

Abstract
Neuronal necrosis occurs during early phase of ischemic insult. However, our knowledge of neuronal necrosis is still inadequate. To study the mechanism of neuronal necrosis, we previously established a Drosophila genetic model of neuronal necrosis by calcium overloading through expression of a constitutively opened cation channel mutant. Here, we performed further genetic screens and identified a suppressor of neuronal necrosis, CG17259, which encodes a seryl-tRNA synthetase. We found that loss-of-function (LOF) CG17259 activated eIF2α phosphorylation and subsequent up-regulation of chaperons (Hsp26 and Hsp27) and autophagy. Genetically, down-regulation of eIF2α phosphorylation, Hsp26/Hsp27 or autophagy reduced the protective effect of LOF CG17259, indicating they function downstream of CG17259. The protective effect of these protein degradation pathways indicated activation of a toxic protein during neuronal necrosis. Our data indicated that p53 was likely one such protein, because p53 was accumulated in the necrotic neurons and down-regulation of p53 rescued necrosis. In the SH-SY5Y human cells, tunicamycin (TM), a PERK activator, promoted transcription of hsp27; and necrosis induced by glutamate could be rescued by TM, associated with reduced p53 accumulation. In an ischemic stroke model in rats, p53 protein was also increased, and TM treatment could reduce the p53 accumulation and brain damage.
Introduction
As the second most prevalent disease worldwide, brain ischemia causes massive damage to the cells in the brain. However, there is still much progress needs to be made for neuroprotection in clinics. Cell death in brain ischemia is primarily induced by glutamate through the excessive excitation of NMDA receptors, which leads to the dysregulation of calcium homeostasis1, 2. In treatments of ischemic stroke, prevention of calcium entry by antagonizing the NMDA receptors has been intensively investigated. However, several NMDA receptor antagonists have failed in clinical trials, likely due to their interference with normal functions of the NMDA receptors3. Therefore, identifying key regulators downstream of calcium entry may be desirable for drug targeting.
In response to an insult, both survival and death signals are activated and their balance determines cell fate4. For neuronal cell death, different insults may activate either apoptotic or necrotic pathways, and these pathways may positively and negatively crosstalk with each other5, 6. For instance, as apoptotic initiators, caspases may have anti-necrotic function through the degradation of the AMPA receptors7. Conversely, as a necrotic trigger, calpain can cleave caspase-38 and apoptosis regulatory proteins, such as apoptosis protease-activating factor-1, Bcl-xL, Bax, Bid and p535. Due to this complexity, identifying key targets that regulate both apoptosis and necrosis is important.
Previously, we have generated a calcium-overload-induced necrosis model in Drosophila via neuron-specific expression of a constitutively opened cation channel, the glutamate receptor 1 Lurcher mutant (GluR1Lc)9. Here, we performed genetic screens and identified a new suppressor of neuronal necrosis, CG17259. We found loss-of-function (LOF) CG17259 induced up-regulate small chaperons (Hsp26 and Hsp27) and autophagy. These degradation pathways might coordinate to degrade p53, which was accumulated in necrotic neurons. In mammalian neurons under a necrotic stress or in a rat stroke model, tunicamycin had protective effect, likely through up-regulation of hsp27, similar to the flies. Together, our research provides the genetic evidence for the role of LOF CG17259 in neuronal necrosis, likely through activation of the eIF2α signaling, small chaperones and autophagy to degrade p53.
Results
Genetic screens identified a new suppressor of neuronal necrosis
Previously, we have established a genetic model of neuronal necrosis9. In this Drosophila model, neuronal necrosis was induced by the specific expression of a constitutively open glutamate receptor 1 channel (GluR1Lc) in neurons to overload calcium. The fly contains a neuron-specific promoter A ppl-Gal4, UAS- G luR1 Lc, and tub-Gal80 ts (simplified as “AG” flies). At 18 °C, the AG flies developed normally because Gal4 was suppressed by Gal80ts. When placed at 30 °C, the Gal80ts lost its function and permitted the expression of GluR1Lc, which resulted in calcium overload and neuronal necrosis9. We performed genetic screens using the deficiency lines that cover most of the Drosophila genome (the deficiency kit from Bloomington Drosophila Stock Center). By screening suppressors of the AG fly lethality, we identified nine deficiency lines. Here, the Df(2 L)ED206 line was further investigated (Fig. 1a). To narrow down the genes in the flanking region of Df(2 L)ED206, we tested P-element insertion-mediated mutants in this region and found that loss of CG17259 (CG17259 KG03126) strongly rescued the AG flies (Fig. 1a). By qRT-PCR, we confirmed that the transcript of CG17259 was reduced by approximately 40% in the heterozygous CG17259 mutant (CG17259 +/−) (Fig. S1). Because the homozygous mutant of CG17259 was embryonic lethal, we also tested a CG17259 RNAi line (CG17259 RNAi), its effect on the transcript of CG17259 was confirmed (Fig. S1). The CG17259 RNAi rescued AG lethality (Fig. 1a). At the cellular level, loss of CG17259 (CG17259 +/−) reduced neuronal necrosis in the larval ventral nerve cord, as inferred from propidium iodide (PI) staining (Fig. 1b). At the subcellular level, neurons from the brain of adult AG flies showed mitochondrial swollen (Fig. 1c red arrow) and vacuole formation (Fig. 1c blue arrow), these are typical features of necrosis10. While, CG17259 +/− showed rescue effect in the AG fly brains (Fig. 1c). Together, these results suggest that CG17259 is a suppressor of neuronal necrosis.
Figure 1.
