

Abstract
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder with genetic and clinical heterogeneity. The interplay of de novo and inherited rare variants has been suspected in the development of ASD. Here, we applied whole exome sequencing (WES) on 19 trios from singleton Saudi families with ASD. We developed an analysis pipeline that allows capturing both de novo and inherited rare variants predicted to be deleterious. A total of 47 unique rare variants were detected in 17 trios including 38 which are newly discovered. The majority were either autosomal recessive or X-linked. Our pipeline uncovered variants in 15 ASD-candidate genes, including 5 (GLT8D1, HTATSF1, OR6C65, ITIH6 and DDX26B) that have not been reported in any human condition. The remaining variants occurred in genes formerly associated with ASD or other neurological disorders. Examples include SUMF1, KDM5B and MXRA5 (Known-ASD genes), PRODH2 and KCTD21 (implicated in schizophrenia), as well as USP9X and SMS (implicated in intellectual disability). Consistent with expectation and previous studies, most of the genes implicated herein are enriched for biological processes pertaining to neuronal function. Our findings underscore the private and heterogeneous nature of the genetic architecture of ASD even in a population with high consanguinity rates.
Introduction
Delineating the genetic architecture of Autism Spectrum Disorder (ASD) is like finding the way in a labyrinth. Despite the rapid advancements in genetic methods, our ability to identify a common pathway for neuropsychiatric disorders with a therapeutic or diagnostic potential is lagging behind1. This is best exemplified by ASD, for which delivering a near complete genetic picture is hindered by many factors. Examples of such factors include: (i) extensive clinical and genetic heterogeneity (in excess of 600 genes have been implicated in ASD, thus far)2; (ii) the absence of generalizable genetic risk factors as most of the mutations are extremely rare or private in nature; (iii) the variability in diagnosis and data analysis methodology; and finally (iv) the precise function of many of ASD-candidate genes and therefore their possible impact on the central nervous system remains largely undetermined3, 4. All these factors present a substantial challenge, yet incremental progress has and continues to be made towards identifying ASD-risk loci.
Over the past 15 years, efforts were centered on interrogating the role of structural alterations through the study of chromosomal abnormalities and copy number variations (CNVs) in ASD individuals. Key advances have emerged from such studies, for instance, the identification of cytogenetic abnormalities at multiple loci with chromosomes 15q11–13 and 16p11.2 being the most frequent5, as well as recurrent CNVs involving genes like, NRXN1, SHANK3 and PARK2 6. However, a causal relationship is often hard to prove for a number of reasons: (i) such events normally span large regions making it difficult to discern the role of single genes in the disorder; (ii) apparently identical alterations have variable phenotypic outcomes or expressivity; (iii) some of these changes are observed in typically developing individuals; (iv) very low rate of replication, as recurrent individual CNVs were observed only in less than 1% of the cases7.
In the more recent years and with the advent of next-generation sequencing (NGS) technologies, the focus shifted towards investigating the role of inherited and de novo point mutations. This move was triggered by the observation that potentially risk-conferring structural alterations occurred only in (1–20%) of the cases7 and that they probably require other genetic or non-genetics factors for the development of the disease. The increasing availability of whole-genome and –exome sequencing platforms to researchers have accelerated the identification of additional risk factors in previously recognized as well as unrecognized ASD-associated genes8–18. However, most of these studies have focused on the role of either de novo or inherited variants3, 8, 9, 11–16, 19, 20 and much fewer were designed to interrogate both types10, 18, 21, 22. As there is a growing appreciation for the important part transmitted and de novo mutations play in ASD, more studies assessing both types of events within a given cohort are needed to understand their precise role.
In this study, we implemented whole exome sequencing (WES) in an attempt to identify risk genes/rare variants in a cohort of 19 case-parent trios from singleton families with ASD from Saudi Arabia. Enrichment for inherited causes are predicted in highly inbred populations17 as are sporadic (de novo) mutations in families with apparently “unaffected” parents8–10, 12, 13, 18, 20, 23, 24. We therefore, devised an analysis pipeline that enabled us to detect de novo as well as inherited variants in autosomal or X-linked genes (Fig. 1). Using this approach, rare variants were ascertained in 17 of the 19 trios (~90%).We found that most of the probands carried at least 2 unique rare variants occurring in 47 different genes. To the best of our knowledge 15 of the identified genes were not implicated before in ASD and the remaining were previously reported in ASD or other neuropsychiatric/neurodevelopmental disorders. Examples include PRODH2 and KCTD21 (implicated in schizophrenia), USP9X and SMS (implicated in intellectual disability), TRIM9 and KDM5B (known-ASD genes), and others such as AGL and MOGS (candidate genes involved in energy metabolism and immune responses, respectively).
Figure 1.
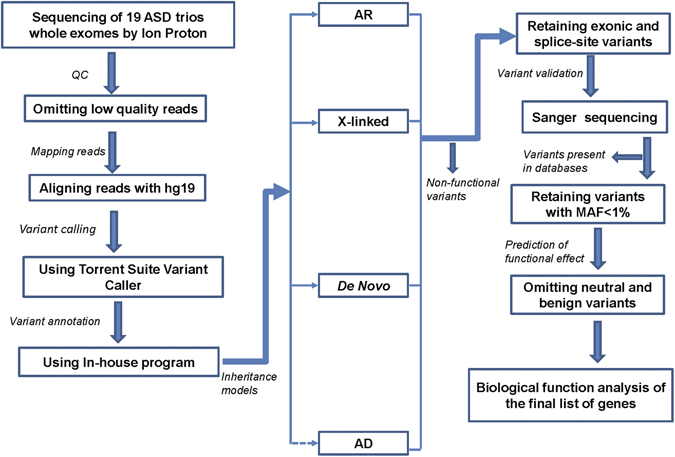
Schematic illustrating the steps of WES data analysis pipeline employed in this study. The multi-step analysis procedure involves eliminating low quality reads, variant mapping to human reference genome (hg19), variant calling and annotation, grouping variants according to inheritance models followed by variant prioritization and validation, and finally biological functions analysis was carried out on the final list of genes. AR, autosomal recessive; AD, autosomal dominant; QC, quality control. Dashed arrow indicates that the model was applied when all three main models failed to discover candidate variant(s).
Results
Sequencing and Alignment Quality
Supplementary Table S1 summarizes the quality of the sequencing and the read mapping steps. It shows that the sequencing achieved good coverage of the target regions (98% average total coverage at 1X and 95% average total coverage at 20X) with enough depth (223X; i.e. each base in the target region is covered by 223 reads on average). We detected an average of 29120.333 total variants comprised of 27466.807 Single Nucleotide Variants (SNVs) and 1653.526 indels per exome (Supplementary Table S2). After comparing the trios, a list of candidate variants was sent to Sanger sequencing for confirmation. The frequency of the confirmed variants in international databases (ExAC, 1000 Genomes Project) and the local the Saudi Human Genome Program Database (SHGP; 2379 local ethnically matching exomes) was also considered. We counted 29 false positives of which 20 were SNVs and 9 were indels as a result of low quality calls.
Reported familial relations history was assessed with three different relatedness tests (described in the methods). The results confirm correct parenthood for all our trios (Supplementary Table S3) as higher scores for parent-child pair were observed by all three methods. For our dataset, the results using the A jk statistics and shared homozygosity were more confirmative than the ones generated by KING program.
Detected SNVs and Indels
The analysis framework (described in methods) developed in this study allows for comprehensive investigation of the genetic architecture of ASD (Fig. 1). It is designed to capture not only de novo variants, but also those with autosomal or X-linked mode of transmission in unknown or previously described ASD-linked genes. This analysis approach is more suited for complex disorders with high genetic heterogeneity and it may offer much needed explanation for the increased ASD heritability15.
Using the 4 main inheritance models (refer to methods) a number of variants was detected in each sample except two (ASD-52 and ASD-55). From the variants that have passed all filtering steps, 44 were considered to be deleterious by at least one prediction tool and were confirmed as being true positives for presence of the variant by Sanger sequencing. The majority of the validated variants were missense, while the remaining were either frameshifts, splice-site changes or nonsense and only one small deletion was detected (Fig. 2A). As for the mode of transmission, 3 were de novo, 4 were autosomal dominant, 21 were found in X-linked genes, and the remaining 19 were inherited in an autosomal recessive manner (Fig. 2B). Only 9 of the verified variants were previously reported (in dbSNP with MAF < 1%) (MOGS, rs370842409; ITIH2, rs748626881; OR6C6, rs748626881; GPKOW, rs782015404, TRIM9, rs748897524; NEB rs375412223; BOC, rs752313669; SSTR3,rs577113986; PRODH2,rs370842409), while the remaining 38 variants were novel. Altogether, we have identified 47 rare variants in 47 different genes including 15 ASD-candidate genes (Tables 1 and 2).
