

Abstract
The effects on the drug efflux of 3,3′,4,4′,5-pentachlorobiphenyl (PCB-126), the most toxic of all coplanar polychlorinated biphenyls (Co-PCBs), were examined in KB-3 cells expressing human wild-type and mutant P-glycoprotein in which the 61st amino acid was substituted for serine or phenylalanine (KB3-Phe61). In the cells expressing P-glycoproteins, accumulations of vinblastine and colchicine decreased form 85% to 92% and from 62% to 91%, respectively, and the drug tolerances for these chemicals were increased. In KB3-Phe61, the decreases in drug accumulation were inhibited by adding PCB-126 in a way similar to that with cyclosporine A: by adding 1 μM PCB-126, the accumulations of vinblastine and colchicine increased up to 3.3- and 2.3-fold, respectively. It is suggested that PCB-126 decreased the drug efflux by inhibiting the P-glycoprotein in KB3-Phe61. Since there were various P-glycoproteins and many congeners of Co-PCBs, this inhibition has to be considered a new cause of the toxic effects of Co-PCBs.
INTRODUCTION
Among the group of polychlorinated biphenyls (PCBs), coplanar PCBs (Co-PCBs) are known to be highly toxic and indestructible congeners [1, 2]. Co-PCBs had been produced as useful artificial lubricants for the chemical industry and in the manufacture of a variety of daily-use products until their production and use were prohibited. These chemicals have thoroughly polluted the environment, and may have accumulated via the food chain and bioaccumulation in humans and animals, especially in their lipid-rich organs [2, 3, 4, 5]. In the environment, 3,3′,4,4′,5-pentachlorobiphenyl (PCB-126) which is the most toxic congener of Co-PCBs is also highly polluting the environment [1, 6]. Like dioxin, the mixture of Co-PCBs or the metabolites of Co-PCBs seems to produce either estrogenic or antiestrogenic effects [7, 8]. Co-PCBs were the agonists for arylhydrocarbon receptors (AhR) [1, 9], and they inhibited the estrogenic response via crosstalk between AhR and estrogen receptors [10, 11]. Actually, Co-PCBs have acted as environmental endocrine disruptors, exerting adverse biological effects even at a very low dose [6, 12]. They also disturbed normal fetal and neonatal development, producing malformations and the onset of cancer [4, 6, 13, 14].
Drug transport systems such as P-glycoprotein are present on the cell membrane to exclude toxic xenobiotics [15] and to transport metabolites such as steroid hormones [16]. P-glycoprotein is capable of transporting a variety of structurally unrelated chemicals [17] and can induce multidrug resistance during cancer chemotherapy [18, 19]. The P-glycoprotein-mediated transport of chemicals was inhibited by other chemicals. Cyclosporine A, for example, competed with vinblastine for a site to bind to P-glycoprotein, and inhibited the transport of this substrate [20]. The most hydrophobic steroid hormone, progesterone, blocked vinblastine transport, although progesterone itself was not transported. In a series of steroid hormones, it was revealed that the more hydrophobic the congeners, the more potent their interference with the transport of other chemicals [21]. One of the congeners of Co-PCBs, 3,3′,4,4′-tetrachlorobiphenyl (PCB-77), was not transported by human P-glycoprotein, but accumulated abundantly in cells as can be expected due to its lipophilic nature [22]. Co-PCBs easily enter cells and may accumulate in the cell membrane due to their lipophilicity. P-glycoprotein in the cell membrane may be exposed to Co-PCBs, which may modify the transport of other drugs and metabolites in the same way as do hydrophobic steroid hormones and cyclosporine A, thus altering the effects of those substances.