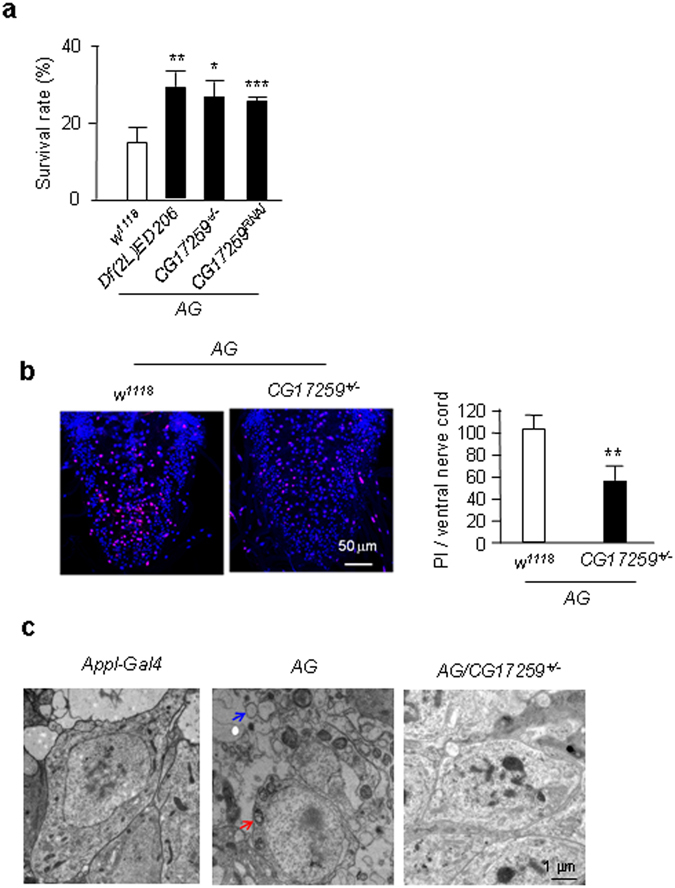
Deficiency screen indentifies a gene as a suppressor of neuronal necrosis. (a) Effect of LOF CG17259 on AG fly survival. Df(2 L)ED206 is a deletion mutant; CG17259 +/− represents the heterozygous mutant of CG17259 KG03126 and CG17259 RNAi represents the UAS-CG17259 RNAi fly. Trial n = 5. For all AG survival experiments, 60–150 flies were tested for each trial. Throughout all bar graphs, error bars are mean + standard deviation (s.d.); white bars represent control; gray bars represent no statistical difference; and black bars represent statistical significant difference from the control (ANOVA for group comparisons followed by post-hoc Tukey test; unpaired t-test for comparison of two data sets). Asterisk *for p < 0.05, **for p < 0.01 and ***for p < 0.001. (b) Neuronal necrosis in AG flies determined by Propidium Iodide (PI) staining. The larval ventral nerve cord was stained with PI, and the statistic result is shown on the bar graph. 10 larvae were examined for each genotype. (c) The subcellular characteristic of AG fly brain. Representative images from the transmission electron microscope are shown, with the genotype of flies indicated on the micrograph. Red arrow points to a swollen mitochondria; and blue arrow points to a vacuole.
CG17259 induced the eIF2α branch of unfolded protein response (UPR)
CG17259 encodes a seryl-tRNA synthetase, an essential enzyme, which catalyzes the ligation of serine to its cognate tRNA, and thereby affects the fundamental building blocks of protein synthesis11. In this respect, CG17259 +/− may increase errors in protein synthesis and induce UPR and ER stress. On the ER membrane, three distinct receptors are responsible to UPR, including inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase (PKR)-like ER kinase (PERK)12. The IRE1 branch can be quantified by increase of a spliced form of Xbp1 mRNA (Xbp1 sp)13. We found that full length Xbp1 (Xbp1 FL) was unaltered and the Xbp1 sp was undetectable in the CG17259 +/− flies (Fig. 2a), suggesting the IRE1 branch was not activated. Next, we tested the PERK branch, which had been shown to be independent from ER stress and can be detected by phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α)14, 15. Indeed, phosphorylated eIF2α level was increased in the CG17259 +/− flies (Fig. 2b). If ER stress was induced in the AG flies, protein synthesis of GluR1Lc might be reduced, however, the level of GluR1Lc was unaltered (Fig. 2c). In addition, severe ER stress may induce apoptosis through the activation of ASK116, which, in turn, activates TRAF2-dependent c-Jun N-terminal kinase (JNK) signaling17. Using an in vivo reporter of JNK activation in Drosophila 18, the JNK signaling was not activated (Fig. S2); and no cell death was detected in the larval eye discs of CG17259 +/− flies (Fig. S3). These results together suggest that no severe ER stress was induced in the CG17259 +/− flies. To test the functional importance of eIF2α phosphorylation, we studied GADD34, a regulatory subunit of protein phosphatase 1 (PP1) that dephosphorylates eIF2α19, 20. The result showed that the loss of Gadd34 (Gadd34 e02638) increased eIF2α phosphorylation and suppressed the lethality of AG flies (Fig. 2d and e). On the other hand, GOF of Gadd34 (Gadd34 G18907) had the opposite effect (Fig. 2f). Together, these results suggest that loss of CG17259 is likely to induce the PERK/ eIF2α signaling branch to suppress neuronal necrosis in the AG flies.
Figure 2.

Loss of CG17259 induced eIF2α phosphorylation. (a) The quantitative RT-PCR for Xbp1 FL is shown. The DNA gel with migrations of Xbp1 FL and Xbp1 sp are indicated. Xbp1 sp is not detected. Trial n = 8. (b) The Western blot shows the level of phosphorylated form of eIF2α (eIF2α-p) in the CG17259 mutant flies. The eIF2α-p antibody recognizes the S51 phosphorylation site of eIF2a. Actin is shown as the protein loading control. Trial n = 3. The full length gels of all Western blots were shown at the end of the supplementary information. (c) The Western blot shows the level of GluR1Lc in the genotype of flies indicated. Actin is shown as the protein loading control. Trial n = 3. (d) The Western blot shows the level of eIF2α-p in the Gadd34 mutant flies. Actin is shown as the protein loading control. Trial n = 3. (e) Survival rate of AG flies was shown with the genotype of flies indicated. Trial n = 4.