Figure 2.

Effect and mode of transmission of the validated variants. Pie charts illustrating the distribution of the confirmed variants found in all probands according to their effect (A) or mode of transmission (B). AR: autosomal recessive. AD: autosomal dominant.
Table 1.
Summary of the genes with confirmed rare variants detected in this study.
| Gene | OMIM/HGNC | Gene function | Biological process(s) | PANTHER Protein Class | Human disease | Reported link to ASD | Autism databases | Reported link to other neurological disorders | |
|---|---|---|---|---|---|---|---|---|---|
| SFARI Gene | Autism KB | ||||||||
| ACE2 | 300335/13557 | Angiotensin converting enzyme 2 | Cell signaling cascade (Renin angiotensin system) | NA | NI | Yes | No | No | No |
| AGL | 610860/321 | Glycogen debranching enzyme necessary for glycogenolysis | Mitochondrial function/Energy metabolism | Glycosyltransferase/glycosidase | Glycogen storage disease IIIa and b | No | No | No | No |
| APC2 | 612034/24036 | Tumor suppressor involved in microtubules stabilization | Neuronal function/Development | NA | a Sotos syndrome-like features | Yes | No | Yes | Patients with Sotos syndrome-like features also had intellectual disability |
| ARSH | 300586/32488 | Arylsulfatase H | Unknown | NA | NI | Yes | No | Yes | No |
| ASB9 | 300890/17184 | E3 ubiquitin-ligase involved in regulating the levels of creatine kinase B | Protein quality control | NA | NI | Yes | No | Yes | No |
| ATP2B3 | 300014/816 | The plasma membrane Ca2+ ATPase acts as a route for cellular calcium extrusion | Cell signaling cascade (Calcium homeostasis) | Cation transporter/ion channel/hydrolase | X-linked congenital cerebellar ataxia | Yes | No | Yes* | Possible association with ID |
| AVPR2 | 300538/897 | Arginine-vassopressin receptor 2 | Water homeostasis | G-protein coupled receptor | Nephrogenic diabetes insipidus | Yes | No | Yes | In dizygotic twins with nephrogenic diabetes insipidus and intellectual disability (ID) |
| BOC | 608708/17173 | An atypical receptor for Sonic Hedgehog regulating synapse formation | Neuronal function/Development | Immunoglobulin superfamily cell adhesion molecule | NI | Candidate gene in a patient with 3q13.31 microdeletion presenting with neurodevelopmental phenotype and autism | No | Yes | No |
| CELSR2 | 604265/3231 | Involved in axonal guidance | Neuronal function/Development | G-protein coupled receptor/cadherin | NI | No | No | No | SZ |
| CEP152 | 613529/29298 | Maintenance of genomic integrity and centrosome duplication | Cell division or differentiation | Signaling molecule | Seckel syndrome and primary microcephaly | Yes | No | Yes | No |
| CRY1 | 601933/2384 | Circadian clock gene | Sleep regulation | DNA photolyase/lyase | NI | Sleep problems are common in autistic children | No | Yes | Possible association with unipolar depression |
| CXORF30/CFAP47 | NA/6708 | Unknown | Unknown | NA | NI | Yes | No | Yes | No |
| DDX26B/INTS6L | NA/27334 | Unknown | Unknown | NA | NI | No | No | No | No |
| DNAJC13 | 614334/30343 | Component of the endosomal recycling system | Protein quality control | NA | Parkinson’s disease | No | No | No | Parkinson’s disease |
| DUSP3 | 600183/3069 | Dual-specificity phosphatase | Cell signaling cascade (MAPK signaling) | NA | NI | Yes | No | Yes* | No |
| FGF5 | 165190/3683 | Growth factor with neurotrophic activity | Neuronal function/Development | Growth factor | NI | Yes | No | Yes | No |
| FLNA | 300017/3754 | Actin-binding protein that may contribute to dentrites development | Neuronal function/Development | Non-motor actin binding protein | NI | Yes | No | Yes | No |
| GLT8D1 | NA/24870 | Unknown | Unknown | NA | NI | No | No | No | No |
| GPKOW | NA/30677 | Component of the human spliceosome | Gene regulation (mRNA splicing) | NA | NI | Yes | No | Yes | No |
| HSPBP1 | 612939/24989 | Hsp70-binding protein involved in protection against stress-induced damage | Protein quality control | NA | NI | Autism with FX mutation and chr15 dup | No | Yes | Autism with FX mutation and chr15 dup |
| HTATSF1 | 300346/5276 | Transcription-splicing factor | Gene regulation (mRNA splicing) | NA | NI | No | No | No | No |
| IDS | 300823/5389 | Lysosomal degradation of glycosami-noglycans | Protein quality control | Hydrolase | Mucopolysa-ccharidosis type II | Yes | No | Yes | No |
| ITIH2 | 146640/6167 | Plasma protease inhibitor involved in the stabilization of the extracellular matrix | Unknown | NA | NI | Yes | No | Yes | No |
| ITIH6 | NA/28907 | Unknown | Unknown | NA | NI | No | No | No | No |
| KCTD21 | NA/27452 | Cullin-3 adaptor protein required for HDAC1 ubiquitination and degradation | Gene regulation/Protein quality control | Enzyme modulator | Medulloblastoma | Yes | No | Yes | SZ |
| KDM5B | 605393/18039 | A demethylase involved in genome stability maintenance, and cell fate determination | Gene regulation/Cell differentiation | Zinc finger transcription factor | NI | De novo mutations identified in ASD patients | Yes* | Yes | Non-syndromic ID |
| MAN1B1 | 604346/6823 | ER-located endoplasmic enzyme involved in targeting misfolded glycoproteins for ER-associated degradation | Protein quality control | Chaperone | Congenital disorders of glycosylation | Yes | No | Yes | ID |
| MAOB | 309860/6834 | Deamination of a number of neurotransmitters | Neuronal function/Development | DNA methyltransferase; oxidase; DNA binding Protein | NI | No | Yes | Yes# | SZ |
| MOGS | 601336/24862 | Normal glycans processing | Immune responses | NA | Congenital disorders of glycosylation type II B | No | No | No | No |
| MXRA5 | 300938/7539 | Proteoglycan involved in extracellular matrix remodeling and cell-cell adhesion | Neuronal function/Development | Immunoglobulin superfamily cell adhesion molecule | NI | Mutations identified in three unrelated multiplex ASD families | No | Yes | No |
| NEB | 161650/7720 | Actin regulator | Neuronal function/Development | NA | Nemaline myopathy 2 | No | No | No | Mutations in this gene were identified in patients exhibiting ID and epilepsy with other symptoms |
| NGF | 162030/7808 | Trophic factor that has a role in behavior and brain development | Neuronal function/Development | Neurotrophic factor | Hereditary sensory and autonomic neuropathy type V | Yes | No | Yes | No |
| NLRP2 | 609364/22948 | Part of the inflammasome complex in astrocytes and acts as an inhibitor of NF-κB | Immune responses | Transcription co-factor/Nucleic acid binding | Beckwith-Wiedemann syndrome | Yes | No | Yes | No |
| NT5DC1 | NA/21556 | Unknown | Unknown | NA | NI | No | No | No | Bipolar disorder |
| OR6C65 | NA/31295 | Olfactory receptor | Olfaction | NA | NI | Impaired olfaction was reported in ASD | No | Yes | No |
| PDK3 | 300906/8811 | Negative regulator of pyruvate dehydrogenase | Mitochondrial function/Energy metabolism | Protein kinase | X-linked dominant Charcot-Marie-Tooth disease | Yes | No | Yes | No |
| PLP1 | 300401/9086 | Major constituent of the CNS myelin | Neuronal function/Development | Myelin protein | Pelizaeus-Merzbacher disease and spastic paraplegia type 2 | No | No | No | Microdeletion of Xq22 encompassing PLP1 in female patients with ID and behavioral problems |
| PRODH2 | 616377/17325 | Catalyzes proline degradation | Neuronal function/Development | Oxidase | NI | No | No | No | SZ |
| RPS6KA6 | 300303/10435 | Ribosomal protein S6 kinase A6 | Cell signaling cascade (MAPK signaling) | Non-receptor serine/threonine protein Kinase;transfer/carrier protein/annexin/calmodulin | NI | Yes | No | Yes* | Possible role in nonspecific X-linked ID |
| SEMG2 | 182141/10743 | Human seminal plasma protein | Semen function | NA | NI | Yes | No | Yes | No |
| SMS | 300105/11123 | Synthesis of spermine that has a modulatory effect on NMDAR | Neuronal function/Development | Transferase | X-linked intellectual disability (Snyder-Robinson type) | Yes | No | Yes | X-linked intellectual disability (Snyder-Robinson type) |
| SSTR3 | 182453/11332 | G protein-coupled receptor somatostatin receptor 3 with a role in synapse formation | Neuronal function/Development | G-protein coupled receptor | NI | Yes | No | Yes | No |
| SSX3 | 300325/11337 | Unknown | Unknown | NA | NI | Yes | No | Yes | No |
| SUMF1 | 607939/20376 | Activator of steroid sulfatases | Neuronal function/Development | NA | NI | Structural variants detected in ASD | No | Yes | SZ and multiple sclerosis |
| TRIM9 | 606555/16288 | E3 ubiquitin-ligase that plays a role in axonal branching | Neuronal function/Development | Ubiquitin-protein ligase | NI | Identified as a putative causative gene in one ASD case | No | No | Atypical psychosis |
| USP9X | 300072/12632 | Deubiquitinating enzyme | Neuronal function/Development | NA | X-linked intellectual disability | Yes | No | Yes# | X-linked intellectual disability |
| ZNF630 | 300819/28855 | Unknown | Unknown | NA | NI | Yes | No | Yes | No |
*Nonsynonymous variant(s) were reported. #Synonymous variant(s) were reported. Schizophrenia: SZ. Intellectual disability: ID. Fragile-X: FX. NI: Not identified. NA: Not available. For more details refer to supplementary information.