Though P-glycoprotein has a low specificity for substrates, it was found to contain many mutations of amino acids that modulated substrate specificity [23, 24]. Such mutations might be involved in the modification of binding and transporting mechanisms. The toxicity of drugs was reduced in the transformant cells expressing P-glycoprotein, probably due to an enhancement of the drug efflux. These drug tolerances varied among the transformant cells in which the His 61 position (His61) in the predicted transmembrane domain 1 of human P-glycoprotein was replaced with a variety of amino acids [24]. Accordingly, the effect of Co-PCBs might differ among the mutants of P-glycoproteins. The possibility of a modification in P-glycoprotein by binding with Co-PCBs must be examined. Here, we investigated the effect of PCB-126 on the efflux of vinblastine and colchicine in KB-3 cells transfected with multidrug resistant (MDR1) genes which code both wild and His61 mutant P-glycoproteins.
MATERIALS AND METHODS
Chemicals
For radioactive tracers, we used [G-3H]-vinblastine sulphate (470 GBq/mmol) (Amersham Pharmacia Biotech, NJ, USA) and [3H]-colchicine (2,830 GBq/mmol) (Perkin Elmer Japan Co Ltd, Tokyo, Japan). As the representative of Co-PCBs, PCB-126 (Kanto Kagaku, Tokyo, Japan) was used, and cyclosporine A (Sigma, Mo, USA) was used as the inhibitor of P-glycoprotein.
Cell culture
The transformant cells of human cultured cells, that is, KB3-1 expressing with wild-type human P-glycoprotein (KB3-His61) and the mutant P-glycoprotein, in which His61 was substituted with serine (KB3-Ser61) or phenylalanine (KB3-Phe61), were prepared as reported in [24]. As the control, KB3-1 transfected with the transfection vector pHaMAIRESneo (KB3-Vec) was also used. These cells were maintained in medium 199 supplemented with 10% fetal calf serum (FCS) in an atmosphere of 5% CO2 at 37°C, and the transformant cells were maintained in the same medium with appropriate reagents; KB3-His61 and KB3-Ser61 were maintained with 100 μM vinblastine and KB3-Phe61 with 60 μM colchicine.
Drug tolerance of cells
The cells were seeded in 96-well microplates at a density of 105/well in a 500-μL medium identical to that of the maintenance culture except for varied concentrations of vinblastine and colchicine, and then incubated for 4 days. The viability of these cells was then measured by a colorimetric assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (Wako Co Ltd, Osaka, Japan). The inhibition dose for 50% cell growth (ID50) was calculated as the index for drug tolerance.
Drug accumulation and its inhibition
For a determination of the cellular accumulation of the chemicals, coverslips with a 24-well multidish (Nalgen Nunc International, Ill, USA) were used. Cells were seeded at a density of /well in a 750-μL maintenance medium on the dish and incubated in 5% CO2 at 37°C. After 4 days, the medium was replaced with a fresh medium without colchicine, and the cells were incubated for 6 hours. The medium was again replaced with a 750-μL fresh medium containing either 11 nM [3H]-vinblastine (5.16 kBq/mL) or 11 nM [3H]-colchicine (31.1 kBq/mL). The coverslips were removed from the 24-well dish after a 1-, 2-, and 3-hours incubation. These cells were washed 3 times with PBS, and treated with a lysing solution (cell culture lysis reagent, Promega, Wis, USA) overnight. Radioactivity was then measured by a liquid scintillation counter. The accumulation was indicated as pmol/well.
To examine the effect of PCB-126 on drug accumulation, PCB-126 and the inhibitor of p-glycoprotein, cyclosporine A, were added to the incubation medium. One hundred pM to 107 μM PCB-126 or cyclosporine A was added to the medium, and the accumulations of vinblastine and colchicine were measured.
Statistics
The student's -test was employed to examine the statistical significance of the accumulation between cell groups.