CG17259 upregulated small chaperones (Hsp26 and Hsp27) and autophagy
Phosphorylation of eIF2α may activate transcription of chaperones, which help to degrade misfolded proteins in proteasome or lysosome21. Indeed, we found that mRNA levels of Hsp26 and Hsp27 were elevated; while larger chaperons such as Hsp70 and Hsp83 were unchanged (Fig. 3a). To confirm the increase of Hsp26/Hsp27 at protein level, we generated rabbit polyclonal antibodies and found that both Hsp26 and Hsp27 protein levels were increased in CG17259 +/− flies (Fig. 3b). The heterozygous mutant of Hsp26 (Hsp26 −/+) and Hsp27 (Hsp27 −/+) were shown. These protein levels in the heterozygous mutant of Hsp26 −/+ and Hsp27 −/+ were much lower in the than in the wild type (w 1118) flies (Fig. 3b), and the transcription levels of Hsp26/Hsp27 in the heterozygous mutant of Hsp26 and Hsp27 were equally reduced (Fig. 3c). These results suggested that the antibodies were specific to detect the Hsp26 and Hsp27 proteins. Moreover, the immunostaining data showed that Hsp26 and Hsp27 were primarily localized in the cytosol/mitochondria and nucleus, respectively (Fig. S4). Functionally, GOF of either protein rescued AG fly lethality. In contrast, the LOF of the proteins increased lethality (Fig. 3d). These results indicate that Hsp26 and Hsp27 play a protective role against neuronal necrosis. To test whether Hsp26 and Hsp27 acted downstream of CG17259, we generated double-heterozygous mutants, CG17259 +/−; Hsp26 +/− and CG17259 +/−; Hsp27 +/−. The results showed that the protective effect of CG17259 +/− against AG lethality was abolished in flies with either the Hsp26 +/− or Hsp27 +/− mutant background (Fig. 3e), suggesting Hsp26 and Hsp27 function downstream of CG17259.
Figure 3.

Characterization of the small chaperones and autophagy. (a) Transcription level of Hsp26 and Hsp27 in the CG17259 mutant detected by qRT-PCR. The RNAs were collected from the fly heads. The relative mRNA level of Hsp26, Hsp27, Hsp70 and Hsp83 in the wild type fly (w 1118) was defined as 1. The means ± SD of relative transcripts. Trial n = 3. (b) The Western blot to detect the protein level of Hsp26 and Hsp27 in the CG17259 mutant flies. The heterozygous mutants of hsp26 and hsp27 serve as controls to indicate the specificity of the antibodies. Trial n = 3. (c) qPCR for Hsp26 and Hsp27. The heterozygous mutant of Hsp26−/+ and Hsp27−/+ lines contain a third chromosome balancer (short bristle) and they are homozygous lethal. The relative levels of transcript of Hsp26 and Hsp27 to the wild type (w 1118) are shown. Trial n = 3. (d) Survival of AG flies under the genetic background indicated. Trial n = 6. (e) Survival of AG flies under the genetic background indicated. Trial n = 5. (f) Autophagy activation determined by the in vivo reporter (mCherry-Atg8a, the red channel). In the fed condition, the fat bodies of wild type (w 1118) showed minimum activation of autophagy, but autophagy was activated under the starvation condition. This response serves as a positive control for autophagy activation. In the CG17259 mutant flies, autophagy was activated under the fed condition. Trial n = 2. (g) Survival of AG flies under different autophagy mutant backgrounds. The RNAi and mutant of the Atg genes (as indicated as LOF) all showed enhancing effect on lethality of AG flies; whereas up-regulation of several Atg genes (GOF) all showed a rescue effect on lethality of AG flies. Trial n = 5.
Downstream of eIF2α signaling may induce autophagy in various organisms, including Drosophila 12, 22. It has been reported that autophagy is activated in the CG17259 +/− flies 22. To replicate this result, we used an in vivo reporter of autophagy, mCherry-Atg8a, and observed that its fluorescent puncta were increased in CG17259 +/− flies (Fig. 3f). Similarly, this autophagy activation was detectable by a lysotracker staining (Fig. S5). Because role of autophagy on neuronal necrosis is unclear, we focused on the functional study in the AG flies. The result showed that targeting genes in the autophagy pathway by RNAi enhanced AG fly lethality; and inhibition of Atg1 activity by GOF of dTor 23, 24 had a similar effect (Fig. 3g). In contrast, GOF of several Atg genes rescued fly lethality (Fig. 3g). In addition, the protective effect of CG17259 +/− was abolished under the LOF background of Atg4 (Fig. 3g), suggesting CG17259 functions upstream of autophagy. These results together suggest that autophagy functions downstream of CG17259 +/−, and it is necessary for the protective role of LOF CG17259 against neuronal necrosis in Drosophila.
Down regulation of P53 reduced neuronal necrosis in Drosophila
The protective effect of small chaperons and autophagy suggests accumulation of misfolded proteins or damaged organelles in neuronal necrosis. To test whether massive amounts of misfolded proteins were generated in the AG flies, we preformed immunohistochemical staining and Western blotting to detect the global level of ubiquitinated proteins. Surprisingly, we observed no elevation of ubiquitinated proteins in AG fly heads (Figs S6 and S7). This result indicates that a specific protein may be accumulated in the necrotic neurons. Of note, phosphorylation of eIF2α inhibits p53 accumulation and apoptosis in mouse fibrosarcoma cells25. Moreover, by searching the database of protein interactions in Drosophila, both Hsp26 and Hsp27 interact directly with p5326, 27. To confirm these interactions, we conducted co-immunoprecipitation (co-IP) using the Hsp26 and Hsp27 antibodies. Because the wild type (Appl-Gal4) flies had very low level of p53, we were unable to detect p53 protein after IP with the anti-Hsp26 or anti-hsp27 (Fig. 4a). However, in the flies with neuron-specific overexpression of Hsp26 and Hsp27 (Appl > Hsp26 and Appl > Hsp27), both Hsp26 and Hsp27 antibodies could pull down p53 (Fig. 4a). This result suggests that Hsp26/Hsp27 may bind with p53.
Figure 4.

