

Abstract
The extensive environmental adaptability of the genus Paenibacillus is related to the enormous diversity of its gene repertoires. Paenibacillus sp. SSG-1 has previously been reported, and its agar-degradation trait has attracted our attention. Here, the genome sequence of Paenibacillus sp. SSG-1, together with 76 previously sequenced strains, was comparatively studied. The results show that the pan-genome of Paenibacillus is open and indicate that the current taxonomy of this genus is incorrect. The incessant flux of gene repertoires resulting from the processes of gain and loss largely contributed to the difference in genomic content and genome size in Paenibacillus. Furthermore, a large number of genes gained are associated with carbohydrate transport and metabolism. It indicates that the evolution of glycometabolism is a key factor for the environmental adaptability of Paenibacillus species. Interestingly, through horizontal gene transfer, Paenibacillus sp. SSG-1 acquired an approximately 150 kb DNA fragment and shows an agar-degrading characteristic distinct from most other non-marine bacteria. This region may be transported in bacteria as a complete unit responsible for agar degradation. Taken together, these results provide insights into the evolutionary pattern of Paenibacillus and have implications for studies on the taxonomy and functional genomics of this genus.
Introduction
The genus Paenibacillus was designated in 1993 and was then composed of 11 species originally belonging to the genus Bacillus 1. Novel species of this genus have been rapidly discovered, and currently, more than 150 named species have been identified. Members of this genus are facultative anaerobic, endospore-forming, motile and rod-shaped bacteria. Most of these bacteria are Gram positive, while others are Gram negative or variable2–4. The genus Paenibacillus is physiologically, biochemically and morphologically diverse and is present in various environments, such as soil5, spring water6, insect larvae7 and human feces4. Corresponding to its adaptability to a wide range of environment, the specific biological characteristics of these bacteria vary. This group initially received attention because of the excellent ability to promote plant growth in some species, such as Paenibacillus polymyxa, via several mechanisms, including phosphate solubilisation and nitrogen fixation8. Paenibacillus larvae has been reported as one of two bacterial species pathogenic for American Foulbrood (AFB), a fatal, globally spread epizootic disease7. These bacteria have also been noted for their ability to hydrolyze a variety of carbohydrates, including cellulose, starch, and xylan9, 10. Whole-genome sequencing has provided insights into the molecular mechanisms of these unique attributes. Djukic et al. identified toxic proteins of Paenibacillus larvae based on genome searching and compared the pathogenic mechanisms of two genotypes, ERIC I and ERIC II11. Dsouza et al. comparatively analyzed Antarctic and temperate species of Paenibacillus and identified traits of Paenibacillus darwinianus that enable it to withstand extremely cold environments12. Recently, Xie et al., using 35 newly sequenced or previously reported Paenibacillus genomes, systematically studied the mechanisms of plant growth promotion13. However, the reason these strains exhibit broad environmental adaptability remains unknown.
Comparative genomics revealed that the diversity of the gene repertoires endows these microorganisms with various metabolic activities and extensive adaptabilities to the environment14–18. In this context, the key unit of microbial evolution is not the genome of an individual bacterium but, rather, the pan-genome of a prokaryote species. Moreover, comparative genomics indicated that the gene repertoires of species are dynamic, and the incessant flux reflects expansion through horizontal gene transfer (HGT), gene duplication, the potential de novo emergence of genes, and contraction via gene loss and genome reduction19. Many obligate endosymbionts of insects lack genes considered essential in other bacteria, and the genome size of some bacterial symbionts is even reduced to 150 kb20. By contrast, some prokaryotic organisms have enormous gene sets of as many as 13 Mb for responding to complex environments21, 22. Bacteria of the Paenibacillus genus show the extensive environmental adaptability and the broad range of genome size23, 24. However, research on adaptability of this genus based on genome dynamics is not available. To date, more than 100 Paenibacillus strains, covering a variety of habitats, have been sequenced, and these large available genome data make it possible for a comprehensive understanding of the adaptability of these microbes to environment.
Additionally, our lab has isolated the soil bacterium Paenibacillus sp. SSG-1, which exhibits agar degradation25. We comprehensively examined the characters of Aga1, an agarase gene in Paenibacillus sp. SSG-1, and proposed that this gene was acquired through HGT26. However, we subsequently found that the HGT event was involved in not only Aga1 gene but also a large region. In the present study, we sequenced the genome of Paenibacillus sp. SSG-1, conducted a comparative genomic analysis of SSG-1 using 76 previously sequenced strains (75 Paenibacillus spp. and 1 bacillus subtilis), and constructed a pan-genome for Paenibacillus. The 75 selected Paenibacillus spp. were originated from various niches, such as diverse soil, water, plant and human isolates, and their genomes are available in the public database. The present study examined the effects of genome dynamics on the adaptability of Paenibacillus species to disclose the evolution pattern of the genus Paenibacillus. Moreover, the HGT event endowing Paenibacillus sp. SSG-1 with the agar-degrading trait was further investigated.
Results
Genome features of Paenibacillus sp. SSG-1
The genome of Paenibacillus sp. SSG-1 was sequenced. The genomic features of SSG-1 are shown in Fig. 1 and Table 1, and those of other genus members are shown in Dataset S1. The SSG-1 genome comprises a 7.56 Mb chromosome, which is larger than most other members of the genus (4.05~8.82 Mb, mean 6.50 Mb). A similar result was observed in protein-coding gene number, for which SSG-1 has 6,659 genes and other strains have 3,668 to 7,522 genes (mean 5,476). The GC content of SSG-1 is 0.5305, while that of other strains is 0.4134 to 0.6327 (mean 0.4879). The result from scanning the SSG-1 genome shows that the GC content is unevenly distributed and undergoes relatively dramatic changes in some location, and that abundant insert sequences (IS) and transposon sequences (TE) are present in SSG-1 genome. It suggests that a large number of HGT events occurred in the evolution of SSG-1.