RESULTS AND DISCUSSION
Table 1 indicates the drug tolerances of KB3-Vec, KB3-His61, KB3-Ser61, and KB3-Phe61 in the medium with vinblastine or colchicine. The ID50 increased in all the cell groups expressing P-glycoprotein (KB3-MDR1s), KB3-His61, KB3-Ser61, and KB3-Phe61, except for KB3-Vec. As for vinblastine, the ID50 increased around 10-fold in all KB3-MDR1s except for KB3-Vec, and the order of drug tolerances was KB3-Ser61> KB3-Phe61 > KB3-His61. With colchicine, the ID50 increased around 5-fold in KB3-His61 and KB3-Ser61, and 98-fold in KB3-Phe61. The increase in the ID50 for colchicine in KB3-Phe61 was extremely high, indicating a special relation between the side chain of Phe61 and the relatively smaller substrate, colchicine. Taguchi et al showed an inverse relation between the bulk of the side chain of the 61st amino acid in P-glycoprotein and the molecular weight of the drug; the P-glycoprotein, which has a large side chain in the 61st amino acid, indicated less sensitivity to the smaller molecular weight of the chemicals, and vice versa [24].
Table 1.
Drug tolerance for vinblastine and colchicine in KB3-Vec and KB3-MDR1s, and the ratios for the tolerance of KB3-Vec to KB3-MDR1s (KB3-MDR1s/KB3-Vec).
| KB3-MDR1s | ||||
|---|---|---|---|---|
| KB3-Vec | KB3-His61 | KB3-Phe61 | KB3-Ser61 | |
| Vinblastine (ID50, nM) | 3.4 | 26 | 30 | 40 |
| (KB3-MDR1s/KB3-Vec) | (1) | (7.6) | (8.8) | (11.8) |
| Colchicine (ID50, nM) | 8.8 | 46 | 859 | 43 |
| (KB3-MDR1s/KB3-Vec) | (1) | (5.2) | (97.6) | (4.9) |
Note. Inhibition dose for 50% cell growth (ID50) by adding chemicals was expressed as the index of drug tolerance.
Figure 1a shows the vinblastine uptake as a function of time in KB3-Vec and of reductions in the uptake in KB3-MDR1s. The uptakes of vinblastine in KB3-Vec hyperbolically increased during a 3-hour incubation as reported in [22], and the accumulations after that incubation in KB3-His61, KB3-Ser61, and KB3-Phe61 were reduced by 85%, 88%, and 92%, respectively, compared to KB3-Vec. Thus, under such conditions, most of the drug might have been transported to an extracellular site by the P-glycoproteins which were expressed by transfection with each MDR1 gene. These excretions might contribute to the increase in ID50 in KB3-MDR1s.
Figure 1.
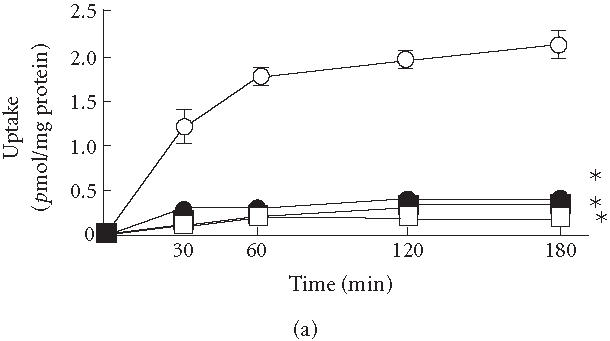
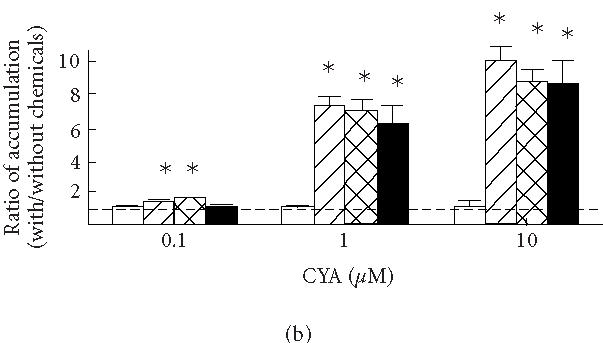
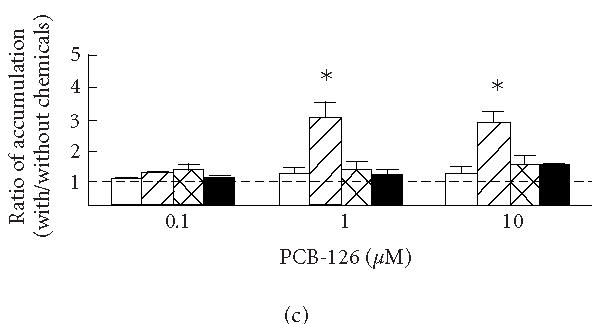