Characterization of p53 in the AG flies. (a) Co-immunoprecipitation of Hsp26/Hsp27 with p53. The homogenized fly heads were immunoprecipitated with anti-Hsp26 or anti-Hsp27 antibodies and probed with anti-Hsp26, anti-Hsp27, anti-p53 antibody, with anti-IgG as a control. The input of actin is the protein loading control. The genotypes of flies are indicated on the image. Trial n = 2. (b) Accumulation of p53 in the AG adult flies. The control (AG flies at 18 °C) p53 protein level is set as 1, and the indicated genotype flies relative to the control are shown. Trial n = 3. (c) Accumulation of p53 in the larval AG brain. The images show immunostaining with anti-p53 and anti-GluR1 antibodies. The nuclei are stained with DAPI. The result showed that the 53 protein (the red channel) was lower in the control (Appl-Gal4) ventral nerve cord of larval flies; whereas p53 protein level was higher in the AG flies, especially in neurons with higher GluR1 expression (the green channel). Trial n = 2. (d) Effect of p53 on AG fly survival. The survival rate of indicated genotype of flies is shown. Trial n = 4.
In the AG fly brains, the p53 protein was increased after induction of neuronal necrosis (Fig. 4b); and p53 protein tended to accumulate in neurons with higher GluR1Lc expression in the larval ventral nerve cord (Fig. 4c). These results suggest that p53 protein level was altered in the necrotic neurons.
Functionally, overexpression of p53 enhanced the AG fly lethality and LOF p53 (p531 is a LOF mutant of p53) rescued the AG fly (Fig. 4d). The double mutants of CG17259 and p53 showed no additive effect (Fig. 4d), suggesting they may function in the same pathway. Whereas p53 1 reduced the lethality of AG/dTor flies (Fig. 4d), indicating that inhibition of autophagy enhanced the p53 toxicity. This result is consistent with a report suggesting that degradation of p53 depends on the autophagy pathway in cancer cells28. Together, these results suggest that p53 may play a functional role in neuronal necrosis.
Down regulation of p53 reduced necrosis in mammalian neurons
To test the effect of activation PERK/eIF2α signaling in mammalian neurons, mouse cortical neurons were pretreated with tunicamycin (TM), an inhibitor of N-linked glycosylation, known to activate the PERK/eIF2α signaling29. In primary neuron cultures, TM treatment showed a protective effect against necrosis induced by glutamate (Fig. S8). However, p53 protein level was too low to quantify by Western blot analysis in the primary neuron cultures. Therefore, we turned to SH-SY5Y cells, a human neuroblastoma cell line. In these cells, p53 protein was detectable, and glutamate treatment resulted in an increase of p53 protein (Fig. 5a and b). Importantly, TM pretreatment resulted in a transcriptional activation of Hsp27, which is the homolog of Drosophila Hsp26/hsp27 (Fig. S9). Furthermore, TM treatment protected against the glutamate toxicity (Fig. 5a and b), and reduced p53 accumulation (Figs. 5c and d). In addition, TM pretreatment enhanced autophagy because the marker of autophagosomes in mammalian cells, LC3-II30, was increased (Fig. 5c). Mdm2 is a key E3 ligase for ubiquitination and proteasomal degradation of p5331. However, the Mdm2 protein level was unaltered upon TM treatment (Fig. 5c), suggesting that the main degradation pathway for p53 was likely through autophagy in these cells. Consistent with this notion, enhanced autophagy by rapamycin reduced the level of p53 protein (Fig. S10). On the other hand, inhibition of autophagy by 3-MA resulted in the accumulation of p53 protein (Fig. S11). Together, these results suggest that TM treatment mimics the LOF CG17259.
Figure 5.

Effect of ER stress on neuronal necrosis and stroke. (a) Effect of TM on glutamate-induced cell death in SH-SY5Y cells. The cell death was determined by PI staining. The statistic result is shown on the bar graph. Trial n = 4. (b) Effect of TM on glutamate-induced cell death in SH-SY5Y cells. The cell death was determined by the ATP assay. Trial n = 4. (c) Protein level of p53 and the active state of autophagy. The result showed that TM treatment reduced p53 accumulation upon glutamate treatment and autophagy was activated as shown by the increased LC3-II level. The Mdm2 protein level shows no change. Trail n = 3. (d) Immunofluorescent staining of p53 in SH-SY5Y cells. DAPI labels DNA. The bar graph shows the percentages of p53 positive cells from 3 independent experiments. (e) Effect of TM (dissolved in DMSO) on the rat MCAO model. Injection of DMSO is the sham control. The white areas of TTC-stained images show the levels of brain damage. The quantitative data is shown in the bar graph. Five rats were tested for each condition. (f) Protein level of p53 detected by Western blot under conditions indicated. Trial n = 3.
To test the potential protective effect of TM against p53 accumulation in vivo, we examined an intraluminal middle cerebral artery occlusion (MCAO) model in rats, in which neuronal necrosis is the predominant form of cell death, especially in the ischemic core32. Six hours after intraperitoneal injection of TM (0.4 mg/kg), we performed permanent focal occlusion32, 33. The infarction volume was assessed by triphenyl-tetrazolium chloride (TTC) staining in the coronally-sectioned brain slices. The defective brain area was not stained by TTC and had a white color34. Our results demonstrated that TM treatment significantly reduced infarct volume (Fig. 5e). Moreover, p53 accumulated after stroke, whereas TM pretreatment reduced the increase in p53 accumulation (Fig. 5f). This result suggests that reducing p53 by eIF2α signaling pathway may be a novel strategy for preconditioning neurons against ischemic insult.
In conclusion, loss of CG17259 may induce the eIF2α signaling pathway, which further activates the chaperon and autophagy pathways. Necrotic stress results in accumulation of p53, which is recognized and degraded by Hsp26/Hsp27 and the autophagy pathway. A graphic summary is shown (Fig. 6).
Figure 6.

Model of CG17259 function in neuronal necrosis. The model of CG17259 functions against neuronal necrosis. Loss of CG17259 may induce eIF2α signaling pathway, and further activates the chaperon and autophagy pathways. The chaperons (Hsp26/Hsp27) capture the accumulated p53 generated from necrotic stress and degrade them in the autophagy pathway.
