

Abstract
Background
Myelofibrosis (MF) is the most aggressive Philadelphia-negative chronic myeloproliferative neoplasm (MPN) with high morbidity and mortality due to thrombo-hemorrhagic complications and leukemic transformation. MF is characterized by profound alterations of megakaryocytopoiesis, with consequent abnormalities in platelet number and function. We recently showed that the overexpression of the oncoprotein PKCepsilon plays a key role in the aberrant differentiation of MF megakaryocyte clone and that its levels correlate with disease burden. Moreover, our group previously demonstrated that PKCepsilon is over-expressed in platelets from patients with acute myocardial infarction (MI) and accounts for their increased reactivity. On these bases, we investigated here the activation state and PKCepsilon expression of MF platelets, testing potential correlations with thrombotic risk and disease aggressiveness.
Methods
Platelets were isolated from peripheral blood samples of MF patients and healthy donors (HDs). Patients were stratified according to the IPSS/DIPSS risk category and history of cardiovascular events. Platelet activation was assessed by flow cytometry. PKCepsilon mRNA and protein levels were determined by real time-PCR and western blot.
Results
MF platelets circulate in an activated status and display significantly higher levels of PKCepsilon compared to HDs. In MF patients, PKCepsilon platelet levels were associated with high-risk disease as well as with a positive history of major cardiovascular events.
Conclusions
PKCepsilon is configuring as the common denominator of neoplastic transformation and thrombus formation in MF. Overall, our data pinpoint PKCepsilon as a potential novel biomarker of disease aggressiveness and thrombotic risk in this hematologic neoplasm.
Keywords: Myelofibrosis (MF), PKCepsilon, thrombotic risk
Introduction
Myelofibrosis (MF), either arising as de novo disease (PMF) or evolving from a previous polycythemia vera (PV) or essential thrombocythemia (ET) (named post PV/ET MF) is a chronic myeloproliferative neoplasm (MPN) characterized by clonal expansion of hematopoietic progenitors. Together with ET and PV, MF belongs to the so called “classic” Philadelphia-negative MPN and it is characterized by abnormal differentiation of the megakaryocytic (MK) clone, leading to aberrant platelet production and cytokine release in the marrow environment, which eventually drive reactive fibroblast proliferation and fibrosis (1).
MF is the most aggressive Philadelphia-negative MPN, with high morbidity and mortality due to thrombo-hemorrhagic complications and leukemic transformation. The pathogenesis of thrombosis in MPN is complex and based on the interplay of both clinical and biological disease-related factors. Abnormalities of blood cells arising from the clonal proliferation of hematopoietic stem cells involve quantitative and qualitative modifications, which characterize the switch of these cells from a resting to a procoagulant phenotype (2). It is well known that platelets play a pivotal role in thrombus formation and, in case of MPN, they tend to show increased expression of surface activation markers, enhanced aggregation and release of activation products, resulting in a pro-thrombotic phenotype (2).
Platelet activation, and subsequent thrombus formation, is a complex event regulated by several molecular pathways (3-5). Interestingly, proteins belonging to protein kinase C family (PKCs), mediate several platelet functions and show a dual controlling role in thrombus formation, balancing the pro-aggregatory and procoagulant properties of thrombi (6). Among PKC isoforms, we have previously demonstrated that PKCepsilon is over-expressed in platelets from myocardial infarction (MI) patients compared to subjects with stable coronary artery disease and healthy controls. We also showed that PKCepsilon-expressing platelets display higher levels of surface activation markers and enhanced adhesion to sub-endothelial collagen, thus suggesting that higher levels of this kinase account for platelets’ increased reactivity (7).
PKCepsilon has been extensively investigated for its role in cell proliferation and apoptosis, being recognized as an oncoprotein promoting tumorigenesis, cancer survival and invasiveness (8). Our publications pinpoint to PKCepsilon as relevant modulator of hematopoietic progenitors’ differentiation pathways, since its over-expression has been linked to a significant impairment of MK differentiation up to a complete block of maturation, as described in acute myeloid leukemia blasts (9-13). More in details, we recently demonstrated that PKCepsilon plays a crucial role in PMF aberrant megakaryocytopoiesis. Indeed, we found that PKCepsilon is over-expressed in MF MK malignant clone and its levels correlate with disease burden. Moreover, in vitro pharmacological inhibition of PKCepsilon restores a proper megakaryo- and thrombopoiesis (14).
