 355-361. [DOI] [PubMed] [Google Scholar]
- Dermitzakis E.T., Reymond, A., Lyle, R., Scamuffa, N., Ucla, C., Deutsch, S., Stevenson, B.J., Flegel, V., Bucher, P., Jongeneel, C.V., et al. 2002. Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature 420: 578-582. [DOI] [PubMed] [Google Scholar]
- Dhalluin C., Carlson, J.E., Zeng, L., He, C., Aggarwal, A.K., and Zhou, M.M. 1999. Structure and ligand of a histone acetyltransferase bromodomain. Nature 399: 491-496. [DOI] [PubMed] [Google Scholar]
- Durrin L.K., Mann, R.K., Kayne, P.S., and Grunstein, M. 1991. Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell 65: 1023-1031. [DOI] [PubMed] [Google Scholar]
- Ellmeier W., Sawada, S., and Littman, D.R. 1999. The regulation of CD4 and CD8 coreceptor gene expression during T cell development. Annu. Rev. Immunol. 17: 523-554. [DOI] [PubMed] [Google Scholar]
- Gardiner-Garden M. and Frommer, M. 1987. CpG islands in vertebrate genomes. J. Mol. Biol. 196: 261-282. [DOI] [PubMed] [Google Scholar]
- Gilbert N., Boyle, S., Fiegler, H., Woodfine, K., Carter, N.P., and Bickmore, W.A. 2004. Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell 118: 555-566. [DOI] [PubMed] [Google Scholar]
- Hardison R.C. 2000. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 16: 369-372. [DOI] [PubMed] [Google Scholar]
- Hassan A.H., Neely, K.E., and Workman, J.L. 2001. Histone acetyltransferase complexes stabilize swi/snf binding to promoter nucleosomes. Cell 104: 817-827. [DOI] [PubMed] [Google Scholar]
- Hecht A., Strahl-Bolsinger, S., and Grunstein, M. 1996. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature 383: 92-96. [DOI] [PubMed] [Google Scholar]
- Horak C.E. and Snyder, M. 2002. ChIP-chip: A genomic approach for identifying transcription factor binding sites. Methods Enzymol. 350: 469-483. [DOI] [PubMed] [Google Scholar]
- Jacobson R.H., Ladurner, A.G., King, D.S., and Tjian, R. 2000. Structure and function of a human TAFII250 double bromodomain module. Science 288: 1422-1425. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis, C.D. 2001. Translating the histone code. Science 293: 1074-1080. [DOI] [PubMed] [Google Scholar]
- Karolchik D., Baertsch, R., Diekhans, M., Furey, T.S., Hinrichs, A., Lu, Y.T., Roskin, K.M., Schwartz, M., Sugnet, C.W., Thomas, D.J., et al. 2003. The UCSC Genome Browser Database. Nucleic Acids Res. 31: 51-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer L.J., Yan, L., Hanke, J.H., and Kavathas, P.B. 1997. Appropriate developmental expression of human CD8 β in transgenic mice. J. Immunol. 159: 4907-4912. [PubMed] [Google Scholar]
- Kieffer L.J., Greally, J.M., Landres, I., Nag, S., Nakajima, Y., Kohwi-Shigematsu, T., and Kavathas, P.B. 2002. Identification of a candidate regulatory region in the human CD8 gene complex by colocalization of DNase I hypersensitive sites and matrix attachment regions which bind SATB1 and GATA-3. J. Immunol. 168: 3915-3922. [DOI] [PubMed] [Google Scholar]
- Kornberg R.D. and Lorch, Y. 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285-294. [DOI] [PubMed] [Google Scholar]
- Kuo M.H. and Allis, C.D. 1998. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20: 615-626. [DOI] [PubMed] [Google Scholar]
- Kurdistani S.K. and Grunstein, M. 2003. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 4: 276-284. [DOI] [PubMed] [Google Scholar]
- Kurdistani S.K., Tavazoie, S., and Grunstein, M. 2004. Mapping global histone acetylation patterns to gene expression. Cell 117: 721-733. [DOI] [PubMed] [Google Scholar]
- Lander E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K, Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Larsen F., Gundersen, G., Lopez, R., and Prydz, H. 1992. CpG islands as gene markers in the human genome. Genomics 13: 1095-1107. [DOI] [PubMed] [Google Scholar]
- Liang G., Lin, J.C., Wei, V., Yoo, C., Cheng, J.C., Nguyen, C.T., Weisenberger, D.J., Egger, G., Takai, D., Gonzales, F.A., et al. 2004. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. 101: 7357-7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.X. and Leonard, W.J. 1997. Signaling from the IL-2 receptor to the nucleus. Cytokine Growth Factor Rev. 8: 313-332. [DOI] [PubMed] [Google Scholar]