Discussion
Activation of eIF2α signaling plays an important role in the protection of neurons
By genetic screens using AG fly lethality, we identified a novel suppressor of neuronal necrosis, LOF CG17259. CG17259 encodes a seryl-tRNA synthetase and functions in ligation of serine to its cognate tRNA. Therefore, LOF CG17259 may affect protein synthesis and induce cytoplasmic protein folding defects and/or ER stress35. ER stress initiates through three distinct sensors in the ER membrane, including PERK, ATF6 and IRE112. Each signaling branch has both overlapping and distinct functions. For example, PERK phosphorylates eIF2α to reduce overall protein translation and promote cell survival36. Whereas the IRE1 branch reduces protein synthesis by promoting the degradation of mRNA and activates JNK, which may, in turn, induce apoptosis37. Our data demonstrated that the IRE1 branch was not activated in LOF CG17259, because transcription of Xbp1 sp and JNK pathway were not activated. In contrast, and the PERK/eIF2α branch was up-regulated in the LOF CG17259 flies. Consistent with our data, activation of the PERK/eIF2α signaling branch has been implicated in the treatment of various neurodegenerative diseases. For instance, treatment with salubrinal, an inhibitor of eIF2α dephosphorylation, can rescue neurodegeneration in α-synuclein transgenic mice or ischemic stroke in rats38, 39. Further, we found that autophagy was activated in LOF CG17259. The coupling of the PERK/eIF2α signaling branch with autophagy has been well documented to protect neurons40. Our research is consistent with these results from the literature. Additionally, our research provides an additional mechanism by which the eIF2α signaling pathway affects neuron survival.
Small chaperones regulate p53 protein abundance
Our results showed that the rescue effect of CG17259 −/+ was abolished by the mutants of Hsp26/Hsp27, and overexpression of Hsp26 or Hsp27 was sufficient to rescue AG flies, suggesting Hsp26/Hsp27 are down stream of LOF CG17259. The small chaperones of Drosophila Hsp26/Hsp27 are likely to have a similar function to that of mammalian Hsp27, which is known to protect neurons under various pathological conditions, including ischemic stroke41. The protective mechanisms of Hsp27 may involve the suppression of the formation of actin aggregates, activation of the NF-κB pathway, or direct inhibition of components in the apoptotic machinery42–45. The mammalian Hsp27 may share the combined function of Drosophila Hsp26/Hsp27, because it localizes in both cytosol and nucleus upon phosphorylation; while, it mainly localizes in the nucleus upon dephosphorylation45. Our data showed that the Drosophila Hsp26 and Hsp27 distributed in cytosol or nucleus, respectively. For functional study, our data suggests that Hsp26/Hsp27 and p53 may function in the same pathway, because the rescue effect of p53 1 and CG17259 −/+ was not additive and Hsp26/Hsp27 protein could pull down p53. Although the co-IP data was obtained under the Hsp26/Hsp27 overexpression condition, the interaction between Hsp26/Hsp27 and p53 has been reported by other studies26, 27.
Autophagy regulates neuronal necrosis likely through degradation of p53
The autophagy pathways can be further classified into autophagy (in this text macroautophagy refers to autophagy) and chaperone-mediated autophagy (CMA). Autophagy requires the formation of autophagosomes and the function of Atg genes. In contrast, the CMA pathway degrades proteins in lysosomes and does not require Atg genes46. Our data suggested that autophagy was activated in the LOF CG17259 flies; up-regulation of autophagy rescued the AG lethality and down-regulation of autophagy had the opposite effect. Because LOF p53 rescued the enhancing death effect of LOF autophagy, it is possible that degradation of accumulated p53 was dependent on autophagy in the AG flies. Consistent with our data, the increase in the level of p53 protein has been observed in embryonic fibroblasts in Atg7 −/− or Atg5 −/− mice47.
Accumulation of p53 plays a key role in neuronal necrosis; and p53 may function at the convergent code for both neuronal apoptosis and necrosis
Function of p53 in apoptosis has been well documented48–50. Upregulation of p53 has been linked to neuronal cell death in numerous models of injuries and diseases51, including excitotoxicity52. The absence of p53 protects neurons from a wide variety of toxic insults, including focal ischemia53, ionizing radiation54 and MPTP-induced neurotoxicity55. In response to various types of stress, p53 promotes apoptosis through either transactivation of specific target genes50 or transcription-independent pathways49. As a transcription factor, p53 upregulates proapoptotic genes, such as Bax, Noxa and PUMA56. In addition, p53 can interact with Bcl2 family proteins, such as Bax and Bak, to induce permeabilization of the outer mitochondrial membrane57, 58. Whether p53 is involved in neuronal necrosis is unclear. In support of its involvement in necrosis, p53 may physically interact with cyclophilin D (CypD), a component of the mitochondrial permeability transition pores and trigger the opening of the pores and necrosis59. In addition, the formation of the p53-CypD complex occurs during brain ischemia/reperfusion insult59. Here, we provide the genetic and cell biology evidence indicating that p53 is involved in neuronal necrosis. In SH-SY5Y cells, we showed that p53 was accumulated upon cells treated with glutamate; and this accumulation was prohibited by TM treatment, which enhanced Hsp27 transcription. Similarly, the increased level of p53 in MCAO rat brain was down-regulated by TM treatment. Together, these results indicate conserved function of p53 in neuronal necrosis. In fact, protective effect of TM against neurodegeneration has been widely reported60–62. The difference is that our study evaluated potential down-stream function of TM to degrade p53 in neuronal necrosis. How does p53 trigger both apoptosis and necrosis? We propose that mild p53 accumulation likely induces apoptosis, whereas the additional accumulation of p53 promotes necrosis. This hypothesis requires further investigation however.
Potential clinical applications
The inhibition of p53 transcriptional activity by pifithrin α or its mitochondrial targeting by pifithrin μ protects the brain in rodent models of stroke63–65. However, p53 also benefits animal survival under hypoxic conditions66. Thus, administration of pifithrins may interfere with the normal function of p53 and thereby produce side effects. An alternative way to target p53 may be to aim to reduce the accumulation of p53. Our research suggests that the promotion of eIF2α signaling may activate endogenous mechanisms (activation of small chaperones and autophagy) to degrade p53.