(a) Uptake of vinblastine in KB3-1 expressing with wild-type human P-glycoprotein (KB3-His61, filled circles), mutant P-glycoprotein in which His61 was replaced with phenylalanine (KB3-Phe61, open squares) and serine (KB3-Ser61, filled squares), and transfected with transfection vector (KB3-Vec, open circles). (b) The ratios between the accumulations of vinblastine with and without cyclosporine A (CYA) and (c) PCB-126 (with/without CYA or PCB-126) in KB3-Vec (open columns), KB3-His61 (cross-hatched columns), KB3-Phe61 (hatched columns), and KB3-Ser61 (filled columns). , compared to KB3-Vec. Means and SD of 4 experiments.
Figures 1b and 1c show the effects of cyclosporine A (b) and PCB-126 (c) on the accumulations of vinblastine after a 3-hours incubation in KB3-Vec and KB3-MDR1s; the ratios between the accumulations with and without cyclosporine A or PCB-126 in each cell group were compared. An increase in the ratios indicates an increased inhibition against the drug efflux due to the addition of cyclosporine A or PCB-126. The ratios were increased by adding more than 1 μM cyclosporine A in all KB3-MDR1s, while there was no clear increase in the ratio in KB3-Vec. The ratios in KB3-His61, KB3-Ser61, and KB3-Phe61 increased 7.0-, 6.3-, and 7.5-fold, respectively, by adding 1 μM cyclosporine A, and those accumulations increased 9.2-, 9.1-, and 10.3-fold, respectively, by adding 10 μM cyclosporine A. Though the increase in the ratio was less than 2-fold by adding 0.1 μM cyclosporine A, those in KB3-Phe61 and KB3-His61 were increased significantly. The ratio in KB3-Phe61 was relatively higher than in the other cell groups, though the difference among KB3-MDR1s was less than 20%. By adding 0.1, 1 and 10 μM PCB-126, the ratio in KB3-Phe61 increased 1.4-, 3.3-, and 2.8-fold, respectively, thus revealing an inhibition of the drug efflux by the addition of PCB-126. PCB-126 might have had an effect on the mutant P-glycoprotein in KB3-Phe61 thus inhibiting the drug efflux. The ratios in KB3-His61 and KB3-Ser61 also increased around 1.5-fold, but their effect was no different from that in KB3-Vec.
Figure 2a shows the colchicine uptake as a function of time in KB3-Vec and of reductions in the uptakes in KB3-MDR1s. The colchicine uptake displayed a somewhat linear function compared to the vinblastine uptake in all cell groups. The accumulations after a 3-hours incubation in KB3-His61, KB3-Ser61, and KB3-Phe61 were reduced, respectively, by 75, 62, and 91% compared to that in KB3-Vec. Thus, under those conditions, KB3-Phe61 demonstrated the highest ability to transport colchicine, while KB3-Ser61 showed the lowest.
Figure 2.