Collectively, data from our lab suggest that PKCepsilon may be the common denominator of the neoplastic and pro-thrombotic phenotype underlying MF.
Based on this hypothesis, herein we evaluated PKCepsilon in MF platelets and its potential correlation with patients’ clinical features.
Methods
Study population
This study was performed according to the Declaration of Helsinki and the protocol approved by the Ethical Committee of Parma University Hospital. After written informed consent, ~15 mL of peripheral blood from 19 MF patients (both primary and post-PV/ET MF) and 15 healthy donors (HD) were collected in 3.8% sodium citrate tubes. The diagnosis was established according to the World Health Organization (WHO) criteria (15). Only treatment-naïve patients were recruited. Patient data were retrospectively collected from clinical charts. Prognostic risk category was assessed according to the International Prognostic Scoring System (IPSS) (16) for newly diagnosed patients or according to the dynamic IPSS (DIPSS) (17) for patients with ongoing disease at the time of sample collection.
Platelet activation assay
To test platelet activation, aliquots of whole blood samples were stained with anti-CD62p, anti-CD154, anti-PAC1 monoclonal antibody (all from BD Pharmingen™, BD Biosciences, Franklin Lakes, New Jersey, USA) and analyzed by flow cytometry, as described elsewhere (18). Briefly, 5 µL of whole blood was diluted 1:100 in PBS and incubated with mAb anti CD62p-FITC or mAb anti CD154-PE or mAb anti PAC1-FITC and after 20 min of incubation at RT, samples were fixed by 400 mL of 2% buffered paraformaldehyde. Analysis was performed by an Epics XL flow cytometer (Beckman Coulter, Fullerton, CA, USA) and the Expo ADC software (Beckman Coulter).
Platelet purification
Platelet were purified from whole blood sample as we previously described (7). Blood samples were centrifuged at 160 g for 20 min at RT, to obtain platelet rich plasma (PRP). Platelets were then purified by negative separation using magnetic beads coated with anti-CD45 antibodies (Dynabeads, Invitrogen, Carlsbad, CA, USA), to deplete nucleated cells. Purified platelets were tested for purity by anti-CD41 staining and flow cytometry analysis (only samples where CD41+ cells, 98% were used), and the pelleted for RNA and protein extraction.
RNA extraction and real time PCR
Expression of PKCepsilon mRNA was analyzed as previously described (7). Briefly, purified platelets were treated with an appropriate amount of TRIzolTM (Invitrogen) for cell lysis and then processed for RNA extraction, following manufacturer’s protocol. RNA pellets were washed and resuspended in DEPC-treated water for quantification by spectrophotometer. Equal quantities of RNA for each sample were retro-transcribed with Malone Murine Leukemia Virus Reverse Transcriptase (Promega, Madison WI, USA) according to manufacturer’ instructions. Two mL of 1:1 cDNA dilution was used to perform real-time PCR with GoTaqH qPCR master mix (Promega) and 200 nM of each primer in Applied Biosystems StepOne real-time machine (Applied Biosystems, Carlsbad, CA, USA). Each reaction was performed in triplicate and mean Ct values were considered for quantitation. Relative gene expression was analysed using comparative Ct experiment software subset following manufacturer’s instructions. The sequences of primers used for PCR were previously reported (4).
Protein extraction and western blot
PKCepsilon protein expression was analyzed as previously described (7). Briefly, purified platelets were treated with an appropriate amount of cell lysis buffer (50 mM Tris-HCl, pH 7.4; 1% NP-40; 0.25% sodium deoxycholate; 150 mM NaCl; 1 mM EDTA; 1 mM PMSF; 1 mM Na3VO4; 1 mM NaF) supplemented with fresh protease inhibitors and protein concentration was determined using BCATM protein assay kit (Pierce, Rockford, IL, USA). Fifty mg of proteins from each sample were then migrated in 5% SDS-acrylamide gels and blotted onto nitrocellulose filters. Blotted filters were blocked and incubated with specific primary antibodies diluted as described in manufacturers’ protocols. Specifically, rabbit polyclonal anti-PKCe (Upstate, Lake Placid, NY, USA) and mouse monoclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Millipore, Billerica, Massachusetts, USA-Darmstadt, Germania) were diluted 1:5,000. Filters were washed and further incubated for 1.5 hours at room temperature with 1:5000 peroxidase-conjugated anti-rabbit or with 1:2,000 peroxidase-conjugated anti-mouse IgG (Pierce) in the primary antibody working solution at RT. Proteins were resolved by a chemiluminescence detection method (ECL Supersignal West Pico Chemiluminescent Substrate detection system, Pierce) and densitometric analyses were performed by using the ImageJ software system.