Figure 1.
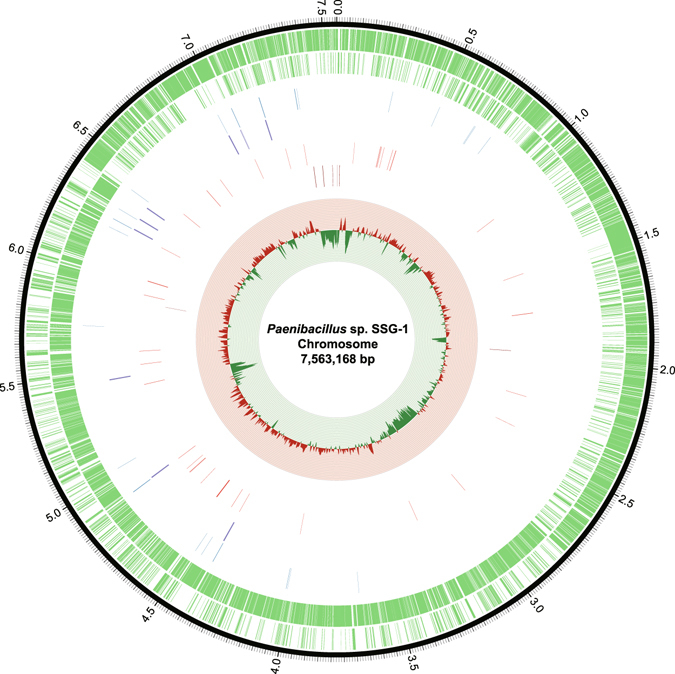
Circular diagrams of the Paenibacillus sp. SSG-1 chromosome. Information from the outermost circle to the innermost circle provide the following data: (1) position in megabases (black); (2) forward strand CDSs (green); (3) reverse strand CDSs (green); (4) tRNAs (blue); (5) rRNAs (purple); (6) repeats (red); (7) ISs and TEs (dark red); and (8) deviation of the GC content per 5000 bp compared with the global genome (positive: red; negative: green; wave range: −0.1714~0.0832).
Table 1.
General features of Paenibacillus sp. SSG-1 genome.
| Category | Number |
|---|---|
| Genome size (nt) | 7,563,168 |
| G + C content (%) | 0.5305 |
| Protein-coding gene | 6,812 |
| Genes with assigned function | 5,827 |
| tRNA | 87 |
| rRNA | 25 |
| sRNA | 48 |
| Tandem repeat | 231 |
| Transposon | 20 |
| IS element | 17 |
Pan-genome analysis reveals great difference in Paenibacillus
In order to ensure the reliability of subsequent analysis, we implemented a filtering process for all downloaded gene sets (see section “Materials and Methods” for more information) and performed BUSCO assessment27 of filtered gene sets (together with Paenibacillus sp. SSG-1). The average of completeness of the gene sets is 96.5% (based on bacillales_odb9 lineage dataset) (Table S1), which indicates that the datasets are highly reliable and suitable for the subsequent analyses.
Using the reciprocal best hit method28, the homology relationship of genes of species were constructed. Among 76 Paenibacillus spp., 416,204 coding genes were clustered in 69,882 gene families. The number of gene families in strains ranges from 3,288 to 6,934 (mean 5,244), which largely consistent with the number of genes, indicating that most genes do not have multiple copies. The number of common gene family is 599, and genes clustered in common gene families occupies 8.64% to 18.22% (mean 11.69%) of total genes in each strain (Figure S1). The number of strain unique gene families ranges from 9 to 1469 in each strain, and genes clustered in unique gene families occupies 0.18% to 29.82% (mean 9.26%). Among 69,882 gene families, only 599 (0.86%) are shared by all 76 Paenibacillus spp., but up to 37,557 (53.74%) are uniquely found in one strain (Fig. 2F), indicating that there is the great difference in these genomes. In more detail, we selected five high-quality genomes of Paenibacillus polymyxa to investigate intra-species difference. Total 6,052 gene families were identified, and 3,650 (60.31%) are shared by the five strains (Fig. 2C). Although the five strains are the same species, about 20% of the gene families in their genomes are not conserved. Similarly, we chose five high-quality genomes of different species to investigate inter-species difference. The result shows the difference among the five strains greatly increased. Total 12,601 gene families were identified, but only a very small part (1,423, 10.99%) is conserved (Fig. 2D). All of the above results indicate that the difference in the genomes of Paenibacillus is very large. Unlike Nocardiopsis spp. which showed the largest proportion of genes are conserved (43.1%)17, Paenibacillus spp. have only a small portion of the conserved genes and more genes are varied.
Figure 2.
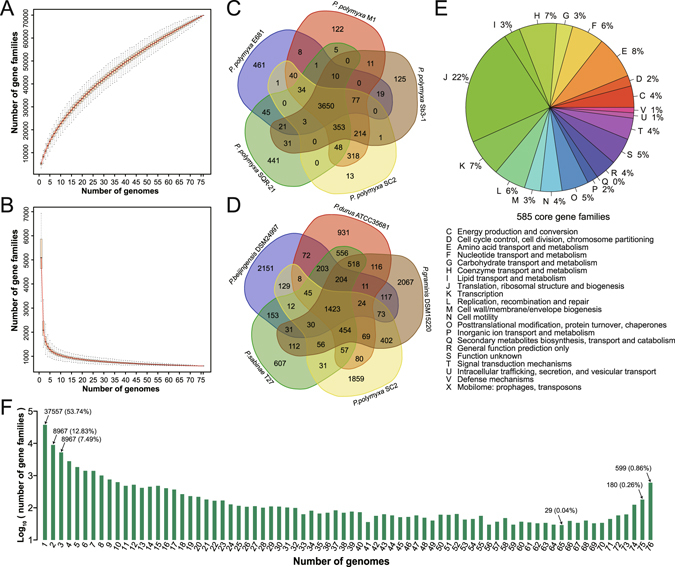
Pan-genome analysis of Paenibacillus. (A) The pan-genome accumulation curve. (B) The core-genome accumulation curve. (C) Venn diagram of the gene families in five Paenibacillus polymyxa strains. (D) Venn diagram of the gene families in five different species of Paenibacillus. (E) Functional distribution of the gene families in the core-genome. (F) Distribution of gene families shared in different numbers of genomes.
