

Abstract
Peroxisome proliferator-activated receptors (PPARs) were discovered over a decade ago, and were classified as orphan members of the nuclear receptor superfamily. To date, three PPAR subtypes have been discovered and characterized (PPARα, β/δ, γ). Different PPAR subtypes have been shown to play crucial roles in important diseases and conditions such as obesity, diabetes, atherosclerosis, cancer, and fertility. Among the most studied roles of PPARs is their involvement in inflammatory processes. Numerous studies have revealed that agonists of PPARα and PPARγ exert anti-inflammatory effects both in vitro and in vivo. Using the carrageenan-induced paw edema model of inflammation, a recent study in our laboratories showed that these agonists hinder the initiation phase, but not the late phase of the inflammatory process. Furthermore, in the same experimental model, we recently also observed that activation of PPARδ exerted an anti-inflammatory effect. Despite the fact that exclusive dependence of these effects on PPARs has been questioned, the bulk of evidence suggests that all three PPAR subtypes, PPARα, δ, γ, play a significant role in controlling inflammatory responses. Whether these subtypes act via a common mechanism or are independent of each other remains to be elucidated. However, due to the intensity of research efforts in this area, it is anticipated that these efforts will result in the development of PPAR ligands as therapeutic agents for the treatment of inflammatory diseases.
INTRODUCTION
A group of nuclear receptors, termed peroxisome proliferator-activated receptors (PPARs), was identified during the last decade [1]. These receptors, which belong to the steroid hormone receptor superfamily, bind to and are activated by fatty acids, eicosanoids, and numerous structurally dissimilar xenobiotics, known collectively as peroxisome proliferators (see [2]; Figure 1 and Table 1).
Figure 1.

Chemical structures of representative PPAR agonists. PPARα: Wy-14,643, PPAR β/δ: L-165041, and PPARγ: Rosiglitazone.
Table 1.
Relative PPAR subtype-specificity of selected agonists.
| Agonist | PPAR subtype | ||
|---|---|---|---|
| α | β/δ | γ | |
| Wy-14,643 | +++ | −−− | + |
| L-165041 | −−− | +++ | + |
| Rosiglitazone | −−− | −−− | +++ |
Three related PPAR isotypes have been identified to date; PPAR α, PPAR β/δ, and PPAR γ [3, 4]. Studies have documented the existence of human forms of PPARα (h PPARα; [5, 6]) and PPARγ (hPPAR γ; [7]). The tissue distribution pattern of hPPAR α mRNA is similar to that of the rat PPAR α. In both species PPAR α is highly expressed in brown adipose tissue, skeletal muscle, liver, heart, and kidney, while expressed at low levels in the brain and lung [6, 8]. The principal site of expression of PPAR γ is the adipose tissue, but this receptor is also expressed, albeit at lower levels, in many other tissues and cell types such as the retina, some parts of the immune system, mammary and colonic epithelium [9]. PPAR δ subtype is found in higher amounts than PPAR α and PPAR γ in almost all tissues examined, except the adipose tissue [10]. PPARδ is expressed ubiquitously in the rat cerebellum, thalamus and cerebellar cortex [11], and specific PPARδ agonists, and to a much lesser extent those of PPARγ, stimulated oligodendrocyte differentiation in vitro [12].
The chromosomal localization of the human and mouse PPAR genes has been identified [3]. The human PPARα (h PPARα) was mapped on chromosome 22 [3]. The hPPAR γ gene is located on chromosome 3 and the hPPAR δ has been assigned to chromosome 6 [3]. In the mouse, PPAR γ is located on chromosome 6, while PPAR α and PPAR δ are found on chromosomes 15 and 17, respectively [3].
DNA binding properties of PPARs
The DNA binding domain is the most conserved domain among all nuclear hormone receptors, and is the hallmark of this superfamily of receptors. The DBD is formed by two zinc finger-like motifs folded in a globular structure that can recognize a DNA target composed of 6 nucleotides [3]. In most cases, nuclear hormone receptors bind as dimers to two copies of such a core motif, which constitute a functional hormone response element [3]. The spacing of the two motifs and their relative orientation determine which receptors bind to a given hormone response element [3].
Peroxisome proliferator-activated receptor response element (PPRE) is defined as a direct repeat of two core recognition motifs AGGTCA spaced by one nucleotide [3]. PPAR interacts with the upstream extended core hexamer of the PPRE [3]. The fact that some tissues express more than one PPAR isotype suggests the presence of PPAR isotype-specific PPRE recognition [3]. PPREs are classified into three functional groups: strong, intermediate, and weak elements. The PPAR DNA binding activity is modulated by, among other factors, the isotype of the 9-cis retinoid X receptor (RXR) heterodimeric partner [3]. Heterodimerization with RXRγ leads to binding to a strong element, while heterodimerization with RXRα favors binding to weak elements [3].
Ligand-binding properties of PPARs
Compounds that trigger the expression of a reporter gene, when added to a cell culture medium, have been identified as PPAR activators. Studies revealed that these compounds selectively bind to various PPAR isotypes (Figure 1 and Table 1). It has been demonstrated that the prostaglandin 15-deoxy--prostaglandin J2 and the antidiabetic thiazolidinediones are ligands for the PPARγ [17, 18, 19]. Additionally, leukotriene B4 and several known peroxisome proliferating agents including Wy-14,643 and fibrates bind specifically to PPAR α [20, 21]. Fatty acids and eicosanoids also bind to PPARα, with varying degrees of specificity [17, 22, 23]. A novel series of fibrates has recently been described as specific ligands for the PPAR δ subtype [4].
BIOLOGICAL ROLES OF PEROXISOME PROLIFERATOR-ACTIVATED RECEPTORS
Although the focus of this review is on the involvement of PPARs in inflammatory diseases, it is important that we present a brief overview of the plethora of other functions controlled by these receptors. A PPAR subtype-specific role has been discovered in several important human diseases and pathological conditions [2, 4, 10, 19]. These receptors have been implicated in aging [24, 25], aging-related diseases [25, 26], inflammation and immunity [2, 10, 27, 28, 29], as well as in obesity [30, 31, 32, 33] and responsiveness to insulin [34]. Furthermore, PPARs have been reported to play a significant role in the regulation of fertility [2, 10, 35], and in cancer [2, 10, 36, 37, 38, 39].