Statistical analysis
Data were expressed as mean ± standard deviation (SD) or median (and range), and statistical significance was assessed by t-test, Mann-Whitney and Chi square tests as applicable.
Correlation between PKCε levels and variables such as age and white blood cell count was analyzed by Spearman Rank-order correlation test. A P<0.05 was considered statistically significant. Statistical analysis was performed by GraphPad Prism (GraphPad Software Inc., La Jolla, CA 92037, USA).
Results
MF patient population
Nineteen MF patients were recruited. Nine out of 19 (47%) were male. Thirteen out of 19 (69%) had PMF, while 5 out of 19 (26%) had post-PV MF and one (5%) post-ET MF. Median age was 69 (range, 30–86) years. Complete blood cell (CBC) counts at the time of sample collection are reported. Patients were checked for JAK2, MPL and CALR mutational status. Patient population included low (n=2), intermediate-1 (n=6), intermediate-2 (n=4) and high (n=7) IPSS/DIPSS risk patients. History of major cardiovascular events [which included: acute MI, stroke/TIA, peripheral artery thrombosis, deep venous thrombosis, as listed in (2)] is reported for each patient. Clinical data are summarized in Table 1.
Table 1. Clinical and molecular profile of MF patients.
| Patient code | Diagnosis | Gender | WBC (109/ìL) | Hb (g/dL) | PLT (109/ìL) | JAK2 V617F | MPL/CALR | IPSS/DIPSS | CV |
|---|---|---|---|---|---|---|---|---|---|
| MF1 | PMF | F | 1,160 | 7.9 | 65,000 | Pos | – | High | CV− |
| MF2 | PMF | M | 33,680 | 11.4 | 286,000 | Pos | – | Int-2 | CV− |
| MF3* | PMF | M | 5,740 | 7.8 | 112,000 | Pos | – | High | CV+ (AMI) |
| MF4* | PMF | F | 4,460 | 10.1 | 290,000 | Neg | – | Int-1 | CV− |
| MF5* | PMF | M | 52,450 | 13.4 | 174,000 | Pos | – | High | CV+ (PAT) |
| MF6* | PMF | F | 10,500 | 13.9 | 732,000 | Pos | – | Int-1 | CV− |
| MF7* | postPV MF | F | 52,760 | 15.0 | 204,000 | Pos | – | High | CV− |
| MF8* | PMF | M | 10,130 | 14.9 | 705,000 | Pos | – | Low | CV− |
| MF9* | postET MF | F | 8,960 | 11 | 639,000 | Neg | MPL neg CALR type1 | Low | CV− |
| MF10* | postPV MF | F | 30,680 | 13.2 | 202,000 | Pos | – | High | CV+ (DVT) |
| MF11* | PMF | F | 7,210 | 12.6 | 238,000 | Neg | – | Int-1 | CV+ (stroke) |
| MF12 | PMF | M | 12,210 | 13.8 | 207,000 | Pos | – | Int-1 | CV− |
| MF13 | PMF | M | 7,800 | 8.8 | 506,000 | Pos | – | High | CV+ (TIA) |
| MF14 | PMF | F | 8,000 | 11 | 752,000 | Neg | MPL neg CALR neg | Int-2 | CV− |
| MF15 | postPV MF | F | 2,890 | 7.9 | 106,000 | Pos | – | High | CV− |
| MF16 | PMF | F | 25,080 | 10.7 | 223,000 | Neg | – | Int-2 | CV− |
| MF17 | postPV MF | M | 39,900 | 12 | 202,000 | Pos | – | Int-2 | CV− |
| MF18 | PMF | M | 10,300 | 10.3 | 527,000 | - | – | Int-1 | CV− |
| MF19 | postPV MF | M | 18,970 | 11 | 214,000 | Pos | – | Int-1 | CV− |
*, patients included for correlation studies between platelet PKCepsilon levels and CV events. AMI, acute myocardial infarction; CV, cardiovascular events; CV+, history of cardiovascular events; CV−, no history of cardiovascular events; DIPSS, dynamic international prognostic scoring system; Hb, hemoglobin; IPSS, international prognostic scoring system; PAT, peripheral artery thrombosis; PLT, platelet count; PMF, primary myelofibrosis; postPV MF, post polycythemia vera myelofibrosis; postET MF, post essential thrombocythemia myelofibrosis; TIA, transient ischemic attack; WBC, white blood cell count.