Furthermore, to estimate the size of the pan-genome for the genus Paenibacillus, the number of gene families after continually adding new genome data was analyzed during 1,000 random duplicate tests. The size of the pan-genome rapidly increased with each addition (mean 862 for each increase), even when the final genome was added (Fig. 2A). Similarly, to estimate the size of the core-genome for the genus Paenibacillus, the number of shared gene families after continually adding new genome data was analyzed during 1,000 random duplicate tests. The size of core-genome initially sharply descended and finally relatively stabilized at a minimum of 599 gene families (Fig. 2B). A great substantial change was found in the size of the pan-genome after the addition of new genomes, indicating that the pan-genome of Paenibacillus is open. The open pan-genome suggests that Paenibacillus tend to change genomic content to adapt to the environment.
Core-genome is expected to be important or essential in all Paenibacillus. Thus, we used Cluster of Orthologous Groups (COG) assignment to categorize the functions of gene families in Paenibacillus core-genome (Fig. 2E). The result shows that genes involved in translation, ribosomal structure and biogenesis (Category J) occupies largest portion in core genome, indicating that most of these genes are housekeeping. We used Paenibacillus sp. SSG-1 as the reference and performed GO enrichment analysis of its core genes. Besides housekeeping functions, the significant functions include flagellum assembly (GO:0044780), flagellum-dependent cell motility (GO:0071973) and chemotaxis (GO:0006935) (Table S2). It suggests that mobility is important to Paenibacillus. In complex environments, Paenibacillus could move towards regions where there are higher concentrations of nutrients or other beneficial conditions for survival. With respect to the entire pan-genome of 76 Paenibacillus spp., carbohydrate transport and metabolism (Category G) and transcription (Category K) are most abundant in the COG assigned gene families (Figure S2A). A similar result was obtained from functional distribution of individual gene set (Figure S2B). These results show that a large proportion of expanded genes in genomes of Paenibacillus is responsible for glycometabolism and transcriptional regulation.
Phylogenetic analysis contributes to taxonomy of Paenibacillus
As a molecular marker, 16S rDNA is widely used for strain identification, but the sensitivity of this method for sub-genus distinction is markedly decreased. Because the 16S rDNA sequences of Paenibacillus species show high similarity, the phylogenetic tree based on these sequences is ambiguous with a low value of bootstraps in some nodes (Figure S3). The phylogeny based on the core-genome remarkably overcomes this problem and has become a standard in the last few years.
In the present study, the core-genome phylogeny of the genus Paenibacillus was investigated. The Bacillus subtilis 16829 with latest genome was selected an outgroup. We concatenated 369 aligned single-copy coding sequences and obtained a matrix of 219,777 precise positions to construct a species phylogenetic tree (Fig. 3). The maximum likelihood and Bayesian inference methods generated highly consistent results, indicating the tree is robust. The newly constructed species tree will contribute to correct and improve the current taxonomy of Paenibacillus. The phylogenetic tree shows that P. panacisoli DSM 21345 and P. massiliensis DSM 16942 are likely the same species (16S similarity: 99.53%), P. sp. J14 should belong to P. barengoltzii (16S similarity: 99.32%), and P. peoriae HS311 (16S similarity: 99.49%) and P. sp. UNCCL52 (16S similarity: 99.69%) should belong to P. polymyxa (Fig. 3 and Dataset S2).
Figure 3.
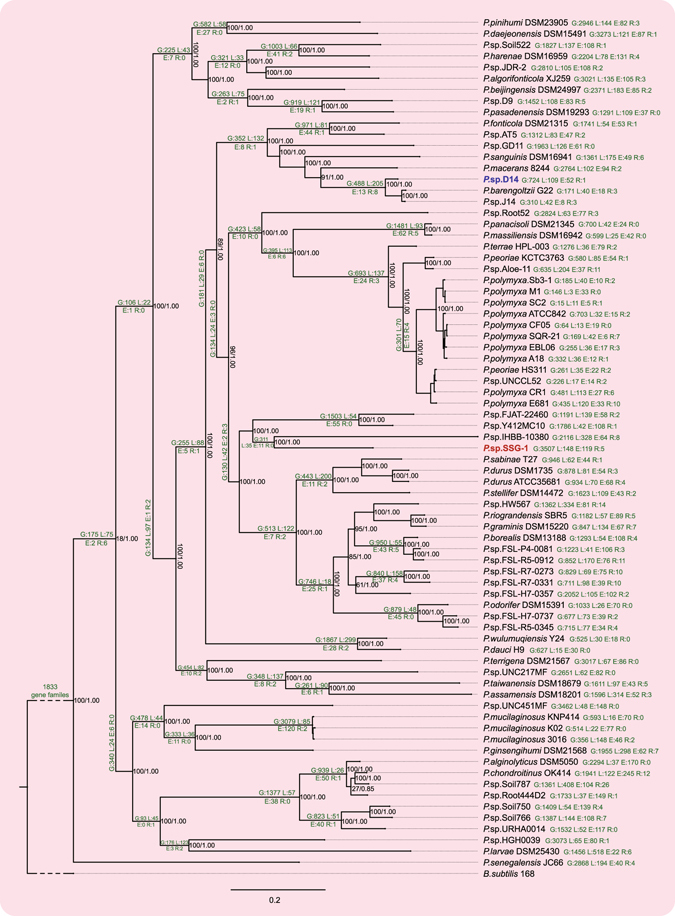
Phylogenetic tree of Paenibacillus. The numbers in nodes denote the bootstrap value (maximum 100) and Bayesian posterior probability (maximum 1.00). The numbers in branches denote the numbers of four categories of the change events (G: gain; L; loss; E: expansion; R: reduction). Numbers in short branches or in crowded nodes are omitted.