Aging
Senescence is associated with a decline in peroxisomal enzyme activities [24, 25]. Our most recent results suggest that peroxisomal decline, as a function of aging, may be the result of a decline in the availability of the obligatory PPARα heterodimer partner RXR α, and not PPAR α itself in senescent animals [40]. The importance of this aging-associated peroxisomal enzyme deficiency lies in the fact that very-long-chain fatty acids are metabolized exclusively in peroxisomes [41]. Thus, PPARs appear to play an important role in maintaining membrane structure and function by regulating fatty acid balance [42, 43, 44].
Obesity and diabetes
There are three variants of PPAR γ; γ1, γ2, and γ3 [3]. Adipose expression of PPAR γ2 mRNA increases in obese human subjects, with a strong positive correlation between the ratio of PPAR γ2 to γ1 and the body mass index [30]. Interestingly, differential expression of PPAR γ in intra-abdominal and subcutaneous adipose tissues in humans may underscore the association of visceral, but not subcutaneous, fat with obesity [31].
The recognition that PPAR γ is the receptor for the antidiabetic thiazolidinediones linked this receptor to glucose homeostasis in rodents and humans [21, 34]. There appears to be an inverse relationship between the level of PPAR γ mRNA and that of nonfunctional insulin receptors in human visceral adipose tissue [31]. PPAR γ ligands improve insulin sensitivity in vivo [32].
Cell cycle control and cancer
A link between PPAR and cancer was first drawn after it became clear that PPAR agonists cause a dramatic increase in the incidence of liver tumors in mice and rats [24, 45]. Cell proliferation and peroxisomal production of H2O2 are two major factors enhanced by peroxisome proliferators and are implicated in liver cancer caused by these chemicals [36, 37]. Alternatively, several studies have shown that agonists of PPARs suppress apoptosis in the liver in a process mediated by PPAR α [46, 47]. Apoptosis appears to be a safeguard to prevent cells with DNA damage from progressing to a tumor [38], where hepatocytes resistant to apoptotic death may represent preferential targets for promotion by PPAR agonists.
The potential role of PPAR in cancer formation in humans is controversial [48]. While two recent studies implicated PPAR γ in promotion and development of colon cancer [31, 39], a third report indicated a possible protective role for PPAR γ agonists against colon cancer in humans [37]. It is evident that mice genetically predisposed to develop polyps in the colon show an increased number of polyps when subjected to PPAR γ agonists orally [31, 39]. Conversely, it was demonstrated that human colon tumor cell lines both in culture and in nude mice respond to PPAR γ agonists with a reduced rate of growth and an increased degree of differentiation [49]. We recently proposed that PPARγ agonists may be beneficial in combating breast cancer in humans [50]. Furthermore, a recent report suggested that inhibiting PPAR δ may be responsible for reducing the incidence of colorectal cancer caused by nonsteroidal anti-inflammatory drugs [51]. Confirmation of the involvement of PPAR δ in colorectal cancer awaits more definitive studies using compounds specific to this PPAR subtype [4].
Fertility
PPAR δ was detected in the uterus during embryo implantation in mice [35]. Similar to the expression pattern of the prostacyclin synthase enzyme, expression of PPAR δ was also induced in the stroma surrounding the implanting blastocyte and became localized in the decidual zone after implantation [35]. Supporting evidence for a PPAR δ-mediated role in fertility was obtained when activators of this PPAR subtype restored implantation in COX2 null mice [35]. This finding is in support of the hypothesis stipulating that prostacyclin and/or its metabolite(s) regulate embryo implantation by activating PPAR δ [4, 46]. PPAR γ also appears to play an important role in embryonic development [10]. Mice deficient in this PPAR subtype showed embryonic lethality, presumably a result of a major default in placental development [10]. PPAR α, however, does not appear to have a vital role in development [10].
MODULATION OF INFLAMMATORY RESPONSES BY AGONISTS OF PPARs
Inflammation
Inflammation is a series of characteristic tissue responses to injury or insult. Elevated tissue levels of tumor necrosis factor alpha (TNFα), Interleukin-1 (IL-1), and Interleukin-6 (IL-6), among other proinflammatory mediators, have been observed in experimental animal models of inflammation as well as in humans following trauma, severe blood loss, or sepsis. Onset of release of these mediators, and their tissue half-life vary significantly, based on the noxious stimuli to which tissue has been exposed [14]. Because of the impact of inflammation, acute or chronic, on the quality of life, intense research is directed toward the discovery of novel therapeutic agents to prevent/reverse these processes. Among the most promising compounds currently pursued toward achieving this goal are the PPAR agonists [4, 52].
Role of PPARs in inflammation
Both PPARα and PPARγ receptor subtypes have been reported to regulate inflammatory responses, both in vivo and in vitro [53, 54]. However, the extent of this regulation, and indeed its direction, are controversial. No published reports are readily available on the involvement of PPARδ in inflammation control. Potential modulation of inflammatory responses by PPARδ agonists has only been recently investigated in our laboratories.
PPARα
The first indication of a role by PPAR in modulating inflammation was evidenced by the demonstration that Leukotriene B4 (LTB4), a potent chemotactic inflammatory eicosanoid [55], binds to PPARα and induces transcription of genes of the ω- and β-oxidation pathways that can catabolize LTB4 itself [20]. PPAR α null mice showed a prolonged inflammatory response when challenged with LTB4 or its precursor, arachidonic acid, possibly due to the absence of stimulation of the catabolic pathways, hence, the increased duration of the inflammation [20]. Furthermore, dietary n-3 fatty acids and clofibrate, which also bind to PPARα, have been reported to accelerate catabolism of LTB4 in granulocytes and macrophages [56, 57]. It is postulated that activation of PPARα by nonsteroidal anti-inflammatory agents contributes to the anti-inflammatory, antipyretic, and analgesic properties of these drugs through stimulation of oxidative pathways involved in the catabolism of eicosanoids [3]. Inhibition of the synthesis of proinflammatory molecules such as IL-6 and prostaglandins also appears to participate in PPARα-mediated control of inflammation, via a decreased activity of NF-κB [58]. Conversely, dietary treatment with PPARα agonists increased lipopolysaccharide-induced plasma TNFα levels, an effect that was significantly diminished in PPARα-deficient mice [59]. These latter results suggest a proinflammatory role of PPARα.