Platelets of MF patients circulate in an activated status
It’s well known that platelet activation status correlates with thrombotic risk in a wide range of diseases (18-20). Despite data from the literature are scant, various authors report that platelets from MPN patients show higher levels of activation markers, suggesting an increased platelet reactivity (2,21-24).
We performed flow cytometric analysis of platelet activation markers finding an increased expression of CD62p, CD154 and PAC-1 in circulating platelets from MF patients as compared to HDs (Figure 1). These data confirm, that platelets of MF patients circulate in an activated status and this may account for increased risk of thrombus formation.
Figure 1.
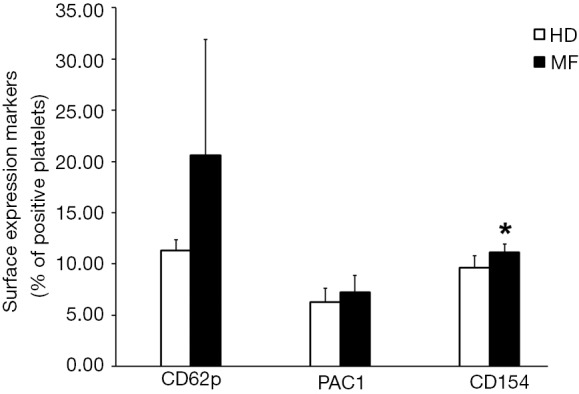
MF platelet activation status. Platelet surface expression of CD62p, PAC1 (from 3 MF patients and 3 HDs) and CD154 (from 5 MF patients and 5 HD). Data are expressed as mean of positive platelets (%) ± SD. *, P<0.05 vs. HDs; t-test statistical analysis. MF, myelofibrosis; HD, healthy donor; SD, standard deviation.
MF platelets are characterized by higher levels of PKCepsilon
We have previously demonstrated that PKCepsilon levels are finely tuned during MK differentiation of primary CD34+ cells from healthy individuals, peaking in early precursors and then progressively declining in differentiated cells (9). In fact, it is known that in human normal platelets, PKC epsilon is nearly absent (25,26). This kinetic of expression can be altered during malignant megakaryopoiesis, as we showed that PKCepsilon is over-expressed in PMF MKs accounting for enhanced proliferation and impaired differentiation (13,14). Since platelets are anucleated cells that retain mRNA and proteins from their MK precursors, we tested whether MF platelets could inherit higher PKCepsilon levels from MF MK. Therefore, we evaluated PKC epsilon mRNA and protein expression in circulating platelets isolated from peripheral blood of 9 MF patients and 10 healthy controls. As expected, MF platelets showed significantly higher mRNA and protein levels of PKCepsilon as compared to HD (1.51±1.39 vs. 0.52±0.39, P=0.0448) (Figure 2), indicating that platelets PKCepsilon mirrors what happens in their precursor cells. Specifically, the failed downregulation of this kinase during the neoplastic differentiation of MF megakaryocytes, lead to the production of platelets with an aberrant expression of PKCepsilon.
Figure 2.

PKCepsilon mRNA and protein expression in MF and HD platelets. (A) Quantitative analysis of PKCepsilon mRNA by real-time PCR in 3 HD and 3 MF. Equal amounts of total cDNA were amplified by real time PCR and PKCepsilon mRNA levels are expressed as PKCepsilon/CD41 ratio. Data are expressed as means ± SD (*, P=0.03; t-test statistical analysis). (B) Representative western blot assay for protein detection of PKCepsilon and GAPDH from MF patient and HD. (C) Densitometric analysis of PKCepsilon protein expression [relative PKCepsilon/GAPDH optical density (OD) values] in 10 HDs and 9 MF patients. Data are expressed as means ± SD (*, P=0.045; t-test statistical analysis). MF, myelofibrosis; HD, healthy donor; SD, standard deviation.