Methods
Fly maintenance and stocks
Flies were raised on standard sucrose/cornmeal medium at constant 18 °C or 25 °C with 12 hours light and 12 hours dark cycle. For CG17259 RNAi lines, transgenic lines were obtained from the w 1118 background using the SympUAST-w vector67 to target −100 to −1 in the 5′-UTR from the translational start site of CG17259. The other RNAi (TRiP) lines were obtained from the Tsinghua Drosophila stock center (Beijing, China). All other fly lines were obtained from The Bloomington Drosophila Stock Center (Bloomington, IN, USA).
Genetic screening
The AG flies were crossed with deficient lines or point mutations or P-element-based mutations or RNAi lines obtained from the Bloomington Drosophila Stock Center, and raised at 18°C. The 3-day-old progeny flies were incubated at 30°C for 13 h, then transferred back to 18°C and after 2 days, their survival rate was recorded.
Antibodies
The following antibodies were used in this study (W: Western blot; IF: immunofluorescence; IP: immunoprecipitation): p53 (sc-6243,W), actin (transgen, HC-201, W); p53 (DSHB, 25F4C, IP and W); eIF2αph (ab32157, W); Hsp26 (our own lab, Rabbit polyclonal, IF, IP and W); Hsp27(our own lab, Rabbit polyclonal, IF, IP and W); p53 (CST, 1C12#2524, W); LC3 (NB100-2331,W); Mdm2(GTX110608,W); p53(sc-126, W); ubiquitin (13-1600, Invitrogen, IF and W).
Transmission Electron Microscope
Heads was collected and fixed overnight on ice in 2.5% glutaraldehyde. The heads were then washed for 3 × 10 min in PB and postfixed on ice for 4 h in 1% osmium tetroxide. The samples were then washed at room temperature 3 × 10 min in H2O and dehydrated for 1 × 10 min in 25, 50, 70, 80, 90% acetone and 2 × 10 min in 100% acetone. The heads were then incubated for 3 × 5 min in propylene oxide and incubated overnight in a 1:1 mixture of propylene oxide and Spurr’s medium. The heads were then immersed in 100% Spurr’s for 2 × 2 h and baked in moulds at 60 °C for 24 h.
PCR primer sets to detect Xbp1FL and Xbp1sp
Xbp1 full Forward: GCAGGCGCTGAGGGCTGTGC
Reverse: GGACACACAGTCGTCAGCGCGTCT
Xbp1 splice Forward: GCAGGCGCTGAGGGCTGTGC
Reverse: AGACGCGCTGACGACTGTGTGTCC.
qRT-PCR
Total RNA was extracted by Trizol reagent (Invitrogen) followed by DNase I treatment using the manufacturer’s standard protocol, then the purity and integrity of total RNA was determined by 1% agarose gel electrophoresis. The concentration of total RNA was measured by Nanodrop. Five μg mRNA was reverse-transcribed into a cDNA library by oligo-dT primer using Revert Aid First Strand cDNA Synthesis Kit (Thermo scientific) based on the manufacturer’s instructions. For qPCR, the final volume of the q-PCR reaction was 25 μl using a Platinum SYBRGreen qPCR SuperMix-UDG Kit (Invitrogen) containing 1μl diluted cDNA sample (1:3). The qPCR was performed in triplicate using the 7500 real time PCR system (ABI). The quantification of target gene was conducted by ΔΔCt method68.
qPCR primer sets for Drosophila genes:
hsp26 Forward: GCTTTCGCTTGTGGATGAACT
Reverse: CCCAGTCCAAGCTCGTAGATG
hsp27 Forward: GGACGGCCATGGAATGATC
Reverse: GCCCTTGGGCAGGGTATACT
hsp70-4 Forward: CCAACCAGCTGGCTGACAA
Reverse: GCACACACCCTCCAGTTCCT
hsp83 Forward: CCGCAATCCCGATGATATCT
Reverse: GTCGTTGGTCAGGGATTTGTAGA
CG17259 Forward: TGACCTCCCCACACGACAA
Reverse: CCGCATTCCCGATCATCTC
GAPDH Forward: CGCAGCGCCATTCTCCTA
Reverse: GACTGCCGCTTTTTCCTTTTC
qPCR primer sets for human gene:
Hsp27 Forward: TGACCTCCCCACACGACAA
Reverse: CCGCATTCCCGATCATCTC.
Immunoprecipitation
The fly heads were homogenized and lysed in the lysis buffer (20 mM Tris-HCl, pH7.5, 100 mM NaCl, 0.5 mM EDTA, pH8.0, 0.5% NP40 and ‘Complete’ Protease Inhibitor Cocktail (Roche)). After centrifugation at 13,000 g for 10 min at 4 °C, the supernatant was incubated with Normal IgG antibody (mouse or rabbit, Invitrogen/Life Technologies) and protein A/G-agarose beads at 4 °C for 2 h. After centrifugation at 13,000 g for 10 min at 4 °C, the supernatant was incubated with the requisite antibody and protein A/G-agarose beads at 4 °C overnight. After washing with washing buffer (20 mM Tris-HCl, pH7.5, 500 mM NaCl, 0.5 mM EDTA, pH8.0, 0.5% NP40) 6 times, the agarose beads were boiled in 1 × SDS loading buffer.
Mammalian cell cultures
SH-SY5Y cells (purchased from Concorde basic medical institute, Beijing, China) were cultured in DMEM supplemented with 15% (v/v) fetal bovine serum and 1%(v/v) penicillin/streptomycin in a 37 °C incubator with 5% CO2. We tested for contamination every two months. Different concentrations of TM were used to pretreat these cells. Twenty-four hours later, glutamate (0.3 M) was applied to trigger PNN for four hours. Cell death was quantified by the ATP assay.
For neuron culture, cortical neurons from 18 day-after-fertilization embryos of mice were collected and cultured following a published protocol69. After 8–11 days in vitro culture, the neurons were treated with 200 μM glutamate for 4 h. Cell death was quantified by the LDH assay.