The postulate that PPARα receptor activation enhances the degradation of lipid-derived proinflammatory mediators, such as LTB4 [20], is not supported by recent findings in our laboratories, using the carrageenan-induced rat paw edema model of inflammation [60]. First, we observed that the PPARα agonist perfluorooctanoic acid (PFOA) produced robust anti-edema effects when administered minutes before carrageenan [60], yet de novo synthesis of enzymes requires a much longer time period, and is therefore expected to take hours following the administration of PPAR agonists. Second, when a sufficient time for de novo enzyme synthesis was allowed by administering PFOA 12 or 24 hours before carrageenan, we did not observe an enhanced anti-edema effect compared to PFOA administered only 30 minutes before carrageenan [60]. Consequently, we speculate that PFOA inhibits the levels of proinflammatory mediators released during the induction of inflammation (Figures 2, 3, and 4).
Figure 2.

Site-specific inhibition of carrageenan-induced paw inflammation by PPAR agonists. The hatched bar indicates the site at which PPAR agonists interfere with the inflammatory process.
Figure 3.
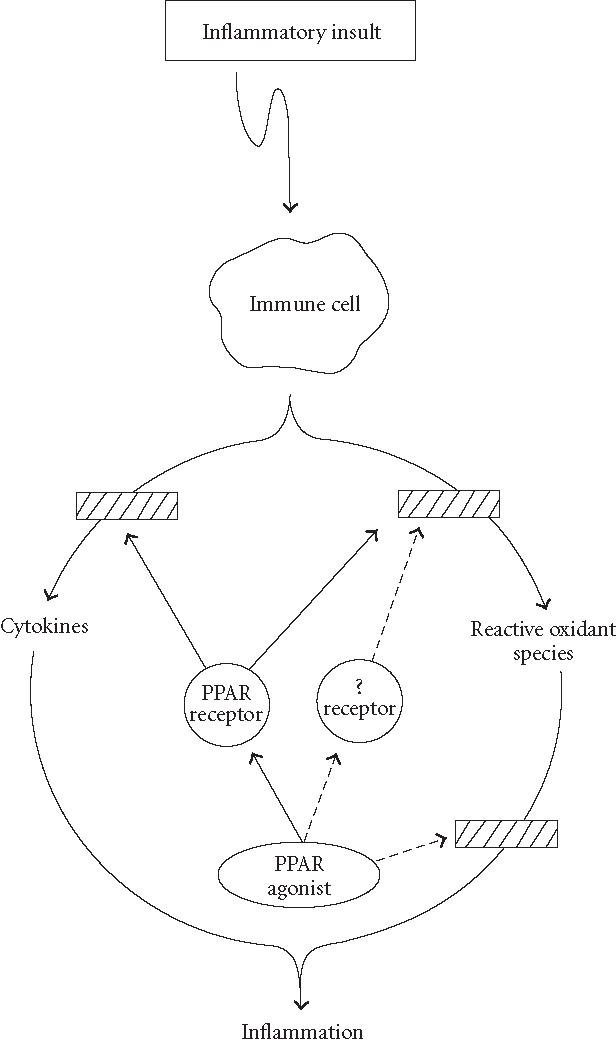
A scheme depicting potential mechanisms via which anti-inflammatory effects are exerted by PPAR agonists. Solid lines represent potential PPAR-mediated effects. Dashed lines represent proposed mechanisms by which PPAR agonists may act via PPAR-independent mechanisms to diminish inflammation.
Figure 4.
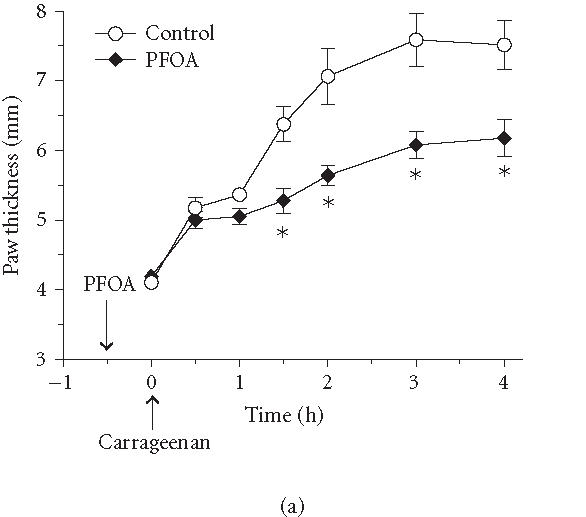

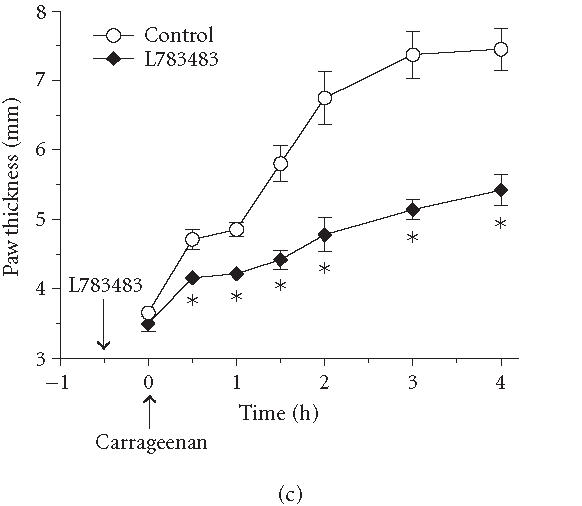
Effect of various PPAR agonists on the temporal profile of paw edema. Rats were treated intraperitoneally with (100 mg/kg) of the PPARα agonist PFOA (a), the PPARγ agonist rosiglitazone (b), or the PPARδ agonist L783484 (c) 30 minutes before induction of inflammation. As indicated by the arrow, carrageenan (1%, intraplantar) was injected at . Carrageenan-induced edema is reflected by an increase in paw thickness. Values represent mean ± SEM, rats per group; .
PPARγ
Recent studies demonstrate that in addition to PPAR α, PPAR γ may also play an important role in inflammation. However, the contribution of PPARγ to the inflammatory response is also unclear [53]. While several studies showed that PPARγ activation blocked the production of proinflammatory mediators [61, 62, 63, 64], other studies proposed that PPARγ has no anti-inflammatory activity, or might indeed exert a pro-inflammatory response [53, 65, 66].