Evolution of glycometabolism is crucial for Paenibacillus survival
Using a parsimony approach, the ancestral gene sets were reconstructed, and events leading to changes in the genomic content were identified and mapped to species tree (Fig. 3). We divided the change events into four categories: 1) gain, which denotes that a gene family was not present in the ancestral gene set and came into beings; 2) loss, which denotes that a gene family was present in the ancestral gene set and missed; 3) expansion, which denotes that the member of a gene family increased; 4) reduction, which denotes that the member of a gene family reduced (Figure S4). The results show the common ancestor of the genus Paenibacillus contained 1,833 gene families, which is larger than the size of core genome. It suggests that massive gain events occurring in every strain result in an increase of their genomic sizes. The occurrence frequency of loss is second, and lineage-specific loss events resulted in a decrease in the size of the core genome. The results indicate that gain and loss largely contributed to the difference in genomic content and genome size in Paenibacillus.
Gained genes are often more important for survival. To understand the roles of gained genes, we further investigated the functional distributions of the recently gained genes in each strain based on COG assignment (Fig. 4). The result shows that the gained genes are most abundant in carbohydrate transport and metabolism (Category G), following by transcription (Category K), corresponding to the functional distribution of pan-genome described above. This observation is different from the previous result that genes involved in regulation and secondary metabolism (Category K, T and Q) disproportionately expanded in larger genomes but genes associated with carbohydrate transport and metabolism (Category G) showed no significantly increase30. Expansion in Category K is not surprising, for increased genome and complex environmental conditions require more sophisticated transcriptional regulation systems30. The proteins encoded by gained genes of Category G can be divided into two parts: carbohydrate transporters and carbohydrate-active enzymes (CAZymes). ATP-binding cassette transporters (ABC transporters) and major facilitator superfamily transporters (MFS transporters) are two types of proteins that are responsible for transporting nutrients, including ions, amino acids and sugars, with representatives in both eukaryotes and prokaryotes31, 32. The phosphotransferase system transporters (PTS transporters) are involved in transporting many sugars into bacteria, and vary with different environments where the different carbon sources are available33. All three types of transporter genes are abundant in gained gene repertoires of Paenibacillus (Dataset S3). The CAZy database is currently the most authoritative CAZyme classification database34. The gained CAZymes of Paenibacillus were classified into 74 glycoside hydrolase families (GHs), 14 glycosyltransferase families (GTs), 7 polysaccharide lyase families (PLs) and 7 carbohydrate esterases families (CEs), covering a broad-spectrum of substrate activity (Dataset S3). The results indicate that Paenibacillus in different habitats evolved different abilities to utilize various carbon sources. For example, Paenibacillus sp. JDR-2 was isolated from the cut stems of sweet gum35, and it gained many xylose transport and metabolism related genes (Dataset S3). Seven strain were isolated from milk, and they gained many genes associated with lactose degradation (Dataset S1 and Dataset S3). Taken together, these observations indicate that the pattern of genome evolution is not fixed, and that the evolution of glycometabolism may be crucial to the survival of Paenibacillus species.
Figure 4.
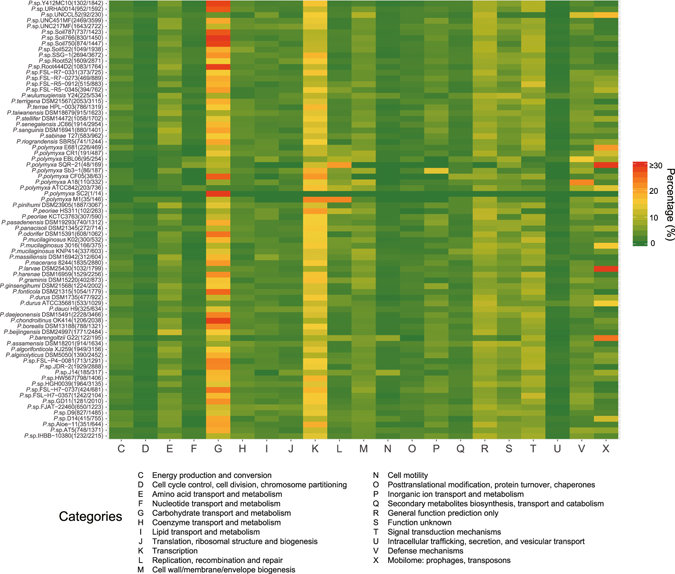
Functional distribution of the gained genes in each strain. The numbers listed after the strain name denote the number of COG assigned genes and total genes.
Agar-degrading traits of Paenibacillus sp. SSG-1 conferred through horizontal gene transfer
Paenibacillus sp. SSG-1 has the ability to degrade agar, a mixture of heterogeneous galactans. However, the agar-degrading traits are usually presented by marine organisms36–38. We proposed that the agarase gene Aga1 together with surrounding genes in SSG-1 were acquired through HGT, but the evidences obtained in the previous study were limited26. In the pan-genome analysis, we found that Aga1 were clustered with two hypothetic agarase genes in Paenibacillus sp. D14 and one of two is virtually identical with Aga1. Therefore, we implemented whole-genome alignment analyses between SSG-1 and D14. Interestingly, an almost identical region was detected in the two genomes and approximately locates at 7.40–7.55 M of the Paenibacillus sp. SSG-1 genome, which contains the comprehensively studied gene of Aga1 (Fig. 5). Moreover, we detected abundant insert sequences and transposon sequences in this matching region (Fig. 1). These results indicate that D14 may also possess agar-degrading capacity and both strains may have acquired this trait through horizontal gene transfer.
Figure 5.