Table 2.
Rare variants identified in each trio.
| Proband ID (Gender) | Mode of Inheritance | Identified Variant | Genotype | CADD (scaled) | ExAC | MAF | ROH | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Base Change | Amino Acid Change | Proband | Other family members | pLI/Z score | SHGP LC database | ExAC/1000 Genomes project | ||||||
| UF | UM | US/(Gender) | |||||||||||
| ASD-9 (M) | AD | AGL | #c.294-2A > T | NA | A/T | A/T | A/A | NA | 24.9 | 0.00/−2.23 | 0.0037 | 0/0 | NA |
| AD | APC2 | #c.1207 + 3G > C | NA | G/C | G/C | G/G | NA | 12.21 | 1.00/4.81 | 0 | 0.0002/0 | NA | |
| ASD-16 (F) | AR | MAN1B1 | c.1897G > T | p.V633F | T/T | G/T | G/T | NA | 26.5 | 0.00/−1.01 | 0 | 0.00001656/0 | chr9:139943549–141071552 |
| AR | KCTD21 | c.152G > A | p.R51H | A/A | G/A | G/A | NA | 34 | 0.29/0.80 | 0 | 0.0001/0 | chr11:74862391–77920563 | |
| AR | DUSP3 | c.439G > A | p.V147I | A/A | G/A | G/A | NA | 24 | 0.74/1.28 | 0 | 0.00002476/0 | chr17:41332822–42248346 | |
| X-linked | CXorf30/CFAP47 | c.8943G > A | p.M2981I | G/A | Hemi (A) | G/G | NA | 13.16 | No score | 0 | 0/0 | NA | |
| X-linked | SSX3 | c.268C > A | p.R90S | C/A | Hemi (A) | C/C | NA | 0.256 | 0.15/−1.08 | 0.0008 | 0/0 | NA | |
| X-linked | AVPR2 | c.739–750 delCGCCGCAGGGGA | p.247-250del | Het | Homo | WT | NA | NA | 0.12/1.61 | 0.0009 | 0.0021/0.000529801 | NA | |
| ASD-17 (M) | AR | CRY1 | c.272G > A | p.W91X | A/A | G/A | G/A | G/A (M) | 40 | 0.00/1.46 | 0 | 0/0 | chr12:99640557–107713511 |
| AR | NLRP2 | c.523T > C | p.W175R | C/C | T/C | T/C | T/T (M) | 0.119 | 0.00/−1.98 | 0.0033 | 0.000008239/0 | NA | |
| X-linked | ACE2 | c.1235C > A | p.A412E | Hemi (A) | Hemi (C) | C/A | Hemi (A) (M) | 25.3 | 1.00/0.98 | 0.0038 | 0/0 | NA | |
| ASD-18 (M) | AR | GLT8D1 | #c.811_812 + 2delGAGT | p.E271K | Homo | Het | Het | NA | 35 | 0.00/0.54 | 0.00168 | 0/0 | chr3:52720080–53269046 |
| AR | DNAJC13 | c.5594A > G | p.N1865S | G/G | A/G | A/G | NA | 18.38 | 1.00/1.04 | 0.00042 | 0/0 | chr3:129800895–133486958 | |
| AR | OR6C65 | c.766delA | p.M256fs | Homo | Het | Het | NA | 27.6 | 0.00/−1.37 | 0.00042 | 0.00001648/0 | chr12:53647373–56089357 | |
| AR | HSPBP1 | c.1016A > C | p.E339A | C/C | A/C | A/C | NA | 26.7 | 0.01/0.79 | 0.00042 | 0/0 | chr19:55525894–56047448 | |
| X-linked | ATP2B3 | c.1678C > G | p.P560A | Hemi (G) | Hemi (C) | C/G | NA | 3.822 | 1.00/3.24 | 0 | 0.0000129/0.00001144 | NA | |
| ASD-19 (M) | X-linked | MXRA5 | c.28C > T | p.L10F | Hemi (T) | Hemi (C) | C/T | NA | 24.1 | 0.38/−1.92 | 0.0012 | 0/0 | NA |
| X-linked | ITIH6 | c.1153A > G | p.S385G | Hemi (G) | Hemi (A) | A/G | NA | 0.411 | 0.00/−3.80 | 0.0021 | 0/0 | NA | |
| De novo | KDM5B | c.2265C > A | p.Y755X | C/A | C/C | C/C | NA | 35 | 0.00/1.99 | 0 | 0.00003295/0 | NA | |
| ASD-21 (M) | AR | NT5DC1 | c.1114T > C | p.S372P | C/C | T/C | T/C | NA | 28.1 | 0.33/−0.27 | 0.0012 | 0/0 | chr6:116442525–116574577 |
| AR | TRIM9 | c.1643G > A | p.R548H | A/A | G/A | G/A | NA | 15.5 | 0.99/4.12 | 0 | 0.0000106/0 | chr14:51225132–51716467 | |
| ASD-24 (M) | X-linked | GPKOW | c.482C > T | p.A161V | Hemi (T) | Hemi (C) | C/T | Hemi (T) (M) | 9.839 | 0.98/0.42 | 0.0037 | 0.00001368/0 | NA |
| X-linked | HTATSF1 | c.894T > G | p.F298L | Hemi (G) | Hemi (T) | T/G | Hemi (G) (M) | 24 | 0.97/1.80 | 0 | 0/0 | NA | |
| De novo | MOGS | c.832_833delAA | p.K278Efs*5 | Het | WT | WT | WT (M) | 27 | 0.00/0.66 | 0 | 0.000008359/0 | NA | |
| ASD-37(M) | X-linked | IDS | c.525T > A | p.D175E | A/A | Hemi (T) | T/A | T/A (F) | 12.51 | 0.98/1.18 | 0.0012 | 0/0 | NA |
| ASD-38 (M) | X-linked | ASB9 | c.310A > C | p.T104P | Hemi (C) | Hemi (A) | A/C | NA | 25.6 | 0.89/0.44 | 0.0008 | 0/0 | NA |
| X-linked | PDK3 | #c.248 + 3A > G | _ | Hemi (G) | Hemi (A) | A/G | NA | 15.91 | 0.88/2.64 | 0.0021 | 0/0 | NA | |
| X-linked | PLP1 | c.328G > A | p.G110S | Hemi (A) | Hemi (G) | G/A | NA | 28.1 | 0.90/2.45 | 0 | 0/0 | NA | |
| ASD-39 (M) | AD | NEB | c.5939T > C | p.L1980S | T/C | T/T | T/C | NA | 25 | 0.00/−4.04 | 0.00084 | 0.0003/0 | NA |
| ASD-40 (M) | AR | BOC | c.1624G > A | p.G542R | A/A | G/A | G/A | NA | 28.6 | 0.00/0.77 | 0 | 0.000008237/0 | chr3:112546306–113377505 |
| AR | SSTR3 | c.724G > C | p.A242P | C/C | G/C | G/C | NA | 25.6 | 0.04/1.45 | 0 | 0.0003/0.000199681 | chr22:32756744–37603744 | |
| ASD-43 (M) | AR | NGF | c.361C > T | p.R121W | T/T | C/T | C/T | NA | 28.3 | 0.70/0.92 | 0.00084 | 0/0 | chr1:114301335–116927468 |
| AR | SUMF1 | c.710A > G | p.Q237R | G/G | A/G | A/G | NA | 26.3 | 0.04/−0.62 | 0 | 0/0 | chr3:439963–4856234 | |
| ASD-52 (M) | No candidate genes | ||||||||||||
| ASD-55 (M) | No candidate genes | ||||||||||||
| ASD-58 (M) | X-linked | MAOB | c.392G > T | p.S131I | Hemi (T) | Hemi (G) | G/T | NA | 22.4 | 0.97/1.38 | 0.00042 | 0/0 | NA |
| X-linked | ZNF630 | c.77A > T | p.N26I | Hemi (T) | Hemi (A) | A/T | NA | 15.35 | 0.00/−1.42 | 0 | 0/0 | NA | |
| De novo | PRODH2 | c.625G > C | p.G209R | G/C | G/G | G/G | NA | 0.461 | 0.00/−0.22 | 0 | 0.000008276/0 | NA | |
| ASD-64 (F) | X-linked | USP9X | c.3803A > G | p.Y1268C | A/G | Hemi (A) | A/G | NA | 25.2 | 1.00/6.35 | 0.00042 | 0/0 | NA |
| X-linked | RPS6KA6 | c.1535A > G | p.Q512R | A/G | Hemi (G) | A/A | NA | 17.14 | 0.74/1.70 | 0.0025 | 0/0 | NA | |
| X-linked | DDX26B/INTS6L | c.1304A > T | p.E435V | A/T | Hemi (T) | A/A | NA | 27.8 | 1.00/1.74 | 0.00084 | 0/0 | NA | |
| ASD-66 (M) | AD | SEMG2 | c.500G > A | p.W167X | G/A | G/G | G/A | NA | 20.4 | 0.00/−2.36 | 0.0058 | 0.000008241/0 | NA |