Reduction of nitric oxide (NO) production by PPAR γ agonists
NO acts not only as a signal molecule mediating various physiological functions, but it also plays an important role in inflammatory processes [67, 68]. Injection of LPS and IFN-γ into rat cerebellum induced the expression of iNOS, which produces NO, in cerebellar granule cells and caused subsequent cell death [69]. In this model, PPARγ agonists reduced iNOS expression and cell death, whereas a selective COX-2 inhibitor had no effect [69]. Furthermore, mesangial cell production of NO was inhibited by PPARγ agonists [70]. These findings suggest that PPARγ regulates the activity of iNOS and activation of this PPAR subtype controls inflammation by diminishing NO production.
Regulation of cytokine production by PPARγ
Cytokines produced by activated macrophages/foam cells, including the macrophage-colony stimulating factor, IL-1, and TNFα, form the basis of the inflammatory component of the atherosclerotic lesions and promote proliferation of smooth muscle cells [3]. Necrosis of macrophages and lipid-loaded foam cells releases their intracellular contents, resulting in an accumulation of extracellular components that form the fibrous cap of the atheromatous lesion [57]. Eventually, the rupture of this plaque leads to the acute arterial obstruction [3]. Many aspects of these pathological processes might be modulated by PPAR γ which is upregulated by oxidized LDL [27, 65]. Furthermore, expression of PPAR γ has indeed been demonstrated in mouse and human atherosclerotic lesions [27, 28]. In contrast to this apparently proatherosclerotic action of PPAR γ ligands, these agonists have been reported to prevent atherosclerotic plaque progression [28]. Further studies are needed to determine the exact role of PPAR γ in the development of atherosclerosis.
Our studies revealed a positive relationship between anti-edema activity of PPARγ agonists in vivo [60] and their ability to activate PPARγ in vitro [4]. Thus, the high affinity PPARγ agonist rosiglitazone, but not the low affinity agonist troglitazone, significantly inhibited paw edema [60]. This suggests that, like the PPARα receptor, activation of the PPARγ receptor leads to anti-inflammatory effects in vivo. Also, as with the PPARα agonists, rosiglitazone was effective only when given prior to, but not after, carrageenan. Therefore, PPARγ also appears to regulate the induction phase of inflammation (Figures 2, 3, and 4).
Role of PPARγ in neurodegenerative and autoimmune diseases
Release of inflammatory mediators has been postulated to play a major role in the etiology of a variety of aging-related neuronal degenerative diseases, such as Alzheimer's disease (AD) [71]. The role of microglial-mediated inflammatory mechanisms in the etiology of AD has achieved prominence owing to recent compelling epidemiological and investigative findings [71]. Epidemiological studies have shown a reduced risk of AD among long-term users of nonsteroidal anti-inflammatory drugs (NSAIDs). The formation of amyloid plaques in AD is accompanied by the recruitment of microglia to these deposits [71]. The interaction of these cells with amyloid fibrils leads to their phenotypic conversion into a reactive phenotype [71]. The activation of microglia results in the elaboration of a diverse array of pro-inflammatory secretory products including cytokines, chemokines, reactive oxygen species, and nitrogen species, as well as of other acute phase proteins [71].
Previously, the anti-inflammatory actions of NSAIDs and their therapeutic benefit in treating AD were attributed to the ability of these drugs to inhibit the cyclooxygenases and PGE2 production [72]. Based on the findings showing that PPARγ ligands prevented the increase in Aβ-stimulated COX-2 expression in microglia and monocytes [71], it was determined that the neuroprotective effect of NSAIDs and PPARγ ligands was not attributable to a reduction in cyclooxygenase activity [71]. This conclusion is supported by the fact that the COX-2-specific inhibitor NS-398 failed to promote neuron survival [71]. Furthermore, extended use of aspirin, a potent COX inhibitor, is not associated with a reduction in the risk of AD [72]. Consequently, it was concluded that microglial COX-2 activity and prostaglandin production are not necessary components in the neuronal death process [71], and that the beneficial effects of NSAIDs in AD are attributable principally to the actions of these drugs as PPAR agonists, rather than via their ability to inhibit cyclooxygenase activity [71].
Several studies have also investigated the role of PPARγ ligands in modifying animal models of autoimmune diseases. In a mouse model of inflammatory bowel disease, thiazolidinediones markedly reduced colonic inflammation [73]. It was consequently proposed that this effect might be a result of a direct effect on colonic epithelial cells, which express high levels of PPARγ and can produce inflammatory cytokines [73]. PPARγ ligands, 15d-PGJ2 and troglitazone, ameliorated adjuvant-induced arthritis with suppression of pannus formation and monouclear cell infiltration in rats [74]. Niino et al [75] examined the effect of a thiazolidinedione on experimental allergic encephalomyelitis and found that these PPARγ agonists attenuated inflammation and decreased clinical symptoms in this mouse model of multiple sclerosis.
PPARδ
No published reports are available on the impact of PPARδ agonists on inflammation. This may, in part, be due to the fact that PPARδ-specific ligands are not readily, commercially available. Preliminary data from our laboratory show that L783483 diminished carrageenan-induced paw edema in rats (Figure 4). Although this compound has PPARγ activity in addition to being a PPARδ agonist [13], the fact that it showed 2-fold efficacy in reducing paw edema compared to the potent PPARγ agonist rosiglitazone (Figure 4) suggests that PPARδ may also play a role in controlling inflammatory responses. It remains to be investigated, however, whether L783483 modulates paw edema if administered after the induction of inflammation by carrageenan, as was the case with agonists of both PPARα and PPARγ (Figure 2).
The development of edema in the rat paw following the intraplantar injection of carrageenan has been described as a biphasic event (see Table 2) [14, 15, 16, 76]. In response to carrageenan-induced paw edema, it has been documented that while cNOS appears to be involved in the early phase of inflammation, iNOS participates in the sustained phase [15, 16]. Early in the inflammatory response to carrageenan, while infilterating neutrophils are not the source of iNOS-derived NO [15, 16], these cells appear to contributeO2− to the early phase of inflammation [15, 16]. Therefore, our findings [60] suggest that anti-inflammatory effects of PPAR agonist are likely due to the ability of these chemicals to interfere with cNOS, but not with iNOS. Given the above studies, the relevance of PPARs and the utility of treatment with PPAR agonists in diseases with inflammatory or autoimmune pathogenesis will likely continue to remain a research focus.
Table 2.
Carrageenan-evoked proinflammatory mediators.
PPAR-independent anti-inflammatory effects of PPAR agonists
Several recent studies report that agonists of both PPARα and PPARγ produce effects that are not mediated by PPARs. However, the nature of these PPAR-independent mechanisms is not understood at this time.