Rat ischemia models
All experiments were approved by the Institutional Animal Care and Use Committee, Peking University; and these experiments were performed in accordance with the relevant guidelines and regulations by the university. For the rat MCAO model, adult male Sprague-Dawley rats (250–270 g, Vital River, Beijing), were housed with a 12 h light and 12 h dark cycle. The number of rats per cage was 3 before surgery and they were raised separately after surgery. A permanent MCAO method was used as described previously70, with the suture-insertion through the left external carotid artery of anesthetized rats. A laser Doppler blood flow monitor was used to ensure successful occlusion (>80% drop from pre-stroke baseline). TM was injected, intraperitoneally, 6 h before the ischemia surgery. The infarct volume was determined 6 h later. The body temperature was maintained with a heated pad, and the infarct volumes were determined by TTC staining (2%, at 37 °C for 30 minutes) and analyzed using the Image J software.
Statistical analyses
One-way ANOVA test was used for group comparison, with Tukey’s post hoc test. Student-t test with 2-tailed pairing was used to compare two data sets. p < 0.05 is considered statically significant.
Electronic supplementary material
Acknowledgements
This work is supported by the Chinese Ministry of Science and Technology (2013CB530700) and National Natural Science Foundation of China (NSFC 91649201) to L.L.
Author Contributions
Y.Lei performed most of the experiments, data analysis and wrote the manuscript; K.L. performed part of the Drosophila genetic study; L.D., L.H. and Y.L. performed part of the cell culture study; L.L. supervised and wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05995-6
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. European journal of pharmacology. 2013;698:6–18. doi: 10.1016/j.ejphar.2012.10.032. [DOI] [PubMed] [Google Scholar]
- 2.Wojda U, Salinska E, Kuznicki J. Calcium ions in neuronal degeneration. IUBMB life. 2008;60:575–590. doi: 10.1002/iub.91. [DOI] [PubMed] [Google Scholar]
- 3.Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? The Lancet. Neurology. 2002;1:383–386. doi: 10.1016/S1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell death and differentiation. 2015;22:58–73. doi: 10.1038/cdd.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puyal J, Ginet V, Clarke PG. Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Progress in neurobiology. 2013;105:24–48. doi: 10.1016/j.pneurobio.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochimica et biophysica acta. 2013;1833:3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Glazner GW, Chan SL, Lu C, Mattson MP. Caspase-mediated degradation of AMPA receptor subunits: a mechanism for preventing excitotoxic necrosis and ensuring apoptosis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20:3641–3649. doi: 10.1523/JNEUROSCI.20-10-03641.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun M, Zhao Y, Xu C. Cross-talk between calpain and caspase-3 in penumbra and core during focal cerebral ischemia-reperfusion. Cellular and molecular neurobiology. 2008;28:71–85. doi: 10.1007/s10571-007-9250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu K, et al. Neuronal necrosis is regulated by a conserved chromatin-modifying cascade. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:13960–13965. doi: 10.1073/pnas.1413644111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Current opinion in cell biology. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Herzog W, Muller K, Huisken J, Stainier DY. Genetic evidence for a noncanonical function of seryl-tRNA synthetase in vascular development. Circulation research. 2009;104:1260–1266. doi: 10.1161/CIRCRESAHA.108.191718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cellular and molecular life sciences: CMLS. 2013;70:2425–2441. doi: 10.1007/s00018-012-1173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plongthongkum N, Kullawong N, Panyim S, Tirasophon W. Ire1 regulated XBP1 mRNA splicing is essential for the unfolded protein response (UPR) in Drosophila melanogaster. Biochemical and biophysical research communications. 2007;354:789–794. doi: 10.1016/j.bbrc.2007.01.056. [DOI] [PubMed] [Google Scholar]
- 14.Kim EJ, Lee YJ, Kang S, Lim YB. Ionizing radiation activates PERK/eIF2alpha/ATF4 signaling via ER stress-independent pathway in human vascular endothelial cells. International journal of radiation biology. 2014;90:306–312. doi: 10.3109/09553002.2014.886793. [DOI] [PubMed] [Google Scholar]
- 15.Rutkowski DT, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nature reviews. Drug discovery. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 17.Nishitoh H, et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Molecular cell. 1998;2:389–395. doi: 10.1016/S1097-2765(00)80283-X. [DOI] [PubMed] [Google Scholar]
- 18.Martin-Blanco E, et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes & development. 1998;12:557–570. doi: 10.1101/gad.12.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. The Journal of cell biology. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nature cell biology. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Y, Hendershot LM. ER chaperone functions during normal and stress conditions. Journal of chemical neuroanatomy. 2004;28:51–65. doi: 10.1016/j.jchemneu.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Arsham AM, Neufeld TP. A genetic screen in Drosophila reveals novel cytoprotective functions of the autophagy-lysosome pathway. PloS one. 2009;4:e6068. doi: 10.1371/journal.pone.0006068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. The Journal of biological chemistry. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 24.Kamada Y, et al. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. The Journal of cell biology. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SK, Kim YS. Phosphorylation of eIF2alpha attenuates statin-induced apoptosis by inhibiting the stabilization and translocation of p53 to the mitochondria. International journal of oncology. 2013;42:810–816. doi: 10.3892/ijo.2013.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhee DY, et al. Transcription factor networks in Drosophila melanogaster. Cell reports. 2014;8:2031–2043. doi: 10.1016/j.celrep.2014.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guruharsha KG, et al. A protein complex network of Drosophila melanogaster. Cell. 2011;147:690–703. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choudhury S, Kolukula VK, Preet A, Albanese C, Avantaggiati ML. Dissecting the pathways that destabilize mutant p53: the proteasome or autophagy? Cell cycle. 2013;12:1022–1029. doi: 10.4161/cc.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan W, et al. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15920–15925. doi: 10.1073/pnas.252341799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. The EMBO journal. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H, Leng RP. UBE4B, a ubiquitin chain assembly factor, is required for MDM2-mediated p53 polyubiquitination and degradation. Cell cycle. 2011;10:1912–1915. doi: 10.4161/cc.10.12.15882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke; a journal of cerebral circulation. 1989;20:84–91. doi: 10.1161/01.STR.20.1.84. [DOI] [PubMed] [Google Scholar]