| ASD-69 (M) | AR | CELSR2 | c.8185G > C | p.D2729H | C/C | G/C | G/C | G/C (M) | 25.8 | 1.00/3.61 | 0 | 0/0 | chr1:109395105–109816176 |
| AR | ITIH2 | c.1863_1864insTATT | p.V622Yfs*17 | Homo | Het | Het | WT (M) | 24.3 | 0.00/−0.95 | 0 | 0.00007417/0 | chr10:3200292–13337588 | |
| AR | CEP152 | c.1718A > C | p.D573A | C/C | A/C | A/C | A/C (M) | 11.88 | 1.00/−1.59 | 0 | 0/0 | chr15:45392075–49284771 | |
| X-linked | ARSH | c.1124T > C | p.I375T | Hemi (C) | Hemi (T) | T/C | Hemi (C) (M) | 22.3 | 0.00/−0.13 | 0 | 0/0 | NA | |
| ASD-73 (M) | AR | FGF5 | c.251C > T | p.S84L | T/T | C/T | C/T | NA | 24.3 | 0.00/−0.58 | 0 | 0/0 | chr4:80977078–88535895 |
| X-linked | SMS | c.424C > T | p.L142F | Hemi (T) | Hemi (C) | C/T | NA | 28.9 | 0.92/2.30 | 0 | 0/0 | NA | |
| X-linked | FLNA | c.7079T > C | p.V2360A | Hemi (C) | Hemi (T) | T/C | NA | 23.2 | 1.00/4.95 | 0.00042 | 0/0 | NA | |
#PredictSNP2 score for splice site variants is “Deleterious” with expected accuracy of 90% or above. UF: unaffected father. UM: unaffected mother. US: unaffected sibling. ROH: region of homozygosity. NA: not applicable. CADD v1.3.
De novo variants
De novo germline mutations arising spontaneously during meiosis have long been known to confer risk of ASD. This knowledge was inferred largely from studies assessing de novo CNVs in ASD cases from simplex or multiplex families25–31, however, structural variants seem to account for a small fraction of the cases28, 32–34, shifting the focus to de novo point mutations and their contribution to risk, which is a line of enquiry we and others have perused8, 9, 11, 13, 20, 24.
Among the 17 probands, 3 (17.6%) were found to carry rare de novo SNVs. These confirmed de novo events were identified in different genes including one with previous association with ASD: two of these events were predicted to result in a premature truncation, and the remaining was a missense rare variant (p.G209R) affecting PRODH2, found in proband ASD-58. The two nonsense variants were: (i) (p.Y755X) affecting KDM5B identified in ASD-19 and (ii) a frameshift insertion of two adenine bases creating a premature stop codon (p.K278Efs*5) in MOGS that was detected in proband ASD-24 (Table 2).
Autosomal variants
Despite the excess and well-recognized importance of de novo mutations, only a fraction of ASD cases could be accounted for by these mutational events8, 17, while cases bearing inherited events couldn’t be fully explained by this class of genetic alteration. The high heritability of ASD15 underscores the need to explore transmitted variants that are often overlooked in many of the existing exome studies, which are mainly focused on de novo variants and not designed to capture inherited variants8, 9, 13, 20, 23, 24. Investigating both de novo and inherited variants has become a favored approach in recent next-generation sequencing based ASD studies10, 11, 14, 16–18.
Considering the high rate of consanguinity in the Saudi population, a recessive model was implemented under which a total of 19 rare variants were identified in different genes (Table 2).
In addition to recessive variants, we interrogated changes transmitting in a heterozygous manner (autosomal dominant). By only considering rare variants that were transmitted to the proband from one heterozygous parent, 4 rare variants were confirmed in 4 different genes (APC2, AGL, NEB and SEMG2). Two of the variants were observed in ASD-9 occurring within a splice site region of APC2 and AGL. One missense variant affecting NEB was found in ASD-39 and a nonsense variant within SEMG2 was found in ASD-66 (Table 2).
X-linked variants
A strong male bias in ASD has been consistently observed over time and across numerous studies in different populations35–37. This provides a strong clue to the potential involvement of sex chromosomes in the etiology of the disorder, which has been supported by the identification of multiple risk loci, particularly on the X-chromosome10, 16, 17, 19.
By setting our analysis approach to identify inherited variants in X-chromosome genes, we detected and confirmed a total of 21 rare variants (19 missense, 1 small deletion and 1 splice site change) and each was found in a different gene (Table 2). Of these variants, 4 were found in genes not previously implicated in ASD nor listed in Autism databases (DDX26B, HTATSF1, ITIH6 and PLP1). The remaining variants existed in genes already reported in ASD or other neurological disorders.
Description of confirmed rare variants per trio
By tailoring our WES analysis pipeline to capture both de novo and inherited variants, the analysis retained at least 2 rare variants affecting different genes for most probands. The variants are discussed in detail in the supplementary data and Supplementary Table S4.
Biological processes over-represented in our study
IPA functional analysis detected significant enrichment for biological processes such as ‘Cell Signaling’, ‘Cell Morphology’ and ‘Cellular Assembly and Organization’ across the entire genes set. As for Diseases and Disorders, the analysis discovered ‘Neurological Disease’, ‘Organismal Injury and Abnormalities’ and ‘Hereditary Disorder’, among other human diseases, to be significantly correlated. Furthermore, ‘Nervous System Development and Function’, ‘Tissue Development’ and ‘Embryonic Development’ were the 3 top ranking functions under the ‘Top Physiological System Development and Function’ category (Supplementary Table S5).