PPARα agonists
In a most recent report [77], treatment of splenocytes with the PPARα agonist Wy-14,643 as well as by other fibrates led to marked increases in Interleukin-4 (IL-4) release. Surprisingly, however, Wy-14,643 also induced IL-4 expression in splenocytes from PPARα knockout mice [77]. Furthermore, GW 7,647, a potent and specific PPARα ligand, did not augment IL-4 under conditions used with Wy-14,643 [77]. These findings suggest that modulating inflammatory responses exerted by Wy-14,643 and by other fibrates in lymphocytes may not be mediated exclusively through PPARα-dependent mechanisms [77].
PPARγ agonists
Numerous studies present evidence for receptor-independent anti-inflammatory effects caused by PPARγ agonists [69, 70, 73, 75]. A study showed that the PPARγ agonist troglitazone enhanced IL-1-induced nitric oxide synthase (NOS) mRNA levels in vascular smooth muscle cells despite the absence of detectable PPARγ levels in these cells [64]. However, these effects were in contrast to those obtained in response to 15-deoxy- prostaglandin J2, the natural ligand for PPARγ [78]. Another study [79] reports that both 15-deoxy- prostaglandin J2 and the thiazolidinedione drugs have anti-inflammatory effects that are independent of PPARγ. Furthermore, in activated microglia, while 15-deoxy- prostaglandin J2 suppressed iNOS promotor activity and decreased its mRNA and protein levels, troglitazone, a specific PPARγ ligand, failed to produce similar effects [80]. These findings further support the conclusion that anti-inflammatory action exerted by 15-deoxy- prostaglandin J2 may involve sites other than PPARγ [80]. Indeed, it has been shown that the two structurally similar thiazolidinediones troglitazone and rosiglitazone cause different conformational changes in the PPARγ, upon binding to this receptor [81]. Further, troglitazone may behave as a partial agonist under certain physiological circumstances, and as a full agonist under others [81].
The exact mechanisms involved in these non-PPAR-mediated effects of PPAR agonists are not well understood. However, it is possible that these agonists act via receptors other than PPARs to affect changes in the synthesis and/or release of pro-inflammatory mediators (Figure 3). Alternatively, PPAR agonists may act via a nonreceptor-mediated mechanism such as antioxidants or free-radical scavengers (Figure 3). The latter potential is supported by experimental findings showing that administering exogenous antioxidants, for example, vitamin E-like compounds, or elements of the endogenous cellular antioxidant defense mechanisms, for example, superoxide dismutase, exhibited protective effects against inflammatory insults [82].
SUMMARY AND CONCLUSIONS
Demonstrating that PPAR agonists diminished inflammatory responses in several experimental models of inflammation has led to a surge in interest in these agonists as potential therapeutic agents to treat inflammatory diseases. Although various postulates have been advanced in an attempt to explain the mode of action of these compounds as anti-inflammatory agents, the exact mechanism by which they act remains elusive. Our recent results strongly suggest that agonists of PPARα and PPARγ interfere with the early phase of inflammation, without influencing its late phase. An unpublished finding from our laboratories shows that activation of PPARδ also elicits an anti-inflammatory response. It is not yet clear, however, whether these three PPAR receptor subtypes share a common mechanism or act independently to control inflammatory processes.
FUTURE DIRECTIONS
The PPAR-independent anti-inflammatory effects of PPAR agonists warrant investigation to localize potential non-PPAR site(s) of action of these compounds. A vast amount of circumstantial evidence implicates oxygen-derived free radicals (especially superoxide and hydroxyl radical) and high-energy oxidants (such as peroxynitrite) as mediators of inflammation [16]. Production of reactive oxygen species (ROS) such as O2−, H2O2, and HO. occurs at the site of inflammation and contributes to tissue damage [16]. Interventions which reduce the generation, and/or the effects of ROS exert beneficial effects in a variety of models of inflammation [83]. Since superoxide dismutase (SOD), but not catalase, the iron-chelator desferrioxamine or serine protease inhibitor, has been shown to attenuate neutrophil-mediated cell injury [84], it is strongly suggested that O2− plays a major role in neutrophil-induced inflammation. Interestingly, SOD mimetics are superior to SOD in attenuating neutrophil-mediated cell injury [84, 85], possibly because of their superior intracellular accessibility and also due to the fact that these compounds, in contrast to SOD, do not react with H2O2, a reaction which is known to result in the formation of HO. via Fenton chemistry [85].
In addition to ROS, it has also been reported that NO. plays a role in the pathophysiology associated with various models of inflammation [15, 86, 87]. In addition to NO., ONOO− is also generated during inflammation [15]. In arthritis, nitrotyrosine levels increase in plasma and synovial fluid [88]. In ileitis [89] and in endotoxin-induced intestinal inflammation, there is immunocytochemical documentation of augmented ONOO− production [90]. The pathophysiological role of NO. and ONOO− in the gastrointestinal damage elicited by endotoxin or chronic inflammation has been the subject of a variety of detailed investigations [91]. The ability of ONOO− to cause severe colonic inflammation has been documented [91], and the production of ONOO− in colitis may be even more pronounced because of the parallel down-regulation of SOD [92], which makes the O2− available for coupling with NO. [92]. It is reported that PPAR agonists decrease NOS activity and the production of NO in experimental inflammation models [29, 62]. However, the question of whether agonists of PPARs act as SOD mimetics, or via other mechanisms, to neutralize ROS deserves an answer and it is currently being planned to investigate this exciting potential.
ACKNOWLEDGMENT
Studies in our laboratories are supported by the National Institutes of Health Grants CA 74384 and AG 18479. Rosiglitazone and L783483 were kind gifts from GlaxoSmithKline and Merck, respectively.