- 34.Rupadevi M, Parasuraman S, Raveendran R. Protocol for middle cerebral artery occlusion by an intraluminal suture method. J Pharmacol Pharmacother. 2011;2:36–39. doi: 10.4103/0976-500X.77113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agashe VR, Hartl FU. Roles of molecular chaperones in cytoplasmic protein folding. Seminars in cell & developmental biology. 2000;11:15–25. doi: 10.1006/scdb.1999.0347. [DOI] [PubMed] [Google Scholar]
- 36.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular cell. 2000;5:897–904. doi: 10.1016/S1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 37.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 38.Colla E, et al. Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sokka AL, et al. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nature reviews. Neuroscience. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- 41.Stetler RA, Gao Y, Signore AP, Cao G, Chen J. HSP27: mechanisms of cellular protection against neuronal injury. Current molecular medicine. 2009;9:863–872. doi: 10.2174/156652409789105561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Concannon CG, Orrenius S, Samali A. Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene expression. 2001;9:195–201. doi: 10.3727/000000001783992605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garrido C, et al. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1999;13:2061–2070. doi: 10.1096/fasebj.13.14.2061. [DOI] [PubMed] [Google Scholar]
- 44.Pivovarova AV, Chebotareva NA, Chernik IS, Gusev NB, Levitsky DI. Small heat shock protein Hsp27 prevents heat-induced aggregation of F-actin by forming soluble complexes with denatured actin. The FEBS journal. 2007;274:5937–5948. doi: 10.1111/j.1742-4658.2007.06117.x. [DOI] [PubMed] [Google Scholar]
- 45.Lanneau D, Wettstein G, Bonniaud P, Garrido C. Heat shock proteins: cell protection through protein triage. TheScientificWorldJournal. 2010;10:1543–1552. doi: 10.1100/tsw.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Todde V, Veenhuis M, van der Klei IJ. Autophagy: principles and significance in health and disease. Biochimica et biophysica acta. 2009;1792:3–13. doi: 10.1016/j.bbadis.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 47.Lee IH, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225–228. doi: 10.1126/science.1218395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding HF, et al. Oncogene-dependent regulation of caspase activation by p53 protein in a cell-free system. The Journal of biological chemistry. 1998;273:28378–28383. doi: 10.1074/jbc.273.43.28378. [DOI] [PubMed] [Google Scholar]
- 49.Gottlieb E, Oren M. p53 facilitates pRb cleavage in IL-3-deprived cells: novel pro-apoptotic activity of p53. The EMBO journal. 1998;17:3587–3596. doi: 10.1093/emboj/17.13.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bates S, Vousden KH. p53 in signaling checkpoint arrest or apoptosis. Current opinion in genetics & development. 1996;6:12–18. doi: 10.1016/S0959-437X(96)90004-0. [DOI] [PubMed] [Google Scholar]
- 51.Morrison RS, Kinoshita Y. The role of p53 in neuronal cell death. Cell death and differentiation. 2000;7:868–879. doi: 10.1038/sj.cdd.4400741. [DOI] [PubMed] [Google Scholar]
- 52.Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA. p53-dependent cell death signaling in neurons. Neurochemical research. 2003;28:15–27. doi: 10.1023/A:1021687810103. [DOI] [PubMed] [Google Scholar]
- 53.Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1994;14:887–891. doi: 10.1038/jcbfm.1994.119. [DOI] [PubMed] [Google Scholar]
- 54.Wood KA, Youle RJ. The role of free radicals and p53 in neuron apoptosis in vivo. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1995;15:5851–5857. doi: 10.1523/JNEUROSCI.15-08-05851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trimmer PA, Smith TS, Jung AB, Bennett JP., Jr. Dopamine neurons from transgenic mice with a knockout of the p53 gene resist MPTP neurotoxicity. Neurodegeneration. 1996;5:233–239. doi: 10.1006/neur.1996.0031. [DOI] [PubMed] [Google Scholar]
- 56.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Molecular cell. 2001;7:683–694. doi: 10.1016/S1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 57.Mihara M, et al. p53 has a direct apoptogenic role at the mitochondria. Molecular cell. 2003;11:577–590. doi: 10.1016/S1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 58.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nature cell biology. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 59.Vaseva AV, et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang X, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy. 2014;10:1801–1813. doi: 10.4161/auto.32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fouillet A, et al. ER stress inhibits neuronal death by promoting autophagy. Autophagy. 2012;8:915–926. doi: 10.4161/auto.19716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hara H, Kamiya T, Adachi T. Endoplasmic reticulum stress inducers provide protection against 6-hydroxydopamine-induced cytotoxicity. Neurochemistry international. 2011;58:35–43. doi: 10.1016/j.neuint.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 63.Leker RR, Aharonowiz M, Greig NH, Ovadia H. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Experimental neurology. 2004;187:478–486. doi: 10.1016/j.expneurol.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 64.Culmsee C, et al. Reciprocal inhibition of p53 and nuclear factor-kappaB transcriptional activities determines cell survival or death in neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:8586–8595. doi: 10.1523/JNEUROSCI.23-24-08586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venna VR, et al. Inhibition of mitochondrial p53 abolishes the detrimental effects of social isolation on ischemic brain injury. Stroke; a journal of cerebral circulation. 2014;45:3101–3104. doi: 10.1161/STROKEAHA.114.006553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng X, Liu X, Zhang W, Xiao W. p53 directly suppresses BNIP3 expression to protect against hypoxia-induced cell death. The EMBO journal. 2011;30:3397–3415. doi: 10.1038/emboj.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giordano E, Rendina R, Peluso I, Furia M. RNAi triggered by symmetrically transcribed transgenes in Drosophila melanogaster. Genetics. 2002;160:637–648. doi: 10.1093/genetics/160.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bustin SA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 69.Dichter, M. A. Rat cortical neurons in cell culture: culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res149, 279–293, doi:0006-8993(78)90476-6 [pii] (1978). [DOI] [PubMed]
- 70.Zuo XL, Wu P, Ji AM. Nylon filament coated with paraffin for intraluminal permanent middle cerebral artery occlusion in rats. Neuroscience letters. 2012;519:42–46. doi: 10.1016/j.neulet.2012.05.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.