Discussion
Dissecting the genetic architecture of an otherwise highly complex disorder such as ASD, comprising diverse forms of genetic alterations (from SNVs to chromosomal aberrations affecting numerous genes/loci), requires employing different approaches. Ideal approaches would be those permitting genetic investigation of ASD cases, in which a polygenic model is assumed (or strongly suspected)7. Therefore, capturing more classes of genetic variation, especially with the emergence of studies identifying multiple mutations in individual ASD patients of which all are predicted to contribute to the etiology of the disease, has become the preferred approach by many researchers. We, as others10, took advantage of NGS to perform comprehensive analysis in parent-child trios designed to detect both de novo and transmitted genetic variants. By considering rare variants with predicted damaging effect, we validated variants in 17 of 19 trios (~90%). We could not comment on whether our detection yield is comparable with other studies or not, due to the lack of published ASD trio exome studies of similar design using the same platform. However, Jiang and colleagues conducted a relatively similar study (utilizing an Illumina platform) in which they have reported successfully detecting medically relevant variants in 50% of their ASD trio families using whole-genome sequencing (WGS)10.
Missense variants constitute the most frequently encountered type of variation (77%) in this study followed by loss of function variants [nonsense (6%), frameshifts (6%) and splice site (9%)] (Fig. 2A). Of the 19 probands only 3 were found to carry de novo events, whereas rare variants in autosomal or X-linked genes were detected in equal number of probands. While most of the previous studies concerned with sporadic autism have focused on assessing the role of de novo variants under the hypothesis that such lesions are more likely to confer risk to ASD than inherited events9, 12, 13, 20, 24, 38, we, on the contrary, observe enrichment for inherited variants. This is unsurprising given the highly inbred nature of the Saudi population in which common ancestry is suspected even in apparently non-consanguineous unions as parents are often unaware of consanguinity in previous generations. Such observation is common in Middle Eastern and North African populations39.
In our study, 32 of the detected variants were observed in genes previously reported in ASD (either in Autism databases or in literature) (Table 1). For instance, SUMF1 40, HSPBP1 41, TRIM9 42, DUSP3 24 and BOC 43. WES detected de novo variants in three genes (Table 2). One missense variant affecting a schizophrenia susceptibility gene (PRODH2)44, 45 and the other two were protein-truncating variants. One was identified in KDM5B. This gene was previously associated with ASD8, 9 and non-syndromic intellectual disability (ID)46 whose function is related to cell cycle control and neural cells differentiation47. The other was a frameshift insertion of two adenine bases creating a premature stop codon in MOGS. This gene encodes an endoplasmic reticulum glucosidase involved in normal immune function48.
By applying the autosomal recessive model, we validated variants in 19 genes (Table 2). One missense variant was observed in MAN1B1, a gene causally linked to non-syndromic intellectual disability49, 50, and another was found in DNAJC13 previously reported to be mutated in patients with Parkinson’s disease51, 52. Two variants were identified in genes with possible, but not confirmed association with mood disorders: CRY1 53–55 and NT5DC1 56, 57. Interestingly, patient (ASD-21) harboring NT5DC1 missense variant displayed symptoms of depression and poor appetite around the age of 5 years. Amongst the autosomal transmitted variants, six were predicted to disrupt a group of genes involved in brain development and/or function namely TRIM9, BOC, SSTR3, NGF, FGF5 and CELSR2 58–65, two were found in genes related to protein homeostasis (KCTD21 66 and HSPBP1 67) and the remaining variants were identified in genes involved in diverse biological process including cell signaling (DUSP3)68, inflammatory responses (NLRP2)69, olfaction (OR6C65)70, maintenance of DNA integrity (CEP152)71, and stabilization of the extracellular matrix (ITIH2)72.
The X-linked model revealed 21 different variants (Table 2). For instance a missense variant was detected in MXRA5, a gene that was reported to be mutated in three multiplex ASD families19. A deletion of 12 nucleotides was detected within exon 2 of AVPR2, defects in this gene cause X-linked congenital nephrogenic diabetes insipidus (NDI) and where detected in dizygotic twins with NDI and ID73. One putative splice site variant was found in PDK3, a gene that encodes an enzyme involved in the shift of energy production site from the mitochondria (oxidative phosphorylation) to the cytoplasm (glycolysis) reported in cancer cells74. The same gene was recently reported to be mutated in a single family with Charcot–Marie–Tooth disease75. However, the patient (ASD-38) reported here to carry PDK3 splice site variant is not affected with Charcot-Marie-Tooth disease, nor does he suffer from any type of neuropathy. Moreover, of the variants detected on the X-chromosome, four were located in genes involved in brain function/development (PLP1 76, MAOB 77, USP9X 78, and FLNA 79, 80) and two were present in genes regulating splicing and/or transcription (HTATSF1 81 and GPKOW 82, 83). Additional rare variants were identified in genes involved in: (i) the hydrolysis of angiotensin II (ACE2)84 or sulfate esters (IDS)85 and (ARSH)86; (ii) or in the ubiquitination and proteasomal degradation of creatine kinase-B (ASB-9)87. Furthermore, two missense variants were detected: one in ATP2B3 and another in RPS6KA6. Both genes participate in signal transduction pathways, however only ATP2B3 was linked to a human disease88, 89. The remaining variants were located in genes of largely unknown function such as (CFAP47, ITIH6 and ZNF630) or in cancer-associated genes namely; INTS6L 90, 91 and SSX3 92.
With regard to the biological functions that might be influenced by the identified genetic changes, we have compared those highlighted by the IPA with the results of the manual literature mining. Cell signaling was among the common biological themes. Typically, categories pertaining to CNS function such as those found by IPA “Nervous System Development and Function” or manual search “Neuronal function/Development” were over-represented. These categories comprised genes serving diverse functions in the CNS such as neurite outgrowth, synaptic function and formation, and neuronal migration (Dataset 1). Aside from the disruption of neuron-specific functions, defects involving basic cellular processes (presented here and elsewhere) represent another commonly encountered theme in ASD, reflecting the clinical and genetic complexity and variability of this condition1. It is noteworthy, that several of the detected rare variants were observed in genes without currently recognized functions/roles making it difficult to propose a functional link to ASD, something that might be revealed by future studies.
One of the interesting findings emerging from this study is the identification of two genes (NLRP2 and MOGS) with established roles in the immune system. The former encodes for NLRP2 protein that plays a vital role in astrocytes innate immunity69 and was found to be mutated in a rare type of imprinting disorder known as Beckwith-Wiedemann syndrome93. The latter gene (MOGS), on the other hand, encodes an endoplasmic reticulum glucosidase involved in normal immune function and is causally linked to congenital disorders of glycosylation type II B clinically characterized by developmental and neurological defects48. It is worth mentioning that immune dysfunction has been documented in ASD and emerging studies propose a role for the immune system in the pathophysiology of autism. In support of this notion, typical behavior and neuropathological symptoms of human ASD were successfully recapitulated in mice born to immune-activated (infected) mothers94.
Another key finding demonstrated here, is the genetic variability not only across trios but within each proband. Defects in genes involved in diverse biological processes were observed in most probands and only few (ASD-40, 43, 58 and 73) that were enriched for variants affecting neuronal function/development. Our findings are in keeping with a growing number of reports suggesting a genetic model of autism whereby cumulative contribution of multiple inherited and de novo variants (multiple hit) in different genes (genetic heterogeneity), including genes associated with other neuropsychiatric disorders (pleiotropy) or involved in diverse biological processes beyond synaptic function (molecular diversity), shape the risk to ASD1, 10, 11, 16, 95
Conclusions
Apart from Yu et al. exome sequencing study, aimed at identifying inherited SNVs in a cohort of consanguineous/multiplex families with ASD including two Middle Eastern multiplex families (one from Saudi Arabia and one from Kuwait)17, the genetic characteristics of ASD in Arab populations remain largely unexplored. This work represents, to the best of our knowledge, a comprehensive exome analysis of trios with ASD from an Arab population. Our primary goal was to identify de novo or rare inherited coding variants of potential clinical relevance through applying WES on case-parent trios from singleton families. Among the advantages of implementing this approach over WGS is that it generates fewer results and therefore is relatively less analytically challenging. Although this approach enabled us to generate a prioritized shortlist of potential deleterious variants, association and extensive functional studies are necessary to identify the disease causing ones with more certainty. Also it is important to consider the possibility of missing candidate variants within coding regions and the fact that neither common variants nor non-coding regions were considered here. In spite of the small number of cases investigated here; our current study is useful in terms of contributing a small, albeit rich data set, revealing new ASD candidate genes that may shed light on potential diagnostic and therapeutic targets.