References
- 1.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 2.Youssef J, Badr M. Peroxisome proliferator-activated receptors: from orphnage to fame in a decade. Saudi Pharm J. 2002;9:1–13. [Google Scholar]
- 3.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20(5):649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 4.Willson T, Brown P, Sternbach D, Henke B. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43(4):527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 5.Sher T, Yi H, McBride O, Gonzalez F. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry. 1993;32(21):5598–5604. doi: 10.1021/bi00072a015. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee R, Jow L, Noonan D, McDonnell D. Human and rat peroxisome proliferator activated receptors (PPARs) demonstrate similar tissue distribution but different responsiveness to PPAR activators. J Steroid Biochem Mol Biol. 1994;51(3-4):157–166. doi: 10.1016/0960-0760(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 7.Greene M, Blumberg B, McBride O, et al. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995;4(4-5):281–299. [PMC free article] [PubMed] [Google Scholar]
- 8.Su J.L, Simmons C, Wisely B, Ellis B, Winegar D. Monitoring of PPAR alpha protein expression in human tissue by the use of PPAR alpha-specific MAbs. Hybridoma. 1998;17(1):47–53. doi: 10.1089/hyb.1998.17.47. [DOI] [PubMed] [Google Scholar]
- 9.Mueller E, Sarraf P, Tontonoz P, et al. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell. 1998;1(3):465–470. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- 10.Michalik L, Wahli W. Peroxisome proliferator-activated receptors: three isotypes for a multitude of functions. Curr Opin Biotechnol. 1999;10(6):564–570. doi: 10.1016/s0958-1669(99)00030-0. [DOI] [PubMed] [Google Scholar]
- 11.Cullingford T, Bhakoo K, Peuchen S, Dolphin C, Patel R, Clark J. Distribution of mRNAs encoding the peroxisome proliferator-activated receptor alpha, beta, and gamma and the retinoid X receptor alpha, beta, and gamma in rat central nervous system. J Neurochem. 1998;70(4):1366–1375. doi: 10.1046/j.1471-4159.1998.70041366.x. [DOI] [PubMed] [Google Scholar]
- 12.Saluja I, Granneman J, Skoff R. PPAR delta agonists stimulate oligodendrocyte differentiation in tissue culture. Glia. 2001;33(3):191–204. [PubMed] [Google Scholar]
- 13.Berger J, Leibowitz M, Doebber T, et al. Novel peroxisome proliferator-activated receptor (PPAR) gamma and PPARdelta ligands produce distinct biological effects. J Biol Chem. 1999;274(10):6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- 14.Azab A, Kaplanski J. A reduction of tumor necrosis factor-alpha in paw exudate of lipopolysaccharide treated rats by nimesulide. Life Sci. 2001;68(14):1667–1675. doi: 10.1016/s0024-3205(01)00960-2. [DOI] [PubMed] [Google Scholar]
- 15.Salvemini D, Wang Z.Q, Wyatt P, et al. Nitric oxide: a key mediator in the early and late phase of carrageenan-induced rat paw inflammation. Br J Pharmacol. 1996;118(4):829–838. doi: 10.1111/j.1476-5381.1996.tb15475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salvemini D, Wang Z.Q, Bourdon D.M, Stern M.K, Currie M.G, Manning P.T. Evidence of peroxynitrite involvement in the carrageenan-induced rat paw edema. Eur J Pharmacol. 1996;303(3):217–220. doi: 10.1016/0014-2999(96)00140-9. [DOI] [PubMed] [Google Scholar]
- 17.Kliewer S, Sundseth S, Jones S, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci USA. 1997;94(9):4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tontonoz P, Singer S, Forman B, et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc Natl Acad Sci USA. 1997;94(1):237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kersten S, Wahli W. Peroxisome proliferator activated receptor agonists. EXS. 2000;89:141–151. doi: 10.1007/978-3-0348-8393-1_9. [DOI] [PubMed] [Google Scholar]
- 20.Devchand P, Keller H, Peters J, Vazquez M, Gonzalez F, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384(6604):39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- 21.Forman B, Chen J, Evans R. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA. 1997;94(9):4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf G. Fatty acids bind directly to and activate peroxisome proliferator-activated receptors alpha and gamma. Nutr Rev. 1998;56(2):61–63. doi: 10.1111/j.1753-4887.1998.tb01693.x. [DOI] [PubMed] [Google Scholar]
- 23.Gustafsson J.A. Fatty acids in control of gene expression. Nutr Rev. 1998;56(2):20–21. doi: 10.1111/j.1753-4887.1998.tb01682.x. [DOI] [PubMed] [Google Scholar]
- 24.Youssef J, Badr M. Biology of senescent liver peroxisomes: role in hepatocellular aging and disease. Environ Health Perspect. 1999;107(10):791–797. doi: 10.1289/ehp.99107791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masters C, Crane D. On the role of the peroxisome in ontogeny, ageing and degenerative disease. Mech Ageing Dev. 1995;80(2):69–83. doi: 10.1016/0047-6374(94)01563-2. [DOI] [PubMed] [Google Scholar]
- 26.Youssef J, Badr M. Peroxisomal alterations in aging and disease. In: Mattson M, editor. Interorganellar Signaling in Age-Related Disease. Amsterdam: Elsevier Press; 2001. pp. 1–28. [Google Scholar]
- 27.Ricote M, Huang J, Fajas L, et al. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1998;95(13):7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marx N, Sukhova G, Murphy C, Libby P, Plutzky J. Macrophages in human atheroma contain PPARgamma: differentiation-dependent peroxisomal proliferator-activated receptor gamma (PPARgamma) expression and reduction of MMP-9 activity through PPARgamma activation in mononuclear phagocytes in vitro. Am J Pathol. 1998;153(1):17–23. doi: 10.1016/s0002-9440(10)65540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spiegelman B.M. PPARgamma in monocytes: less pain, any gain? Cell. 1998;93(2):153–155. doi: 10.1016/s0092-8674(00)81567-6. [DOI] [PubMed] [Google Scholar]
- 30.Vidal-Puig A, Considine R, Jimenez-Linan M, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest. 1997;99(10):2416–2422. doi: 10.1172/JCI119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefebvre A.M, Laville M, Vega N, et al. Depot-specific differences in adipose tissue gene expression in lean and obese subjects. Diabetes. 1998;47(1):98–103. doi: 10.2337/diab.47.1.98. [DOI] [PubMed] [Google Scholar]