Platelet PKCepsilon expression correlates with disease burden and cardiovascular events in MF patients
We then investigated whether the PKCepsilon levels in MF platelets could be associated to specific clinical features of MF patients reflecting disease burden and most frequent complications, such as major thrombotic events. Specifically, we analysed platelet PKCepsilon expression according to the prognostic risk category and the history of thrombotic complication(s).
Nine MF patients were stratified according to (I) IPSS/DIPSS risk category (low/intermediate vs. high risk); (II) history of major cardiovascular events, as listed in Barbui et al. (27) and platelet PKCepsilon was assessed by western blot analysis.
According to what observed for PMF megakaryocytes (14), platelets from high risk patients are characterized by higher expression of PKCepsilon as compared low/intermediate risk patients (3.24±0.24 vs. 0.65±0.61, P=0.0002) (Figure 3A).
Figure 3.

Correlation between PKCepsilon expression with IPSS/DIPSS risk category and cardiovascular events. (A) Protein PKCepsilon expression [relative PKCepsilon/GAPDH optical density (OD) values] in six low/int vs. three high patients. Data are expressed as mean ± SD (*, P=0.0002; t-test statistical analysis). (B) Protein PKCepsilon expression [relative PKCepsilon/GAPDH optical density (OD) values] in 9 MF patients stratified according to a positive (CV+, n=5) or negative (CV−, n=4) history of prior cardiovascular events. PKCepsilon levels for each patient are indicated as dots (CV−) or triangles (CV+); mean PKCepsilon levels for each group (CV+ vs. CV−) are indicated by bars. *, P=0.0417, t-test statistical analysis. SD, standard deviation.
Since we have previously demonstrated that the ectopic expression of PKCepsilon in platelets of MI patients accounts for their increased reactivity (7), we evaluated whether platelets from 9 MF patients with at least one previous major cardiovascular event (CV+, n=4) displayed higher PKC epsilon levels than those with no history of thrombosis (CV−, n=5). We found that platelets from CV+ patients display significantly higher levels of PKCepsilon as compared to CV− (2.51±1.46 vs. 0.71±0.66, P=0.0417) (Figure 3B). Of note, the two patient populations (CV+ and CV−) were homogeneous for WBC counts (24,020±22,131.1 vs. 17,362±19,934; P>0.06 by Mann-Whitney test), age [median age 74 (range, 66–81) vs. 57 (range, 38–78); P>0.06 by Mann-Whitney test] and JAK2 mutational status (JAK2V617F-positive: 75% vs. 60%; P=0.62 by Chi Square test), ruling out the potential confounding effect of well-established pro-thrombotic factors in MPN (23).
On the contrary, platelet PKCepsilon expression does not correlate with age and leucocytosis (Figure 4A,B) and, consistently with what we described for PMF MK (14), we did not observe any significant differences by stratifying patients according to JAK2 mutational status (Figure 4C).
Figure 4.

Correlation of PKCepsilon expression with risk factors for thrombosis. Linear regression plot for correlation analysis between protein PKCepsilon expression [relative PKCepsilon/GAPDH optical density (OD) values] and age (A) and white blood cells count (WBC/mm3) (B) in the nine MF patients tested in the experiments represented in Figure 3. No statistical differences were detected by Spearman Rank-order correlation test. (C) PKCepsilon protein expression [relative PKCepsilon/GAPDH optical density (OD) values] in JAK2V617F-positive (n=6) and JAK2V617F-negative (n=3) patients. Horizontal bars indicate mean PKCepsilon protein levels, which is similar in the two groups (1.98±1.45 for JAK2V617F-positive and 0.65±0.88 for JAK2V617F-negative, P=0.2, t-test statistical analysis).
Collectively these results suggest that PKCepsilon levels in MF platelets are associated with disease aggressiveness and thrombotic complications, leading us to speculate that PKCepsilon may configure as a potential novel biomarker for thrombotic risk and disease severity in MF.
Discussion
MF is characterized by clonal expansion of aberrant MK precursors, defective thrombopoiesis with altered platelet number and function which leads to a pro-thrombotic phenotype.
Although the pathogenesis of thrombosis in MF is complex and multifactorial, quantitative and qualitative changes in mature blood cells arising from the malignant progenitors play a pivotal role in this process. Thus, MK clonal proliferation and thrombosis are deeply interconnected events.