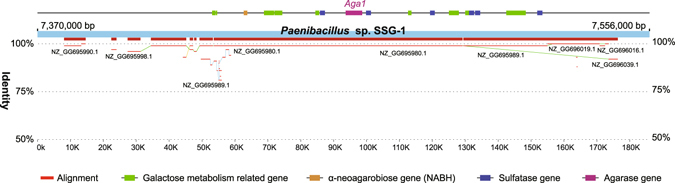
Match of whole-genome alignment between Paenibacillus sp. SSG-1 and D14 in the HGT region.
Subsequently, additional evidence of HGT was obtained. The 7.40–7.55 M region of the Paenibacillus sp. SSG-1 genome shows an obvious difference in GC content (mean GC: 0.4597) compared with the global genome (mean GC: 0.5305). We further calculated the frequency of codon usage in each region of the SSG-1 genome using a sliding window (20 kb window size; 10 kb step size). Then, a 61-dimensional matrix of codon usage frequency was obtained, and we performed the principal component analysis (PCA) for this matrix. The contribution of the principal component one (PC1) and the principal component two (PC2) reach 65.96% and 6.13%, respectively. Therefore, we used the scores of PC1 and PC2 for cluster analysis and result display. The results show that the windows located in 7.40–7.55 M region gathered but deviated from the main cluster (Fig. 6). It indicates that the codon usage bias of the genes located in 7.40–7.55 M region is different from that of most other genes (Fig. 6 and fF S3). Taken together, above results intensively support the inference that the 7.40–7.55 M region was inserted into the genome of Paenibacillus sp. SSG-1 through HGT.
Figure 6.
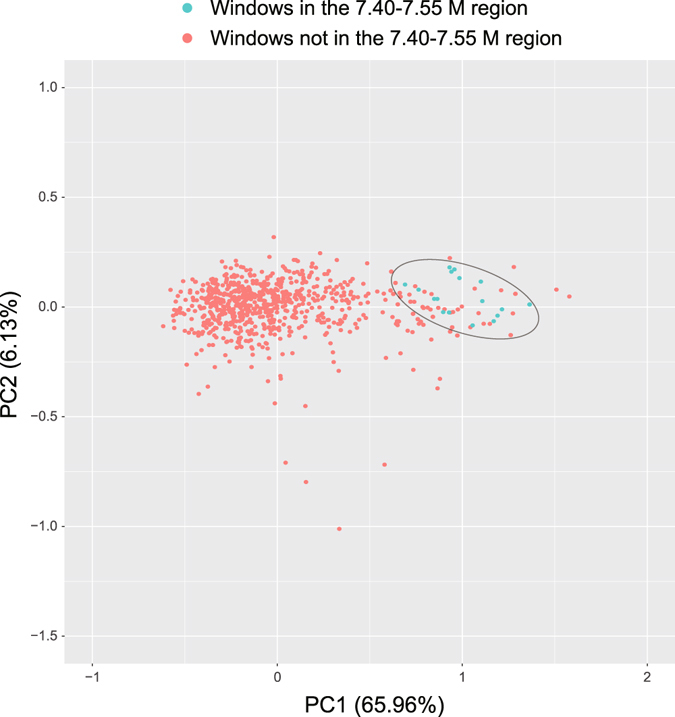
PCA analysis of codon usage frequency of Paenibacillus sp. SSG-1. The X and Y axes represent the scores of the first two principal components and each dot denotes 20 kb genome regions.
Furthermore, we performed a GO enrichment analysis of genes located in the 7.40–7.55 M regions of the Paenibacillus sp. SSG-1 genome, and the significant functions include galactose metabolism (GO:0006012) and sulfuric ester hydrolase activity (GO:0008484) (Table S4). Agar consists of alternating 3-O-linked β-D galactopyranose (G) and 4-O-linked α-L-galactopyranose (LA) linked to sulphate or other groups39. Agar degradation involved multiple process: sulfatases hydrolyze the sulphate ester in agar to transform sulphated agar into agarose; agarases degrade agarose into agaro-oligosaccharides; agaro-oligosaccharides are further hydrolysed by α-neoagarobiose hydrolases (NABH) into the monomers G and LA, and finally utilized by galactose metabolic processes39, 40. Complete pathway and abundant genes related to agar degradation were found in 7.40–7.55 M region (Fig. 5), suggesting that this region as a complete unit responsible for agar degradation merged into the Paenibacillus sp. SSG-1 genome.
Discussion
The genome size of microorganisms influences their extensive adaptabilities to the environment. Under most conditions, strains with larger genome size generally may be more adaptive to complex habitats, as these microorganisms may encode more products for metabolism and stress tolerance41. However, some studies have suggested that the small size of bacterial genomes may be also competitive, reflecting advantages in energy saving and reproductive efficiency42, 43. In summary, there is a balance between maintaining a minimum genome size and facilitating the response to environmental conditions.
The genus Paenibacillus exhibits extensive environmental adaptability and can populate various ecological niches. To our knowledge, this study is the first to investigate the pan-genome of the genus Paenibacillus and explore the evolutionary reason for the wide niche adaptation of these bacteria. The results show that the pan-genome of the genus Paenibacillus is open and theoretically infinite, suggesting that Paenibacillus species tend to acquire new genes to enhance the adaptability. In contrast to increasing pan-genome, the core genome was decreasing, suggesting that the difference in genomic content in Paenibacillus was gradually increasing. Free-living bacteria typically have an open pan-genome whose size shows a tendency toward continuous growth44. Bacteria must change their genetic material to adapt to variable environmental conditions, thus, greater niche diversity reflects larger pan-genomes30, 45. Considering the wide distribution of Paenibacillus, a large pan-genome size corresponds to diverse living conditions.