- 32.Spiegelman B, Flier J. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87(3):377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 33.Brun R, Tontonoz P, Forman B, et al. Differential activation of adipogenesis by multiple PPAR isoforms. Genes Dev. 1996;10(8):974–984. doi: 10.1101/gad.10.8.974. [DOI] [PubMed] [Google Scholar]
- 34.Kliewer S.A, Lenhard J.M, Willson T.M, Patel I, Morris D.C, Lehmann J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83(5):813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 35.Lim H, Gupta R, Ma W.G, et al. Cyclo-oxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARδ. Genes Dev. 1999;13(12):1561–1574. doi: 10.1101/gad.13.12.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cattley R, DeLuca J, Elcombe C, et al. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regul Toxicol Pharmacol. 1998;27(1):47–60. doi: 10.1006/rtph.1997.1163. [DOI] [PubMed] [Google Scholar]
- 37.Karam W.G, Ghanayem B.I. Induction of replicative DNA synthesis and PPAR alpha-dependent gene transcription by Wy-14 643 in primary rat hepatocyte and non-parenchymal cell co-cultures. Carcinogenesis. 1997;18(11):2077–2083. doi: 10.1093/carcin/18.11.2077. [DOI] [PubMed] [Google Scholar]
- 38.Youssef J, Badr M. Enhanced hepatocarcinogenicity due to agonists of peroxisome proliferator-activated receptors in senescent rats: role of peroxisome proliferation, cell proliferation, and apoptosis. ScientificWorldJournal. 2002;2:1491–1500. doi: 10.1100/tsw.2002.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saez E, Tontonoz P, Nelson M, et al. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med. 1998;4(9):1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 40.Chao C, Youssef J, Rezaiekhaleigh M, Birnbaum L, Badr M. Senescence-associated decline in hepatic peroxisomal enzyme activities corresponds with diminished levels of retinoid X receptor alpha, but not peroxisome proliferator-activated receptor alpha. Mech Ageing Dev. 2002;123(11):1469–1476. doi: 10.1016/s0047-6374(02)00086-6. [DOI] [PubMed] [Google Scholar]
- 41.Jakobs B.S, Wanders R.J. Conclusive evidence that very-long-chain fatty acids are oxidized exclusively in peroxisomes in human skin fibroblasts. Biochem Biophys Res Commun. 1991;178(3):842–847. doi: 10.1016/0006-291x(91)90967-c. [DOI] [PubMed] [Google Scholar]
- 42.Zs-Nagy I. The role of membrane structure and function in cellular aging: a review. Mech Ageing Dev. 1979;9(3-4):237–346. doi: 10.1016/0047-6374(79)90102-7. [DOI] [PubMed] [Google Scholar]
- 43.Perichon R, Bourre J.M. Aging-related decrease in liver peroxisomal fatty acid oxidation in control and clofibrate-treated mice. A biochemical study and mechanistic approach. Mech Ageing Dev. 1996;87(2):115–126. doi: 10.1016/0047-6374(96)01705-8. [DOI] [PubMed] [Google Scholar]
- 44.Roth G, Joseph J, Mason R. Membrane alterations as causes of impaired signal transduction in Alzheimer's disease and aging. Trends Neurosci. 1995;18(5):203–206. doi: 10.1016/0166-2236(95)93902-a. [DOI] [PubMed] [Google Scholar]
- 45.Bentley P, Calder I, Elcombe C, Grasso P, Stringer D, Wiegand H.J. Hepatic peroxisome proliferation in rodents and its significance for humans. Food Chem Toxicol. 1993;31(11):857–907. doi: 10.1016/0278-6915(93)90225-n. [DOI] [PubMed] [Google Scholar]
- 46.Roberts R, James N, Woodyatt N, Macdonald N, Tugwood J. Evidence for the suppression of apoptosis by the peroxisome proliferator activated receptor alpha (PPAR alpha) Carcinogenesis. 1998;19(1):43–48. doi: 10.1093/carcin/19.1.43. [DOI] [PubMed] [Google Scholar]
- 47.Bayly A.C, Roberts R.A, Dive C. Suppression of liver cell apoptosis in vitro by the non-genotoxic hepatocarcinogen and peroxisome proliferator nafenopin. J Cell Biol. 1994;125(1):197–203. doi: 10.1083/jcb.125.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seed B. PPARgamma and colorectal carcinoma: conflicts in a nuclear family. Nat Med. 1998;4(9):1004–1005. doi: 10.1038/1990. [DOI] [PubMed] [Google Scholar]
- 49.Sarraf P, Mueller E, Jones D, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med. 1998;4(9):1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- 50.Badawi A, Badr M. Chemoprevention of breast cancer by targeting cyclooxygenase-2 and peroxisome proliferator-activated receptor-gamma (Review) Int J Oncol. 2002;20(6):1109–1122. [PubMed] [Google Scholar]
- 51.He T.C, Chan T, Vogelstein B, Kinzler K. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99(3):335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berger J, Moller D. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–935. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 53.Delerive P, Fruchart J.C, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169(3):453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 54.Chinetti G, Fruchart J.C, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 2000;49(10):497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- 55.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387(6633):620–624. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]
- 56.von Schacky C, Kiefl R, Marcus A, Broekman M, Kaminski W. Dietary n-3 fatty acids accelerate catabolism of leukotriene B4 in human granulocytes. Biochim Biophys Acta. 1993;1166(1):20–24. doi: 10.1016/0005-2760(93)90278-h. [DOI] [PubMed] [Google Scholar]
- 57.Couve A, Koenig C, Santos M. Induction of peroxisomal enzymes and a 64-kDa peptide in cultured mouse macrophages treated with clofibrate. Exp Cell Res. 1992;202(2):541–544. doi: 10.1016/0014-4827(92)90109-l. [DOI] [PubMed] [Google Scholar]
- 58.Poynter M, Daynes R. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273(49):32833–32841. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- 59.Hill M.R, Clarke S, Rodgers K, et al. Effect of peroxisome proliferator-activated receptor alpha activators on tumor necrosis factor expression in mice during endotoxemia. Infect Immun. 1999;67(7):3488–3493. doi: 10.1128/iai.67.7.3488-3493.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor B, Dadia N, Yang C, Krishnan S, Badr M. Peroxisome proliferator-activated receptor agonists inhibit inflammatory edema and hyperalgesia. Inflammation. 2002;26(3):121–127. doi: 10.1023/a:1015500531113. [DOI] [PubMed] [Google Scholar]
- 61.Jiang C, Ting A.T, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391(6662):82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 62.Ricote M, Li A, Willson T, Kelly C.J, Glass C. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391(6662):79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 63.Cuzzocrea S, Wayman N, Mazzon E, et al. The cyclopentenone prostaglandin 15-deoxy-Delta(12,14)-prostaglandin J(2) attenuates the development of acute and chronic inflammation. Mol Pharmacol. 2002;61(5):997–1007. doi: 10.1124/mol.61.5.997. [DOI] [PubMed] [Google Scholar]