Methods
Ethics statement
Patient recruitment and all experimental protocols used in this study were in compliance with the Declaration of Helsinki and were approved by the institution’s relevant committees; Internal Review Board (IRB), Research Ethics committee and Basic Research Committee at King Faisal Specialist Hospital and Research Center. Written informed consent was obtained from subjects before enrollment in an IRB-approved protocol (King Faisal Specialist Hospital and Research Center (KFSHRC) RAC#2080001).
ASD trios selection
A total of 19 simplex (singleton) families were selected for the study (Trio’s analysis). Venous blood samples from parents and affected child were obtained for both DNA and RNA extraction. Of the recruited families, 9 were from consanguineous marriages and 10 were not. Non-consanguineous families were included based on the rationale that in populations with high levels of consanguinity/endogamy (tribal or religious)96, a common lineage is suspected even when couples regard themselves as unrelated as they are often unaware of consanguinity in distant generations39, 97. Diagnosis of ASD was based on the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) criteria (American Psychiatric Association [APA], 2013) and ADI-R, ADOS (which are not yet validated in Arabic). Both parents were present for the interviews and assessment. All selected cases did not have symptomatic ASD secondary to known genetic or metabolic disorders (such as Fragile X syndrome, Tuberous Sclerosis, Rett syndrome, Angelman, Prader-Willi syndrome or Phenylketonuria). Available clinical and demographic information is summarized in Supplementary Table S6.
All probands were negative for any copy number variants (CNV) in previously reported ASD-associated loci/genes listed in Supplementary Table S7. CNV analysis was performed in probands using the Cyto Scan HD array (Affymetrix, Santa Clara, CA,USA). Targeted analysis was carried out using the Chromosome Analysis Suite version Cyto 3.0 using GRC 38/hg19 of the UCSC Genome Browser and the recommended threshold of log2 ratios of more than 0.58 for CNV gains and less than -1 for CNV losses was used.
Whole exome sequencing, data processing and bioinformatics analysis
WES, alignment of reads, and variant discovery
The exomes of the parents and affected children were sequenced on the Ion Proton platforms, using the whole exome AmpliSeq kit. Briefly, 12 separate Exome Primer Pools, AmpliSeq HiFi mix (Life Technologies, Carlsbad, CA, USA) and 100 ng DNA from each sample were used in the amplification step for 10 cycles. The resultant PCR products were then pooled in preparation for primer digestion using FuPa reagent (Life Technologies, Carlsbad, CA, USA). This was followed by a ligation step using Ion P1 and Ion Xpress Barcode adapters. The libraries were then purified and quantified using qPCR and the Ion Library Quantification Kit (Life Technologies, Carlsbad, CA, USA) prior to emulsion on an Ion OneTouch System. The templated Ion Sphere particles were enriched using Ion OneTouch ES (Life Technologies, Carlsbad, CA, USA) and processed for sequencing on the Ion Proton instrument (Life Technologies, Carlsbad, CA, USA). Approximately 15–17 Gb of DNA sequence was generated per sequencing run/sample. Reads were mapped to UCSC Human reference genome (hg19) (http://genome.ucsc.edu/) and variants were identified using the Ion Torrent pipeline (Life Technologies, Carlsbad, CA, USA).
The sequencing targets 293903 amplicons, covering about 19,000 genes (https://www.ampliseq.com/tmpl/view.action?tmplDesignId=50055032). The analysis pipeline is a multi-step process (Fig. 1). First, the NGS reads were subjected to quality control checks for removing any low quality reads. Then the reads were mapped (aligned) to hg19. For the mapping step, we used the program tmap, which is part of the Torrent Suite package. This program is the same as BWA but it is tuned more to the Ion Torrent technology, by including flow signal information in the alignment process. Next, variant calling was performed using the Torrent Suite Variant Caller, which is a GATK-like variant caller, but more tuned to the Ion NGS data. It takes flow signals into account and also recognizes types of sequencing errors common to this platform.
As part of a tertiary data analysis, each trio was further interrogated using four possible inheritance models; autosomal recessive, autosomal dominant, de novo and X-linked. To that end, we compared the variants detected in the affected child to the corresponding positions in the parents DNA. Basically, we grouped the variants into model-specific subsets according the mode of inheritance. For instance, a variant was considered to be de novo if it was present uniquely in the proband and absent in both parents, while if two copies of the variant were inherited by heterozygous parents, the variant was considered to be autosomal recessive (with consideration of compound heterozygosity). On the other hand, the variant was considered autosomal dominant only if it was transmitted to the proband from one heterozygous parent. Moreover, for a variant to be considered as X-linked, it had to occur on the X-chromosome in either a hemizygous (male) or a homozygous (female) state in the proband in reference to the parents.
Variants annotation
ANNOVAR (http://annovar.openbioinformatics.org) software was used to functionally annotate detected variants as previously described98. Briefly, the software was used to perform two types of functional annotations; gene-based and filter-based. The former one identifies the genomic region containing the variant (e.g exonic or intronic), and the latter assesses the frequency of the variant in widely used databases in addition to providing functional prediction scores from a number of tools.
Variants prioritization and Sanger validation
The resulting model-specific set of variants was trimmed by omitting the ones not falling within coding exons or exon/intron boundaries, or non-functional. Then validation by Sanger sequencing was carried out on fresh DNA aliquots prior to assessing the frequency and predicted functional effect of the detected variants. The purpose of this was to avoid overlooking any interesting changes that may have been masked by variant calling/annotation errors. The list of validated variants was further trimmed by omitting those reported in dbSNP, or present in international databases (1000 Genomes project, ExAC) or in the local ethnically matching normal controls database (SHGP 2379 exomes) with a MAF ≥ 1%. Variant deleteriousness was predicted using four different web-based tools: Polyphen-2, SIFT, MutationTaster and CADD as described99–102. For predicting splice-site variants effect, we utilized PredictSNP2 web interface for its improved performance compared to CADD103. In addition, available public Autism genes/variants databases (SFARI Gene; https://gene.sfari.org/autdb/Welcome.do, and AutismKB; http://autismkb.cbi.pku.edu.cn/) were checked for reported SNVs and CNVs in our list of validated genes or bands harboring these genes in case of CNV104, 105. Finally, segregation analysis was carried out in families where DNA from unaffected family members (siblings) was available (ASD-17,-24, -37 and -69).
Relatedness assessment
Three different relationship inference algorithms were employed in this study to verify the self-reported pedigree information. The first is Yang et al. method, which is based on the A jk statistics106. For this method we used the implementation in the VCFtools package (with option –relatedness 1). In the second method, we computed shared homozygosity by comparing the homozygous variants (MAF > 1%) between each pair of individuals and counting the number of overlapping variants. Relatedness was denoted as “Not confirmed” if the number of shared homozygous variants was less than 25 SD above the mean (123 ± 25). The third method estimates kinship as described by Manichaikul and colleagues107 using KING program (http://people.virginia.edu/~wc9c/KING/) and the implementation in VCFtools (option –relatedness2).
Biological functions analysis
In an attempt to gain insight into the biological processes that may potentially be affected by the variants identified herein, we analyzed our final list of genes with validated variants using the core analysis function of Ingenuity Pathway Analysis software (IPA) (IPA®, version 01–07, QIAGEN, Redwood City, https://analysis.ingenuity.com/pa/installer/select). The software identifies the diseases and biological functions that are significantly associated with our genes list. The resulting categories are ranked by p-values calculated using right-tailed Fisher’s exact test.
In parallel, we have grouped gene-sets on the basis of their function (by means of manual literature research) into different categories including: “Neuronal function/Development”, “Mitochondrial function/Energy metabolism”, “Protein quality control”, “Gene regulation”, “Cell signaling cascade”, “Cell division or differentiation”, and “Immune responses” (Table 1).
Electronic supplementary material
Acknowledgements
All NGS library building, sequencing and bioinformatics analysis was performed by the Saudi Human Genome Program (SHGP) at King Abdulaziz City for Science and Technology (KACST) and at KFSHRC. In addition, we would like to thank sequencing and genotyping core facilities in the department of Genetics at KFSHRC for performing sequencing and HDCytoscan.
Author Contributions
Conception of the study and experiment design: N.A.T. Whole exome sequencing data analysis and variant validation: A.O., J.S., A.M., A.T. and A.A. Whole exome sequencing: E.G. and R.A. Supervision of the whole exome sequencing and Sanger sequencing: D.M. Bioinformatics data analysis: M.A., M.E. and S.S. Data interpretation: B.A. and N.A.T. Patients recruitment and assessment: H.A., M.A., M.N., S.A., H.A.A. and A.E. Manuscript drafting: B.A. and N.A.T. D.M. and M.A. contributed to manuscript writing.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-06033-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Bashayer Al-Mubarak, Email: BAl-Mubarak@kfshrc.edu.sa.