- Litt M.D., Simpson, M., Recillas-Targa, F., Prioleau, M.N., and Felsenfeld, G. 2001. Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J. 20: 2224-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Robinson, G.W., Gouilleux, F., Groner, B., and Hennighausen, L. 1995. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc. Natl. Acad. Sci. 92: 8831-8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R., Liu, H., Chen, X., Kirby, M., Brown, P.O., and Zhao, K. 2001. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell 106: 309-318. [DOI] [PubMed] [Google Scholar]
- Loots G.G., Locksley, R.M., Blankespoor, C.M., Wang, Z.E., Miller, W., Rubin, E.M., and Frazer, K.A. 2000. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288: 136-140. [DOI] [PubMed] [Google Scholar]
- Marino-Ramirez L., Spouge, J.L., Kanga, G.C., and Landsman, D. 2004. Statistical analysis of over-represented words in human promoter sequences. Nucleic Acids Res. 32: 949-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megee P.C., Morgan, B.A., Mittman, B.A., and Smith, M.M. 1990. Genetic analysis of histone H4: Essential role of lysines subject to reversible acetylation. Science 247: 841-845. [DOI] [PubMed] [Google Scholar]
- Naya F.J., Stellrecht, C.M., and Tsai, M.J. 1995. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes & Dev. 9: 1009-1019. [DOI] [PubMed] [Google Scholar]
- Ohno H., Takimoto, G., and McKeithan, T.W. 1990. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell 60: 991-997. [DOI] [PubMed] [Google Scholar]
- Olave I., Wang, W., Xue, Y., Kuo, A., and Crabtree, G.R. 2002. Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes & Dev. 16: 2509-2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh T.Y., Ngau, W.C., Cui, K., Landsman, D., and Zhao, K. 2003. Genome-wide mapping of histone modifications. In Chromatin structure and function (22nd Summer Symposium in Molecular Biology, July 30-August 2, 2003). p. 103. Pennsylvania State University, University Park, PA.
- ____. 2004. High-resolution genome-wide mapping of histone modifications. Nat. Biotechnol. 22: 1013-1016. [DOI] [PubMed] [Google Scholar]
- Saunders A., Werner, J., Andrulis, E.D., Nakayama, T., Hirose, S., Reinberg, D., and Lis, J.T. 2003. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301: 1094-1096. [DOI] [PubMed] [Google Scholar]
- Schubeler D., MacAlpine, D.M., Scalzo, D., Wirbelauer, C., Kooperberg, C., van Leeuwen, F., Gottschling, D.E., O'Neill, L.P., Turner, B.M., Delrow, J., et al. 2004. The histone modification pattern of active genes revealed through genomewide chromatin analysis of a higher eukaryote. Genes & Dev. 18: 1263-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semina E.V., Reiter, R., Leysens, N.J., Alward, W.L., Small, K.W., Datson, N.A., Siegel-Bartelt, J., Bierke-Nelson, D., Bitoun, P., Zabel, B.U., et al. 1996. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 14: 392-399. [DOI] [PubMed] [Google Scholar]
- Siu G. 2002. Controlling CD4 gene expression during T cell lineage commitment. Semin. Immunol. 14: 441-451. [DOI] [PubMed] [Google Scholar]
- Strahl B.D. and Allis, C.D. 2000. The language of covalent histone modifications. Nature 403: 41-45. [DOI] [PubMed] [Google Scholar]
- Turner B.M. 2000. Histone acetylation and an epigenetic code. Bioessays 22: 836-845. [DOI] [PubMed] [Google Scholar]
- van der Kuyl A.C., van den Burg, R., Zorgdrager, F., Dekker, J.T., Maas, J., van Noesel, C.J., Goudsmit, J., and Cornelissen, M. 2002. Primary effect of chemotherapy on the transcription profile of AIDS-related Kaposi's sarcoma. BMC Cancer 2: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. and Grunstein, M. 2000. 25 years after the nucleosome model: Chromatin modifications. Trends Biochem. Sci. 25: 619-623. [DOI] [PubMed] [Google Scholar]
- Zhao K., Wang, W., Rando, O.J., Xue, Y., Swiderek, K., Kuo, A., and Crabtree, G.R. 1998. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell 95: 625-636. [DOI] [PubMed] [Google Scholar]