The oncoprotein PKCepsilon has a key role in MF aberrant megakaryocytopoiesis, as we proved to be over-expressed in MF MK and that its levels correlate with high risk disease (13,14). On these bases, and given the assumption that platelets “inherit” mRNA and proteins from their MK progenitors, here we assessed whether circulating platelets from MF patients displayed the same aberrant expression of PKCepsilon as their precursors. Indeed, while PKCepsilon is nearly absent in platelets from healthy volunteers, MF platelets show significantly higher levels of the kinase, both as transcript and protein. Additionally, consistently with our previous data on MF MK, we found that also MF platelet PKCepsilon levels correlate with the clinical risk category.
In fact, platelets from high risk MF patients are typified by significantly higher PKCepsilon protein levels that platelets from low/intermediate risk MF patients.
Additionally, we also proved that, consistently with data from the literature, MF platelets circulate in the blood stream in an activated state, as indicated by flow cytometric analysis of well-established platelet activation markers.
Since we previously demonstrated that PKCepsilon is over-expressed in platelets from MI patients and accounts for their increased reactivity, we then tested whether its levels in MF platelets could be associated with thrombotic risk.
Indeed, we found that platelets from patients at high thrombotic risk for a previous history of major cardiovascular events display significantly higher PKCepsilon protein that those with no history of arterial or venous complications.
Conclusions
Overall our data suggest that PKCepsilon may represent a molecular link between clonal expansion and thrombogenesis in MF. Although further studies are recommended to better define the functional role of PKCepsilon in MF platelet pro-thrombotic phenotype, this study provide the rational to think of PKCepsilon as a potential novel biomarker of disease burden and thrombotic risk in MPNs.
Acknowledgements
This work was supported by Regione Emilia-Romagna Area 1-Strategic Program 2010–2012 and Fondi Locali per la Ricerca 2014-Quota Incentivante-Università degli Studi di Parma.
Ethical Statement: The study was approved by the Ethical Committee of Parma University Hospital (No. 18228 of May 24th, 2013) and written informed consent was obtained from all patients.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Tefferi A, Skoda R, Vardiman JW. Myeloproliferative neoplasms: contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol 2009;6:627-37. 10.1038/nrclinonc.2009.149 [DOI] [PubMed] [Google Scholar]
- 2.Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-84. 10.1182/blood-2013-03-460154 [DOI] [PubMed] [Google Scholar]
- 3.Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med 2008;359:938-49. 10.1056/NEJMra0801082 [DOI] [PubMed] [Google Scholar]
- 4.Rivera J, Lozano ML, Navarro-Núñez L, et al. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica 2009;94:700-11. 10.3324/haematol.2008.003178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lippi G, Franchini M, Targher G. Arterial thrombus formation in cardiovascular disease. Nat Rev Cardiol 2011;8:502-12. 10.1038/nrcardio.2011.91 [DOI] [PubMed] [Google Scholar]
- 6.Strehl A, Munnix IC, Kuijpers MJ, et al. Dual role of platelet protein kinase C in thrombus formation: stimulation of pro-aggregatory and suppression of procoagulant activity in platelets. J Biol Chem 2007;282:7046-55. 10.1074/jbc.M611367200 [DOI] [PubMed] [Google Scholar]
- 7.Carubbi C, Mirandola P, Mattioli M, et al. Protein kinase C ε expression in platelets from patients with acute myocardial infarction. PLoS One 2012;7:e46409. 10.1371/journal.pone.0046409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Totoń E, Ignatowicz E, Skrzeczkowska K, et al. Protein kinase Cε as a cancer marker and target for anticancer therapy. Pharmacol Rep 2011;63:19-29. 10.1016/S1734-1140(11)70395-4 [DOI] [PubMed] [Google Scholar]