Nada Al Tassan, Email: naltassan@kfshrc.edu.sa.
References
- 1.Geschwind DH, Flint J. Genetics and genomics of psychiatric disease. Science. 2015;349:1489–1494. doi: 10.1126/science.aaa8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warrier V, Chee V, Smith P, Chakrabarti B, Baron-Cohen S. A comprehensive meta-analysis of common genetic variants in autism spectrum conditions. Molecular autism. 2015;6:49. doi: 10.1186/s13229-015-0041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hadley D, et al. The impact of the metabotropic glutamate receptor and other gene family interaction networks on autism. Nature communications. 2014;5:4074. doi: 10.1038/ncomms5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaefer GB, Mendelsohn NJ. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genetics in medicine: official journal of the American College of Medical Genetics. 2013;15:399–407. doi: 10.1038/gim.2013.32. [DOI] [PubMed] [Google Scholar]
- 5.Nakai N, Otsuka S, Myung J, Takumi T. Autism spectrum disorder model mice: Focus on copy number variation and epigenetics. Science China. Life sciences. 2015;58:976–984. doi: 10.1007/s11427-015-4891-7. [DOI] [PubMed] [Google Scholar]
- 6.Sener EF. Association of Copy Number Variations in Autism Spectrum Disorders: A Systematic Review. Chinese Journal of Biology. 2014;2014:9. doi: 10.1155/2014/713109. [DOI] [Google Scholar]
- 7.Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Current opinion in genetics & development. 2012;22:229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Iossifov I, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang YH, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. American journal of human genetics. 2013;93:249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krumm N, et al. Excess of rare, inherited truncating mutations in autism. Nature genetics. 2015;47:582–588. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature genetics. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi L, et al. Whole-genome sequencing in an autism multiplex family. Molecular autism. 2013;4:8. doi: 10.1186/2040-2392-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nature reviews. Genetics. 2012;13:537–551. doi: 10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toma C, et al. Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Molecular psychiatry. 2014;19:784–790. doi: 10.1038/mp.2013.106. [DOI] [PubMed] [Google Scholar]
- 17.Yu TW, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuen RK, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nature medicine. 2015;21:185–191. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
- 19.Nava C, et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Translational psychiatry. 2012;2:e179. doi: 10.1038/tp.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez-Barron LT, et al. Genome-wide variant analysis of simplex autism families with an integrative clinical-bioinformatics pipeline. Cold Spring Harbor molecular case studies. 2015;1:a000422. doi: 10.1101/mcs.a000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaelson JJ, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christian SL, et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biological psychiatry. 2008;63:1111–1117. doi: 10.1016/j.biopsych.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacquemont ML, et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. Journal of medical genetics. 2006;43:843–849. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levy D, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Marshall CR, et al. Structural variation of chromosomes in autism spectrum disorder. American journal of human genetics. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poultney CS, et al. Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. American journal of human genetics. 2013;93:607–619. doi: 10.1016/j.ajhg.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szatmari P, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature genetics. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miles JH. Autism spectrum disorders–a genetics review. Genetics in medicine: official journal of the American College of Medical Genetics. 2011;13:278–294. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- 33.Walsh KM, Bracken MB. Copy number variation in the dosage-sensitive 16p11.2 interval accounts for only a small proportion of autism incidence: a systematic review and meta-analysis. Genetics in medicine: official journal of the American College of Medical Genetics. 2011;13:377–384. doi: 10.1097/GIM.0b013e3182076c0c. [DOI] [PubMed] [Google Scholar]
- 34.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. The New England journal of medicine. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 35.Gillberg C, Cederlund M, Lamberg K, Zeijlon L. Brief report: “the autism epidemic”. The registered prevalence of autism in a Swedish urban area. Journal of autism and developmental disorders. 2006;36:429–435. doi: 10.1007/s10803-006-0081-6. [DOI] [PubMed] [Google Scholar]
- 36.Skuse DH. Imprinting, the X-chromosome, and the male brain: explaining sex differences in the liability to autism. Pediatric research. 2000;47:9–16. doi: 10.1203/00006450-200001000-00006. [DOI] [PubMed] [Google Scholar]
- 37.Werling DM, Geschwind DH. Recurrence rates provide evidence for sex-differential, familial genetic liability for autism spectrum disorders in multiplex families and twins. Molecular autism. 2015;6:27. doi: 10.1186/s13229-015-0004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben Halim N, et al. Consanguinity, endogamy, and genetic disorders in Tunisia. Journal of community genetics. 2013;4:273–284. doi: 10.1007/s12687-012-0128-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gai X, et al. Rare structural variation of synapse and neurotransmission genes in autism. Molecular psychiatry. 2012;17:402–411. doi: 10.1038/mp.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishimura Y, et al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Human molecular genetics. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- 42.Butler MG, Rafi SK, Hossain W, Stephan DA, Manzardo AM. Whole exome sequencing in females with autism implicates novel and candidate genes. International journal of molecular sciences. 2015;16:1312–1335. doi: 10.3390/ijms16011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quintela I, et al. Female patient with autistic disorder, intellectual disability, and co-morbid anxiety disorder: Expanding the phenotype associated with the recurrent 3q13.2-q13.31 microdeletion. American journal of medical genetics. Part A. 2015;167:3121–3129. doi: 10.1002/ajmg.a.37292. [DOI] [PubMed] [Google Scholar]
- 44.Chakravarti A. A compelling genetic hypothesis for a complex disease: PRODH2/DGCR6 variation leads to schizophrenia susceptibility. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:4755–4756. doi: 10.1073/pnas.092158299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu H, et al. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3717–3722. doi: 10.1073/pnas.042700699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Athanasakis E, et al. Next generation sequencing in nonsyndromic intellectual disability: from a negative molecular karyotype to a possible causative mutation detection. American journal of medical genetics. Part A. 2014;164A:170–176. doi: 10.1002/ajmg.a.36274. [DOI] [PubMed] [Google Scholar]
- 47.Dey BK, et al. The histone demethylase KDM5b/JARID1b plays a role in cell fate decisions by blocking terminal differentiation. Molecular and cellular biology. 2008;28:5312–5327. doi: 10.1128/MCB.00128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lyons JJ, Milner JD, Rosenzweig SD. Glycans Instructing Immunity: The Emerging Role of Altered Glycosylation in Clinical Immunology. Frontiers in pediatrics. 2015;3:54. doi: 10.3389/fped.2015.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Najmabadi H, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 50.Rafiq MA, et al. Mutations in the alpha 1,2-mannosidase gene, MAN1B1, cause autosomal-recessive intellectual disability. American journal of human genetics. 2011;89:176–182. doi: 10.1016/j.ajhg.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gustavsson EK, et al. DNAJC13 genetic variants in parkinsonism. Movement disorders: official journal of the Movement Disorder Society. 2015;30:273–278. doi: 10.1002/mds.26064. [DOI] [PubMed] [Google Scholar]
- 52.Vilarino-Guell C, et al. DNAJC13 mutations in Parkinson disease. Human molecular genetics. 2014;23:1794–1801. doi: 10.1093/hmg/ddt570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hua P, et al. Cry1 and Tef gene polymorphisms are associated with major depressive disorder in the Chinese population. Journal of affective disorders. 2014;157:100–103. doi: 10.1016/j.jad.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenwasser AM. Circadian clock genes: non-circadian roles in sleep, addiction, and psychiatric disorders? Neuroscience and biobehavioral reviews. 2010;34:1249–1255. doi: 10.1016/j.neubiorev.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Soria V, et al. Differential association of circadian genes with mood disorders: CRY1 and NPAS2 are associated with unipolar major depression and CLOCK and VIP with bipolar disorder. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2010;35:1279–1289. doi: 10.1038/npp.2009.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bigdeli TB, et al. Association study of 83 candidate genes for bipolar disorder in chromosome 6q selected using an evidence-based prioritization algorithm. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2013;162B:898–906. doi: 10.1002/ajmg.b.32200. [DOI] [PubMed] [Google Scholar]