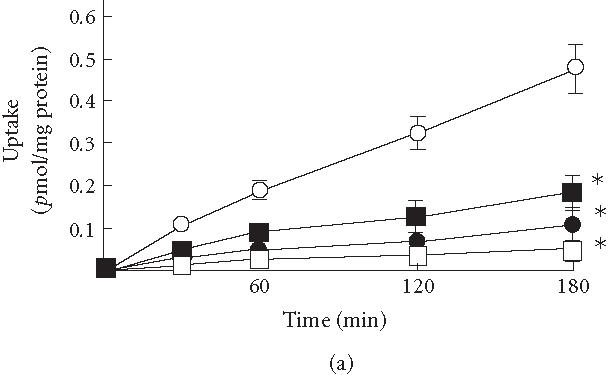
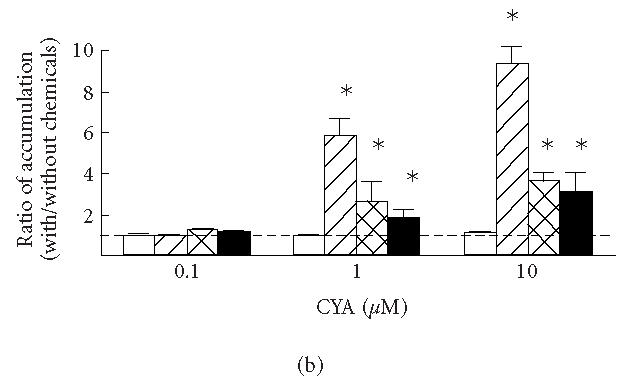
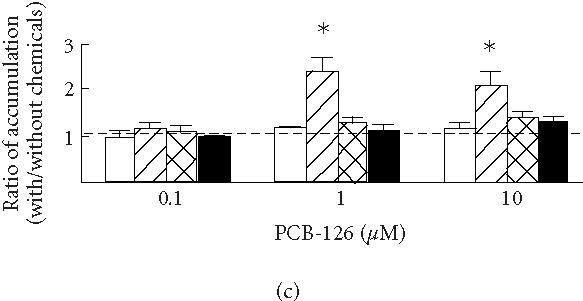
(a) Uptake of colchicine in KB3-1 expressing with wild-type human P-glycoprotein (KB3-His61, filled circles), mutant P-glycoprotein in which His61 was replaced with phenylalanine (KB3-Phe61, open squares) and serine (KB3-Ser61, filled squares), and transfected with transfection vector (KB3-Vec, open circles). (b) The ratios between the accumulations of colchicine with and without cyclosporine A (CYA) and (c) PCB-126 (with/without CYA or PCB-126) in KB3-Vec (open columns), KB3-His61 (cross-hatched columns), KB3-Phe61 (hatched columns), and KB3-Ser61 (filled columns). , compared to KB3-Vec. Means and SD of 4 experiments.
Figures 2b and 2c show the effects of cyclosporine A (b) and PCB-126 (c) on the accumulations during a 3-hours incubation in KB3-Vec and KB3-MDR1s. The ratios between accumulations with and without these chemicals in KB3-His61, KB3-Ser61, and KB3-Phe61 increased 2.6-, 2.0-, and 5.8-fold by adding 1 μM cyclosporine A, and increased 3.6-, 2.9-, and 9.2-fold by adding 10 μM cyclosporine A, respectively, while there was no such effect in KB3-Vec. The cell group which showed the largest reduction in drug accumulation, KB3-Phe61, also showed the largest increase in accumulation when the inhibitor for P-glycoprotein was added. This might be due to an inhibition of the efflux through P-glycoprotein as reported in [21]. These effects of cyclosporine A on drug accumulation were considered due to a modification of P-glycoprotein by its binding [20]. By adding 1 and 10 μM PCB-126, the ratios in KB3-Phe61 increased 2.3- and 2.1-fold, respectively, indicating an inhibition of the drug efflux. The ratios in KB3-His61 and KB3-Ser61 increased slightly, though there was no significant difference from that in KB3-Vec. Thus, PCB-126 might have bound with Phe61 mutant P-glycoprotein to inhibit the efflux of colchicine.