- 9.Gobbi G, Mirandola P, Sponzilli I, et al. Timing and expression level of protein kinase C epsilon regulate the megakaryocytic differentiation of human CD34 cells. Stem Cells 2007;25:2322-9. 10.1634/stemcells.2006-0839 [DOI] [PubMed] [Google Scholar]
- 10.Gobbi G, Mirandola P, Carubbi C, et al. Phorbol ester-induced PKCepsilon down-modulation sensitizes AML cells to TRAIL-induced apoptosis and cell differentiation. Blood 2009;113:3080-7. 10.1182/blood-2008-03-143784 [DOI] [PubMed] [Google Scholar]
- 11.Gobbi G, Masselli E, Micheloni C, et al. Hypoxia-induced down-modulation of PKCepsilon promotes trail-mediated apoptosis of tumor cells. Int J Oncol 2010;37:719-29. [DOI] [PubMed] [Google Scholar]
- 12.Gobbi G, Mirandola P, Carubbi C, et al. Protein kinase C ε in hematopoiesis: conductor or selector? Semin Thromb Hemost 2013;39:59-65. 10.1055/s-0032-1331156 [DOI] [PubMed] [Google Scholar]
- 13.Carubbi C, Masselli E, Martini S, et al. Human thrombopoiesis depends on Protein kinase Cδ/protein kinase Cε functional couple. Haematologica 2016;101:812-20. 10.3324/haematol.2015.137984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masselli E, Carubbi C, Gobbi G, et al. Protein kinase Cɛ inhibition restores megakaryocytic differentiation of hematopoietic progenitors from primary myelofibrosis patients. Leukemia 2015;29:2192-201. 10.1038/leu.2015.150 [DOI] [PubMed] [Google Scholar]
- 15.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391-405. 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- 16.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009;113:2895-901. 10.1182/blood-2008-07-170449 [DOI] [PubMed] [Google Scholar]
- 17.Passamonti F, Cervantes F, Vannucchi AM, et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood 2010;116:2857-8. 10.1182/blood-2010-06-293415 [DOI] [PubMed] [Google Scholar]
- 18.Carubbi C, Masselli E, Gesi M, et al. Cytofluorimetric platelet analysis. Semin Thromb Hemost 2014;40:88-98. [DOI] [PubMed] [Google Scholar]
- 19.Gresele P, Kleiman NS, Lopez JA, et al. Platelets in Thrombotic and Non-Thrombotic Disorders. Pathophysiology, Pharmacology and Therapeutics: an Update. Springer International Publishing, 2017. [Google Scholar]
- 20.Lippi G, Montagnana M, Salvagno GL, et al. Risk stratification of patients with acute myocardial infarction by quantification of circulating monocyte-platelet aggregates. Int J Cardiol 2007;115:101-2. 10.1016/j.ijcard.2005.12.017 [DOI] [PubMed] [Google Scholar]
- 21.Viallard JF, Solanilla A, Gauthier B, et al. Increased soluble and platelet-associated CD40 ligand in essential thrombocythemia and reactive thrombocytosis. Blood 2002;99:2612-4. 10.1182/blood.V99.7.2612 [DOI] [PubMed] [Google Scholar]
- 22.Villmow T, Kemkes-Matthes B, Matzdorff AC. Markers of platelet activation and platelet-leukocyte interaction in patients with myeloproliferative syndromes. Thromb Res 2002;108:139-45. 10.1016/S0049-3848(02)00354-7 [DOI] [PubMed] [Google Scholar]
- 23.Bermejo E, Alberto MF, Meschengieser SS, et al. Assessment of platelet activation in myeloproliferative disorders with complementary techniques. Blood Coagul Fibrinolysis 2004;15:235-40. 10.1097/00001721-200404000-00006 [DOI] [PubMed] [Google Scholar]
- 24.Falanga A, Marchetti M, Vignoli A, et al. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp Hematol 2005;33:523-30. 10.1016/j.exphem.2005.01.015 [DOI] [PubMed] [Google Scholar]
- 25.Buensuceso CS, Obergfell A, Soriani A, et al. Regulation of outside-in signaling in platelets by integrin-associated protein kinase C beta. J Biol Chem 2005;280:644-53. 10.1074/jbc.M410229200 [DOI] [PubMed] [Google Scholar]
- 26.Pears CJ, Thornber K, Auger JM, et al. Differential roles of the PKC novel isoforms, PKCdelta and PKCepsilon, in mouse and human platelets. PLoS One 2008;3:e3793. 10.1371/journal.pone.0003793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbui T, Carobbio A, Cervantes F, et al. Thrombosis in primary myelofibrosis: incidence and risk factors. Blood 2010;115:778-82. 10.1182/blood-2009-08-238956 [DOI] [PubMed] [Google Scholar]