The gene repertoires of bacteria are dynamic, and the incessant flux reflects four major events: gain, loss, expansion and reduction (Figure S4). The events of gain and loss occurred frequently in the evolution of Paenibacillus species. The present results show that the gene repertoires of the genus Paenibacillus are in incessant flux and the genome size of Paenibacillus shows an increasing tendency. As described above, to reduce energy consumption, Paenibacillus species abandoned some dispensable genes while acquiring new characters. The events of gain and loss during evolution largely contributed to the difference in genomic content and genome size. Gained genes are more important for survival. Paenibacillus species gained a large number of genes associated with carbohydrate transport and metabolism, which differ from the most other bacteria30. After a long time of evolution, although the core genome became very small, it still contains a large number of genes associated with flagellar movement. These results indicate that active movement and sugar uptake are the keys to the survival of Paenibacillus. For different habitats, carbon sources which microorganisms can directly use are so diverse that bacteria must evolve their glycometabolism ability to ensure environmental adaptability. Hence, the broad environmental adaptability of Paenibacillus may be caused by abundant glycometabolism-related genes.
The main approaches of gain include horizontal gene transfer (HGT) and gene diversification, and the former is more widespread in bacteria46. The agar-degrading traits of Paenibacillus sp. SSG-1 may largely reflect HGT. The methods used to detect HGT are primarily divided into two categories: parametric and phylogenetic methods47. The parametric method is based on genomic features, and foreign gene regions are typically show differences in GC-content, codon usage, tetra-nucleotide frequency and other attributes. However, these methods are limited by recent HGT events, as the alien features are assimilated with time48. The phylogenetic methods explore the evolutionary histories of genes involved and identify conflicting phylogenies, i.e., considering a well-established species tree and a gene tree, and the inconsistencies in some nodes may indicate occurrences of HGT. In the present study, a large HGT region in Paenibacillus sp. SSG-1 genome was confirmed using various methods. The HGT region endows SSG-1 an agar-degrading capacity which is distinct from most other non-marine bacteria. In addition, a homologous HGT region was observed in Paenibacillus sp. D14. According to the phylogenetic tree, SSG-1 is far from D14 and their closely related strains do not possess this agar-degrading region (Fig. 3). Therefore, we suggested the following hypothetical HGT pathways for these two regions in SSG-1 and D14: first, one common ancestor provided this region separately inserted into SSG-1 and D14’ genomes; second, one of SSG-1 or D14 acquired this region through HGT, and subsequently transferred this genetic material to another strain. In either case, the two agar-degrading regions of SSG-1 and D14 show the same origin during evolution. However, the issue who is the donor or where is the origin of this region still is a puzzle. We attempted to blast this region against the nr/nt, refseq genomic and gss databases using the NCBI online service, but no other remarkable match was observed. Considering the origin of soil-isolated agarase, as previously discussed26, this region is most likely derived from marine organisms.
Conclusions
To our knowledge, this study is the first to investigate the pan-genome of the genus Paenibacillus and explore the evolutionary reason for the wide niche adaptation of these microorganisms. The pan-genome of Paenibacillus is open and members in this genus tend to change their genomic contents to adapt to the environment. The events of gain and loss during evolution largely contributed to the difference in the genomic content and the genome size. The evolution of glycometabolism is a key factor for the environmental adaptability of the genus Paenibacillus. Moreover, the core-genome phylogeny contributed to taxonomy in the genus Paenibacillus.
Materials and Methods
Genome sequencing and data acquisition
For Paenibacillus sp. SSG-1, strain culture and genome DNA extraction were the same as previously reported25. Genome DNA was fragmented, and two DNA libraries were constructed (~500 and ~6,000 bp). Whole-genome sequencing was performed using a high-throughput Illumina HiSeq 2000 sequencing platform at BGI-Shenzhen, Shenzhen, China. After cleaning, the paired-end reads were assembled using SOAPdenovo v2.0449, and 754 Mb of the read sequences represented a 100-fold coverage of the genome. Structures of protein-coding genes, structural RNAs (5S, 16S, 23S), tRNAs and small non-coding RNAs were predicted using NCBI Prokaryotic Genome Annotation Pipeline (PGAP)50. The cleaned sequencing reads were deposited in the SRA database under accession number SRR3948181, and the genome sequence was deposited in GenBank under accession number MBRK00000000.
Other strain genomes and annotation information used in the present study were downloaded from the NCBI refseq genomic database and were summarized in Dataset S1.
Filtering and assessment of gene sets
In order to ensure the reliability of subsequent analysis, the gene sets of all strains were strictly filtered. We considered the screening criteria: 1) protein-coding genes encoding less than 50 aa were filtered, 2) genes which contained an inside stop codon in ORF were filtered, and 3) genes with a high proportion of N (>30%) were also filtered.
The completeness and quality of filtered gene sets was assessed using BUSCO v2.027 based on evolutionarily informed expectations of gene content. We used two BUSCO lineage datasets for our assessments: firmicutes_odb9 containing 232 BUSCOs and bacillales_odb9 containing 526 BUSCOs.
Functional annotation and analysis
Annotation of protein coding sequence was performed by blasting to functional databases, including NCBI non-redundant (NR, filtered unknown and hypothetical proteins), SwissProt51, and enhanced COG database52. Poor alignments were removed, and the highest quality alignments were selected as gene annotations. Gene ontology (GO) terms were assigned using Blast2GO software53. KEGG annotation was implemented using online BlastKOALA54. Carbohydrate-active enzymes34 were classified into families using online dbCAN annotation server55.
GO enrichment analyses were performed using the R package topGO56, and the P-values were calculated using Fisher’s exact test and corrected using Benjamini-Hochberg method. The GO term is significant when corrected P-values ≤ 0.05.
Pan-genome analysis
Proteins of all strains were reciprocally compared using blastp v2.2.31+, and poor alignments were removed. Subsequently, the proteins were classified into families using OrthoMCL v2.0.957 with an inflation index of 1.5. The core, single-copy and unique families were identified according to the classification results. Gene accumulation curves, describing the number of core and pan gene families, with the addition of new comparative genomes were performed using a self-writing R script for parsing OrthoMCL results. This procedure was repeated 1,000 times for every addition of a randomly selected genome to obtain median values.