In all KB3-MDR1s, accumulations of vinblastine were equally reduced, and cyclosporine A inhibited these reductions to almost the same degree among KB3-MDR1s. However, the reductions and inhibitions in the accumulation of colchicine varied among cell groups. Previously, one of the present authors revealed that the cells expressing mutant P-glycoprotein which possessed a larger 61st amino acid gained a tolerance to various chemicals, whereas, in the cells expressing the mutant P-glycoprotein which possessed a smaller 61st amino acid, the tolerance for large chemicals was superior to that for small chemicals [24]. These results were explained as being due to the difference in the bulk of the side chain of the 61st amino acid in P-glycoprotein, rather than to the electron charge or hydrophilicity in the side chain. In this experiment, KB3-Phe61 showed a superior ability for excretion in both vinblastine and colchicine, and the excretion in KB3-Phe61 was more potently inhibited by cyclosporine A and PCB-126 compared to the other cells. These results showed that the Phe61 mutant P-glycoprotein in which the side chain of the 61st amino acid was larger than that in the other mutants was less specific for substrates. Therefore, it might have reacted to a variety of substrates with almost the same affinity. However, the wild type, His61 P-glycoprotein, and Ser61 mutant P-glycoprotein were somewhat specific for substrates, and their affinity was predominant in the larger substrate, vinblastine. The Phe61 mutant P-glycoprotein might have the ability to bind with a variety of chemicals dissimilar to one another in structure and molecular size. Thus, PCB-126 might also bind to the Phe61 mutant P-glycoprotein and inhibit the efflux of vinblastine and colchicine, while being unable to react with the other P-glycoproteins.
The contamination level of Co-PCBs and the concentrations used in this experiment will now be discussed. As a result of their locations on the food chain, several marine mammals showed high levels of contamination with PCBs, that is, up to around 5 mg/kg in their blubber [25]. In humans who consumed high levels of fish, the total content of lipid-adjusted plasma PCBs increased up to 770 μg/kg [26], and PCB-126 accumulated up to 1.4 μg/kg in plasma lipid [4]. Among workers employed in capacitor manufacturing, the concentrations of high- and low-chlorinated PCBs in adipose tissue increased up to 165 and 414 μg/kg, respectively [27]. PCBs are highly lipophilic and may accumulate in the lipid bilayers of the cell membrane. In the present experiment, the effect of Co-PCBs was revealed at levels of 0.1 to 10 μM PCB-126, that is, 30 to 3000 μg/kg of PCB-126. Therefore, in populations exposed to an environment highly contaminated with PCBs, the concentrations of PCBs or Co-PCBs in their cell membranes would probably increase to around the concentrations found in this experiment.
In conclusion, it was indicated that PCB-126 inhibited the drug efflux through the mutant P-glycoprotein in KB3-Phe61. The interaction of PCB-126 with P-glycoprotein was modified by the mutation of the 61st amino acid of the protein. There have been many amino acid mutations that modulated substrate specificity in P-glycoprotein [28]. Therefore, there may be P-glycoproteins which are affected by some congener(s) of Co-PCBs as revealed in this experiment. Though the precise mechanism of toxicity is still unknown, P-glycoprotein plays a role both in the extrusion of xenobiotics and in the transport of steroid hormones [16, 21]. Consequently, not only the toxicities of xenobiotics but also the effects of steroid hormones may be disturbed by Co-PCBs. In particular, the regulation of brain differentiation in an embryo by steroid hormones seems to be influenced by Co-PCBs. In addition to the reported mechanism for toxicities [1, 7, 8, 9, 10, 11, 12], the effect of Co-PCBs on P-glycoprotein has to be considered as a new cause of the toxic effects of these chemicals.
ACKNOWLEDGMENT
This work was partly supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (11839027) and from the Project of the High-Tech Research Center, Azabu University.