- 57.Nurnberger JI, Jr., et al. Identification of pathways for bipolar disorder: a meta-analysis. JAMA psychiatry. 2014;71:657–664. doi: 10.1001/jamapsychiatry.2014.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berry A, Bindocci E, Alleva E. NGF, brain and behavioral plasticity. Neural plasticity. 2012;2012:784040. doi: 10.1155/2012/784040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Courchet J, Polleux F. Sonic hedgehog, BOC, and synaptic development: new players for an old game. Neuron. 2012;73:1055–1058. doi: 10.1016/j.neuron.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 60.Green JA, Gu C, Mykytyn K. Heteromerization of ciliary G protein-coupled receptors in the mouse brain. PloS one. 2012;7:e46304. doi: 10.1371/journal.pone.0046304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guadiana SM, et al. Arborization of dendrites by developing neocortical neurons is dependent on primary cilia and type 3 adenylyl cyclase. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:2626–2638. doi: 10.1523/JNEUROSCI.2906-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qu Y, et al. Genetic evidence that Celsr3 and Celsr2, together with Fzd3, regulate forebrain wiring in a Vangl-independent manner. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E2996–3004. doi: 10.1073/pnas.1402105111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winkle CC, et al. A novel Netrin-1-sensitive mechanism promotes local SNARE-mediated exocytosis during axon branching. The Journal of cell biology. 2014;205:217–232. doi: 10.1083/jcb.201311003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindholm D, et al. Fibroblast growth factor-5 promotes differentiation of cultured rat septal cholinergic and raphe serotonergic neurons: comparison with the effects of neurotrophins. The European journal of neuroscience. 1994;6:244–252. doi: 10.1111/j.1460-9568.1994.tb00267.x. [DOI] [PubMed] [Google Scholar]
- 65.Reuss B, Dono R, Unsicker K. Functions of fibroblast growth factor (FGF)-2 and FGF-5 in astroglial differentiation and blood-brain barrier permeability: evidence from mouse mutants. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:6404–6412. doi: 10.1523/JNEUROSCI.23-16-06404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Z, Xiang Y, Sun G. The KCTD family of proteins: structure, function, disease relevance. Cell & bioscience. 2013;3:45. doi: 10.1186/2045-3701-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rogon C, et al. HSP70-binding protein HSPBP1 regulates chaperone expression at a posttranslational level and is essential for spermatogenesis. Molecular biology of the cell. 2014;25:2260–2271. doi: 10.1091/mbc.E14-02-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patterson KI, Brummer T, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. The Biochemical journal. 2009;418:475–489. doi: 10.1042/BJ20082234. [DOI] [PubMed] [Google Scholar]
- 69.Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61:1113–1121. doi: 10.1002/glia.22499. [DOI] [PubMed] [Google Scholar]
- 70.Olender T, Lancet D, Nebert DW. Update on the olfactory receptor (OR) gene superfamily. Human genomics. 2008;3:87–97. doi: 10.1186/1479-7364-3-1-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalay E, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nature genetics. 2011;43:23–26. doi: 10.1038/ng.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hamm A, et al. Frequent expression loss of Inter-alpha-trypsin inhibitor heavy chain (ITIH) genes in multiple human solid tumors: a systematic expression analysis. BMC cancer. 2008;8:25. doi: 10.1186/1471-2407-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang L, Poke G, Gecz J, Gibson K. A novel contiguous gene deletion of AVPR2 and ARHGAP4 genes in male dizygotic twins with nephrogenic diabetes insipidus and intellectual disability. American journal of medical genetics. Part A. 2012;158A:2511–2518. doi: 10.1002/ajmg.a.35591. [DOI] [PubMed] [Google Scholar]
- 74.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell death & disease. 2013;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kennerson ML, et al. A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. Human molecular genetics. 2013;22:1404–1416. doi: 10.1093/hmg/dds557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nave KA. Myelination and the trophic support of long axons. Nature reviews. Neuroscience. 2010;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- 77.Fagervall I, Ross SB. A and B forms of monoamine oxidase within the monoaminergic neurons of the rat brain. Journal of neurochemistry. 1986;47:569–576. doi: 10.1111/j.1471-4159.1986.tb04537.x. [DOI] [PubMed] [Google Scholar]
- 78.Homan CC, et al. Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth. American journal of human genetics. 2014;94:470–478. doi: 10.1016/j.ajhg.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feng Y, et al. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19836–19841. doi: 10.1073/pnas.0609628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang L, et al. MEK-ERK1/2-dependent FLNA overexpression promotes abnormal dendritic patterning in tuberous sclerosis independent of mTOR. Neuron. 2014;84:78–91. doi: 10.1016/j.neuron.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller HB, Robinson TJ, Gordan R, Hartemink AJ, Garcia-Blanco MA. Identification of Tat-SF1 cellular targets by exon array analysis reveals dual roles in transcription and splicing. RNA (New York, N.Y.) 2011;17:665–674. doi: 10.1261/rna.2462011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aksaas AK, et al. G-patch domain and KOW motifs-containing protein, GPKOW; a nuclear RNA-binding protein regulated by protein kinase A. Journal of molecular signaling. 2011;6:10. doi: 10.1186/1750-2187-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zang S, et al. GPKOW is essential for pre-mRNA splicing in vitro and suppresses splicing defect caused by dominant-negative DHX16 mutation in vivo. Bioscience reports. 2014;34:e00163. doi: 10.1042/BSR20140142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guimond MO, Gallo-Payet N. The Angiotensin II Type 2 Receptor in Brain Functions: An Update. International journal of hypertension. 2012;2012:351758. doi: 10.1155/2012/351758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilson PJ, et al. Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:8531–8535. doi: 10.1073/pnas.87.21.8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Diez-Roux G, Ballabio A. Sulfatases and human disease. Annual review of genomics and human genetics. 2005;6:355–379. doi: 10.1146/annurev.genom.6.080604.162334. [DOI] [PubMed] [Google Scholar]
- 87.Debrincat MA, et al. Ankyrin repeat and suppressors of cytokine signaling box protein asb-9 targets creatine kinase B for degradation. The Journal of biological chemistry. 2007;282:4728–4737. doi: 10.1074/jbc.M609164200. [DOI] [PubMed] [Google Scholar]
- 88.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiology and molecular biology reviews: MMBR. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zanni G, et al. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14514–14519. doi: 10.1073/pnas.1207488109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bohm M, et al. Genetic Variants of DICE1/INTS6 in German Prostate Cancer Families with Linkage to 13q14. Urologia internationalis. 2015;95:386–389. doi: 10.1159/000366229. [DOI] [PubMed] [Google Scholar]
- 91.Filleur S, et al. INTS6/DICE1 inhibits growth of human androgen-independent prostate cancer cells by altering the cell cycle profile and Wnt signaling. Cancer cell international. 2009;9:28. doi: 10.1186/1475-2867-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zendman AJ, Ruiter DJ, Van Muijen GN. Cancer/testis-associated genes: identification, expression profile, and putative function. Journal of cellular physiology. 2003;194:272–288. doi: 10.1002/jcp.10215. [DOI] [PubMed] [Google Scholar]
- 93.Meyer E, et al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome) PLoS genetics. 2009;5:e1000423. doi: 10.1371/journal.pgen.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:12776–12781. doi: 10.1073/pnas.1202556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annual review of genomics and human genetics. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- 96.El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Consanguinity and major genetic disorders in Saudi children: a community-based cross-sectional study. Annals of Saudi medicine. 2008;28:169–173. doi: 10.4103/0256-4947.51726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bittles AH, Black ML. Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(Suppl 1):1779–1786. doi: 10.1073/pnas.0906079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Saudi Mendeliome Group. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome biology16, 134, doi:10.1186/s13059-015-0693-2 (2015). [DOI] [PMC free article] [PubMed]
- 99.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics/editorial board, Jonathan L. Haines. [et al.] Chapter 7, Unit7 20, doi:10.1002/0471142905.hg0720s76 (2013). [DOI] [PMC free article] [PubMed]
- 101.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 102.Sim NL, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic acids research. 2012;40:W452–457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bendl J, et al. PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS computational biology. 2016;12:e1004962. doi: 10.1371/journal.pcbi.1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Basu SN, Kollu R, Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic acids research. 2009;37:D832–836. doi: 10.1093/nar/gkn835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xu LM, et al. AutismKB: an evidence-based knowledgebase of autism genetics. Nucleic acids research. 2012;40:D1016–1022. doi: 10.1093/nar/gkr1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nature genetics. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Manichaikul A, et al. Robust relationship inference in genome-wide association studies. Bioinformatics (Oxford, England) 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.