Phylogenetic analysis
The proteins were aligned using muscle v3.8.42558 per single-copy family, followed by transformation to a CDS alignment using pal2nal v1459. Subsequently, we used Gblocks v0.91b60 to extract unambiguous parts and concatenated all family alignments. We used this alignment for further phylogenetic analysis. RAxML v8.2.761 and MrBayes v3.2.462 were used to construct phylogenetic trees. For RAxML, we selected a GTR substitutions model with an estimated proportion of gamma distribution and invariant sites, and set 1,000 bootstrap iterations for calculation. For MrBayes, we selected the same nucleotide substitution model and set 1,000,000 iterations for MCMC, and trees were sampled every 100 iterations with the first 2,500 samples as burn-in.
The 16S rDNA sequences were obtained according to annotation information. The pairwise identity of 16S rDNA was obtained using blastn and summaried in Dataset S2. The sequences with high integrity (>1,000 bp) were used for phylogenetic analysis. A clustal alignment algorithm and neighbor-joining clustering method were implemented in MEGA v763.
Genomic content change
The ancestral gene sets were reconstructed using Count v10.0464 based on the Wagner parsimony algorithm with a gain penalty of one. Events leading to changes in the genomic content (gain, loss, expansion and reduction) were identified after comparing each node with its latest ancestor.
Inference of horizontal gene transfer
Whole genome alignments were performed using the nucmer script in the MUMmer v3.23 software package65 with Paenibacillus sp. SSG-1 as the reference genome. Alignment maps were drawn using the mapview script in the MUMmer package. Insert and transposon sequences were detected using ISFinder66. The GC content per 5 kb was calculated using self-writing Perl script. The circular map of Paenibacillus sp. SSG-1 was drawn using Circos v0.67.767.
The codon usage analysis was performed according to the following steps: A 20 kb sliding window with 10 kb step size was scanning the genome of Paenibacillus sp. SSG-1, and the codon usage frequencies (FCU, calculated using self-writing Perl script) of the genes located in each window (overlapped >50%) were calculated. Therefore, a 61-dimension matrix was generated, and every dimension described one FCU per codon. We subsequently performed a principal component analysis (PCA) for this matrix using R, and the scores of the first two compositions were used to exhibit the results. In addition, a general comparison of codon usage between the 7.40–7.55 M region with the global genome was also shown in Table S3.
Electronic supplementary material
Acknowledgements
This work was supported by the National Twelfth Five-year Science and Technology support program (2014BAD02B02), the National Natural Science Foundation of China (31272659), the National Infrastructure of Natural Resources for Science and Technology Program of China (NIMR-2014-8) and the Sichuan Science and Technology Bureau (2014GXZ0005 and 2015JPT0032).
Author Contributions
S.Q. and H.X. conducted the experiments, analysed the results. Y.L., M.L., X.C. and D.Q. collected data. H.X., S.Q., Yu C. and Yi C conceived the idea for this project and designed the experiments. H.X., S.Q. and Yi C. wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Hui Xu and Shishang Qin contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-06160-9
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yu Cao, Email: caoyu1984@163.com.
Yi Cao, Email: geneium@scu.edu.cn.
References
- 1.Ash C, Priest FG, Collins MD. Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Proposal for the creation of a new genus Paenibacillus. Antonie Van Leeuwenhoek. 1993;64:253–260. doi: 10.1007/BF00873085. [DOI] [PubMed] [Google Scholar]
- 2.Elo S, et al. Paenibacillus borealis sp, nov., a nitrogen-fixing species isolated from spruce forest humus in Finland. Int. J. Syst. Evol. Microbiol. 2001;51:535–545. doi: 10.1099/00207713-51-2-535. [DOI] [PubMed] [Google Scholar]
- 3.Lee FL, Kuo HP, Tai CJ, Yokota A, Lo CC. Paenibacillus taiwanensis sp nov., isolated from soil in Taiwan. Int. J. Syst. Evol. Microbiol. 2007;57:1351–1354. doi: 10.1099/ijs.0.64764-0. [DOI] [PubMed] [Google Scholar]
- 4.Mishra AK, Lagier JC, Rivet R, Raoult D, Fournier PE. Non-contiguous finished genome sequence and description of Paenibacillus senegalensis sp nov. Stand. Genomic Sci. 2012;7:70–81. doi: 10.4056/sigs.3056450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao M, et al. Paenibacillus beijingensis sp nov., a novel nitrogen-fixing species isolated from jujube garden soil. Antonie Van Leeuwenhoek. 2012;102:689–694. doi: 10.1007/s10482-012-9767-2. [DOI] [PubMed] [Google Scholar]
- 6.Tang QY, et al. Paenibacillus algorifonticola sp. nov., isolated from a cold spring. Int. J. Syst. Evol. Microbiol. 2011;61:2167–2172. doi: 10.1099/ijs.0.025346-0. [DOI] [PubMed] [Google Scholar]
- 7.Neuendorf S, Hedtke K, Tangen G, Genersch E. Biochemical characterization of different genotypes of Paenibacillus larvae subsp larvae, a honey bee bacterial pathogen. Microbiology. 2004;150:2381–2390. doi: 10.1099/mic.0.27125-0. [DOI] [PubMed] [Google Scholar]
- 8.Bloemberg GV, Lugtenberg BJJ. Molecular basis of plant growth promotion and biocontrol by rhizobacteria. Curr. Opin. Plant Biol. 2001;4:343–350. doi: 10.1016/S1369-5266(00)00183-7. [DOI] [PubMed] [Google Scholar]
- 9.Rajesh T, et al. Identification and Functional Characterization of an alpha-Amylase with Broad Temperature and pH Stability from Paenibacillus sp. Appl. Biochem. Biotechnol. 2013;170:359–369. doi: 10.1007/s12010-013-0197-z. [DOI] [PubMed] [Google Scholar]