References
- 1.Safe S.H. Polychlorinated biphenyls (PCBs): environmental impact, biochemical and toxic responses, and implications for risk assessment. Crit Rev Toxicol. 1994;24(2):87–149. doi: 10.3109/10408449409049308. [DOI] [PubMed] [Google Scholar]
- 2.Bonefeld-Jorgensen E.C, Andersen H.R, Rasmussen T.H, Vinggaard A.M. Effect of highly bioaccumulated polychlorinated biphenyl congeners on estrogen and androgen receptor activity. Toxicology. 2001;158(3):141–153. doi: 10.1016/s0300-483x(00)00368-1. [DOI] [PubMed] [Google Scholar]
- 3.Longnecker M.P, Rogan W.J, Lucier G. The human health effects of DDT (dichlorodiphenyltrichloroethane) and PCBS (polychlorinated biphenyls) and an overview of organochlorines in public health. Annu Rev Public Health. 1997;18:211–244. doi: 10.1146/annurev.publhealth.18.1.211. [DOI] [PubMed] [Google Scholar]
- 4.Asplund L, Svensson B.G, Nilsson A, et al. Polychlorinated biphenyls, 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane (p,p'-DDT) and 1,1-dichloro-2,2-bis(p-chlorophenyl)-ethylene (p,p'-DDE) in human plasma related to fish consumption. Arch Environ Health. 1994;49(6):477–486. doi: 10.1080/00039896.1994.9955004. [DOI] [PubMed] [Google Scholar]
- 5.Furst P, Furst C, Wilmers K. Human milk as a bioindicator for body burden of PCDDs, PCDFs, organochlorine pesticides, and PCBs. Environ Health Perspect. 1994;102(suppl 1):187–193. doi: 10.1289/ehp.102-1566908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cogliano V.J. Assessing the cancer risk from environmental PCBs. Environ Health Perspect. 1998;106(6):317–323. doi: 10.1289/ehp.98106317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nesaretnam K, Darbre P. 3,5,3',5'-tetrachlorobiphenyl is a weak oestrogen agonist in vitro and in vivo. J Steroid Biochem Mol Biol. 1997;62(5-6):409–418. doi: 10.1016/s0960-0760(97)00062-9. [DOI] [PubMed] [Google Scholar]
- 8.Jansen H.T, Cooke P.S, Porcelli J, Liu T.C, Hansen L.G. Estrogenic and antiestrogenic actions of PCBs in the female rat: in vitro and in vivo studies. Reprod Toxicol. 1993;7(3):237–248. doi: 10.1016/0890-6238(93)90230-5. [DOI] [PubMed] [Google Scholar]
- 9.Kafafi S.A, Afeefy H.Y, Ali A.H, Said H.K, Abd-Elazem I.S, Kafafi A.G. Affinities for the aryl hydrocarbon receptor, potencies as aryl hydrocarbon hydroxylase inducers and relative toxicities of polychlorinated biphenyls. A congener specific approach. Carcinogenesis. 1993;14(10):2063–2071. doi: 10.1093/carcin/14.10.2063. [DOI] [PubMed] [Google Scholar]
- 10.Connor K, Ramamoorthy K, Moore M, et al. Hydroxylated polychlorinated biphenyls (PCBs) as estrogens and antiestrogens: structure-activity relationships. Toxicol Appl Pharmacol. 1997;145(1):111–123. doi: 10.1006/taap.1997.8169. [DOI] [PubMed] [Google Scholar]
- 11.Wormke M, Castro-Rivera E, Chen I, Safe S. Estrogen and aryl hydrocarbon receptor expression and crosstalk in human Ishikawa endometrial cancer cells. J Steroid Biochem Mol Biol. 2000;72(5):197–207. doi: 10.1016/s0960-0760(00)00030-3. [DOI] [PubMed] [Google Scholar]
- 12.Li M.H, Hansen L.G. Enzyme induction and acute endocrine effects in prepubertal female rats receiving environmental PCB/PCDF/PCDD mixtures. Environ Health Perspect. 1996;104(7):712–722. doi: 10.1289/ehp.96104712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobson J.L, Jacobson S.W. Evidence for PCBs as neurodevelopmental toxicants in humans. Neurotoxicology. 1997;18(2):415–424. [PubMed] [Google Scholar]