- 64.Ricote M, Huang J.T, Welch J.S, Glass C.K. The peroxisome proliferator-activated receptor (PPARgamma) as a regulator of monocyte/macrophage function. J Leukoc Biol. 1999;66(5):733–739. doi: 10.1002/jlb.66.5.733. [DOI] [PubMed] [Google Scholar]
- 65.Tontonoz P, Nagy L, Alvarez J.G, Thomazy V.A, Evans R.M. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93(2):241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 66.Thieringer R, Fenyk-Melody J.E, Le Grand C.B, et al. Activation of peroxisome proliferator-activated receptor gamma does not inhibit IL-6 or TNF-alpha responses of macrophages to lipopolysaccharide in vitro or in vivo. J Immunol. 2000;164(2):1046–1054. doi: 10.4049/jimmunol.164.2.1046. [DOI] [PubMed] [Google Scholar]
- 67.Clark R. The role of PPARs in inflammation and immunity. J Leukoc Biol. 2002;71(3):388–400. [PubMed] [Google Scholar]
- 68.Stamler J. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78(6):931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 69.Heneka M, Klockgether T, Feinstein D. Peroxisome proliferator-activated receptor-gamma ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J Neurosci. 2000;20(18):6862–6867. doi: 10.1523/JNEUROSCI.20-18-06862.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reilly C, Oates J, Cook J, Morrow J, Halushka P, Gilkeson G. Inhibition of mesangial cell nitric oxide in MRL/lpr mice by prostaglandin J2 and proliferator activation receptor-gamma agonists. J Immunol. 2000;164(3):1498–1504. doi: 10.4049/jimmunol.164.3.1498. [DOI] [PubMed] [Google Scholar]
- 71.Combs C, Johnson D, Karlo J, Cannady S, Landreth G. Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20(2):558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaufmann W, Andreasson K, Isakson P, Worley P. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54(3):601–624. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- 73.Su C, Wen X, Bailey S, et al. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest. 1999;104(4):383–389. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kawahito Y, Kondo M, Tsubouchi Y, et al. 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106(2):189–197. doi: 10.1172/JCI9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niino M, Iwabuchi K, Kikuchi S, et al. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J Neuroimmunol. 2001;116(1):40–48. doi: 10.1016/s0165-5728(01)00285-5. [DOI] [PubMed] [Google Scholar]
- 76.Vinegar R, Schreiber W, Hugo R. Biphasic development of carrageenin edema in rats. J Pharmacol Exp Ther. 1969;166(1):96–103. [PubMed] [Google Scholar]
- 77.Cunard R, Ricote M, DiCampli D, et al. Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J Immunol. 2002;168(6):2795–2802. doi: 10.4049/jimmunol.168.6.2795. [DOI] [PubMed] [Google Scholar]
- 78.Hattori Y, Hattori S, Kasai K. Troglitazone upregulates nitric oxide synthesis in vascular smooth muscle cells. Hypertension. 1999;33(4):943–948. doi: 10.1161/01.hyp.33.4.943. [DOI] [PubMed] [Google Scholar]
- 79.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans R.M. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7(1):48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 80.Petrova T.V, Akama K.T, Van Eldik L.J. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2. Proc Natl Acad Sci USA. 1999;96(8):4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Camp H, Li O, Wise S. Differential activation of peroxisome proliferator-activated receptor-gamma by troglitazone and rosiglitazone. Diabetes. 2000;49(4):539–347. doi: 10.2337/diabetes.49.4.539. [DOI] [PubMed] [Google Scholar]
- 82.Cuzzocrea S, Costantino G, Mazzon E, Caputi A.P. Beneficial effects of raxofelast (IRFI 016), a new hydrophilic vitamin E-like antioxidant, in carrageenan-induced pleurisy. Br J Pharmacol. 1999;126(2):407–414. doi: 10.1038/sj.bjp.0702275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salvemini D, Wang Z.Q, Stern M.K, Currie M.G, Misko T.P. Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc Natl Acad Sci USA. 1998;95(5):2659–2663. doi: 10.1073/pnas.95.5.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cuzzocrea S, Riley D.P, Caputi A.P, Salvemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001;53(1):135–159. [PubMed] [Google Scholar]
- 85.Cuzzocrea S, Zingarelli B, Costantino G, Caputi A.P. Beneficial effects of Mn(III)tetrakis (4-benzoic acid) porphyrin (MnTBAP), a superoxide dismutase mimetic, in carrageenan-induced pleurisy. Free Radic Biol Med. 1999;26(1-2):25–33. doi: 10.1016/s0891-5849(98)00142-7. [DOI] [PubMed] [Google Scholar]
- 86.Tracey W.R, Nakane M, Kuk J, et al. The nitric oxide synthase inhibitor, L-NG-monomethylarginine, reduces carrageenan-induced pleurisy in the rat. J Pharmacol Exp Ther. 1995;273(3):1295–1299. [PubMed] [Google Scholar]
- 87.Wei X.Q, Charles I.G, Smith A, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375(6530):408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 88.Kaur H, Halliwell B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation. Nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett. 1994;350(1):9–12. doi: 10.1016/0014-5793(94)00722-5. [DOI] [PubMed] [Google Scholar]
- 89.Miller M.J, Thompson J.H, Zhang X.J, et al. Role of inducible nitric oxide synthase expression and peroxynitrite formation in guinea pig ileitis. Gastroenterology. 1995;109(5):1475–1483. doi: 10.1016/0016-5085(95)90633-9. [DOI] [PubMed] [Google Scholar]
- 90.Chamulitrat W, Skrepnik N.V, Spitzer J.J. Endotoxin-induced oxidative stress in the rat small intestine: role of nitric oxide. Shock. 1996;5(3):217–222. doi: 10.1097/00024382-199603000-00009. [DOI] [PubMed] [Google Scholar]
- 91.Rachmilewitz D, Stamler J.S, Karmeli F, et al. Peroxynitrite-induced rat colitis—a new model of colonic inflammation. Gastroenterology. 1993;105(6):1681–1688. doi: 10.1016/0016-5085(93)91063-n. [DOI] [PubMed] [Google Scholar]
- 92.Seo H.G, Takata I, Nakamura M, et al. Induction of nitric oxide synthase and concomitant suppression of superoxide dismutases in experimental colitis in rats. Arch Biochem Biophys. 1995;324(1):41–47. doi: 10.1006/abbi.1995.9932. [DOI] [PubMed] [Google Scholar]