- 14.Sauer P.J, Huisman M, Koopman-Esseboom C, et al. Effects of polychlorinated biphenyls (PCBs) and dioxins on growth and development. Hum Exp Toxicol. 1994;13(12):900–906. doi: 10.1177/096032719401301213. [DOI] [PubMed] [Google Scholar]
- 15.Thiebaut F, Tsuruo T, Hamada H, Gottesman M.M, Pastan I, Willingham M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA. 1987;84(21):7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ueda K, Okamura N, Hirai M, et al. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J Biol Chem. 1992;267(34):24248–24252. [PubMed] [Google Scholar]
- 17.Ueda K, Cardarelli C, Gottesman M.M, Pastan I. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci USA. 1987;84(9):3004–3008. doi: 10.1073/pnas.84.9.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gottesman M.M, Pastan I. The multidrug transporter, a double-edged sword. J Biol Chem. 1988;263(25):12163–12166. [PubMed] [Google Scholar]
- 19.Endicott J.A, Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem. 1989;58:137–171. doi: 10.1146/annurev.bi.58.070189.001033. [DOI] [PubMed] [Google Scholar]
- 20.Tamai I, Safa A.R. Competitive interaction of cyclosporins with the Vinca alkaloid-binding site of P-glycoprotein in multidrug-resistant cells. J Biol Chem. 1990;265(27):16509–16513. [PubMed] [Google Scholar]
- 21.Barnes K.M, Dickstein B, Cutler G.B. Jr, Fojo T, Bates S.E. Steroid treatment, accumulation, and antagonism of P-glycoprotein in multidrug-resistant cells. Biochemistry. 1996;35(15):4820–4827. doi: 10.1021/bi952380k. [DOI] [PubMed] [Google Scholar]
- 22.Fujise H, Annoura T, Sasawatari S, Ikeda T, Ueda K. Transepithelial transport and cellular accumulation of steroid hormones and polychlorobiphenyl in porcine kidney cells expressed with human P-glycoprotein. Chemosphere. 2002;46(9-10):1505–1511. doi: 10.1016/s0045-6535(01)00273-9. [DOI] [PubMed] [Google Scholar]
- 23.Devine S.E, Ling V, Melera P.W. Amino acid substitutions in the sixth transmembrane domain of P-glycoprotein alter multidrug resistance. Proc Natl Acad Sci USA. 1992;89(10):4564–4568. doi: 10.1073/pnas.89.10.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taguchi Y, Kino K, Morishima M, Komano T, Kane S.E, Ueda K. Alteration of substrate specificity by mutations at the His61 position in predicted transmembrane domain 1 of human MDR1/P-glycoprotein. Biochemistry. 1997;36(29):8883–8889. doi: 10.1021/bi970553v. [DOI] [PubMed] [Google Scholar]
- 25.Fromberg A, Cleemann M, Carlsen L. Review on persistent organic pollutants in the environment of Greenland and Faroe Islands. Chemosphere. 1999;38(13):3075–3093. doi: 10.1016/s0045-6535(98)00514-1. [DOI] [PubMed] [Google Scholar]
- 26.Kosatsky T, Przybysz R, Shatenstein B, Weber J.P, Armstrong B. Fish consumption and contaminant exposure among Montreal-area sportfishers: pilot study. Environ Res. 1999;80(2):S150–S158. doi: 10.1006/enrs.1998.3910. [DOI] [PubMed] [Google Scholar]
- 27.Wolff M.S, Fischbein A, Thornton J, Rice C, Lilis R, Selikoff I.J. Body burden of polychlorinated biphenyls among persons employed in capacitor manufacturing. Int Arch Occup Environ Health. 1982;49(3-4):199–208. doi: 10.1007/BF00377929. [DOI] [PubMed] [Google Scholar]
- 28.Safa A.R, Stern R.K, Choi K, et al. Molecular basis of preferential resistance to colchicine in multidrug-resistant human cells conferred by Gly-185 → Val-185 substitution in P-glycoprotein. Proc Natl Acad Sci USA. 1990;87(18):7225–7229. doi: 10.1073/pnas.87.18.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]