- 10.St John FJ, Rice JD, Preston JF. Paenibacillus sp. strain JDR-2 and XynA(1): a novel system for methylglucuronoxylan utilization. Appl. Environ. Microbiol. 2006;72:1496–1506. doi: 10.1128/AEM.72.2.1496-1506.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Djukic, M. et al. How to Kill the Honey Bee Larva: Genomic Potential and Virulence Mechanisms of Paenibacillus larvae. PLoS ONE9, doi:10.1371/journal.pone.0090914 (2014). [DOI] [PMC free article] [PubMed]
- 12.Dsouza, M., Taylor, M. W., Turner, S. J. & Aislabie, J. Genome-Based Comparative Analyses of Antarctic and Temperate Species of Paenibacillus. PLoS ONE9, 10.1371/journal.pone.0108009 (2014). [DOI] [PMC free article] [PubMed]
- 13.Xie, J. et al. Comparative genomic and functional analysis reveal conservation of plant growth promoting traits in Paenibacillus polymyxa and its closely related species. Scientific Reports6, doi:10.1038/srep21329 (2016). [DOI] [PMC free article] [PubMed]
- 14.Koonin EV, Wolf YI. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008;36:6688–6719. doi: 10.1093/nar/gkn668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bentley S. Sequencing the species pan-genome. Nature Reviews Microbiology. 2009;7:258–259. doi: 10.1038/nrmicro2123. [DOI] [PubMed] [Google Scholar]
- 16.Gan HM, Hudson AO, Rahman AYA, Chan KG, Savka MA. Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genomics. 2013;14:1. doi: 10.1186/1471-2164-14-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li HW, et al. Comparative genomic analysis of the genus Nocardiopsis provides new insights into its genetic mechanisms of environmental adaptability. PLoS ONE. 2013;8:e61528. doi: 10.1371/journal.pone.0061528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smokvina T, et al. Lactobacillus paracasei comparative genomics: towards species pan-genome definition and exploitation of diversity. PLoS ONE. 2013;8:e68731. doi: 10.1371/journal.pone.0068731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puigbo P, Lobkovsky AE, Kristensen DM, Wolf YI, Koonin EV. Genomes in turmoil: quantification of genome dynamics in prokaryote supergenomes. BMC biology. 2014;12:66. doi: 10.1186/s12915-014-0066-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nature Reviews Microbiology. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- 21.Shih PM, et al. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. USA. 2013;110:1053–1058. doi: 10.1073/pnas.1217107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneiker S, et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nature Biotechnology. 2007;25:1281–1289. doi: 10.1038/nbt1354. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, J. et al. Draft Genome Sequence of Paenibacillus sp. Strain DMB5, Acclimatized and Enriched for Catabolizing Anthropogenic Compounds. Genome announcements4, doi:10.1128/genomeA.00211-16 (2016). [DOI] [PMC free article] [PubMed]
- 24.Keita MB, et al. Non-contiguous-Finished Genome Sequence and Description of Paenibacillus camerounensis sp. nov. Microb. Ecol. 2016;71:990–998. doi: 10.1007/s00248-015-0722-4. [DOI] [PubMed] [Google Scholar]
- 25.Song T, et al. Purification and characterization of a novel beta-agarase of Paenibacillus sp. SSG-1 isolated from soil. J. Biosci. Bioeng. 2014;118:125–129. doi: 10.1016/j.jbiosc.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Tao, S. et al. Horizontal Transfer of a Novel Soil Agarase Gene from Marine Bacteria to Soil Bacteria via Human Microbiota. Scientific Reports6 (2016). [DOI] [PMC free article] [PubMed]
- 27.Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, btv351 (2015). [DOI] [PubMed]
- 28.Moreno-Hagelsieb G, Latimer K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics. 2008;24:319–324. doi: 10.1093/bioinformatics/btm585. [DOI] [PubMed] [Google Scholar]
- 29.Belda E, et al. An updated metabolic view of the Bacillus subtilis 168 genome. Microbiology. 2013;159:757. doi: 10.1099/mic.0.064691-0. [DOI] [PubMed] [Google Scholar]
- 30.Konstantinidis KT, Tiedje JM. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA. 2004;101:3160–3165. doi: 10.1073/pnas.0308653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pao SS, Paulsen IT, Saier MH., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones PM, George AM. The ABC transporter structure and mechanism: perspectives on recent research. Cell. Mol. Life Sci. 2004;61:682–699. doi: 10.1007/s00018-003-3336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tchieu JH, Norris V, Edwards JS, Saier MH., Jr. The complete phosphotransferase system in Escherichia coli. J. Mol. Microbiol. Biotechnol. 2001;3:329–346. [PubMed] [Google Scholar]
- 34.Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sawhney N, Crooks C, Chow V, Preston JF, St John FJ. Genomic and transcriptomic analysis of carbohydrate utilization by Paenibacillus sp. JDR-2: systems for bioprocessing plant polysaccharides. BMC Genomics. 2016;17:17. doi: 10.1186/s12864-016-2436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohta Y, et al. Cloning, expression, and characterization of a glycoside hydrolase family 86 beta-agarase from a deep-sea Microbulbifer-like isolate. Appl. Microbiol. Biotechnol. 2004;66:266–275. doi: 10.1007/s00253-004-1757-5. [DOI] [PubMed] [Google Scholar]
- 37.Ohta Y, et al. Purification and Characterization of a Novel α-Agarase from a Thalassomonas sp. Curr. Microbiol. 2005;50:212–216. doi: 10.1007/s00284-004-4435-z. [DOI] [PubMed] [Google Scholar]
- 38.Ma C, et al. Molecular cloning and characterization of a novel β-agarase, AgaB, from marine Pseudoalteromonas sp. CY24. J. Biol. Chem. 2007;282:3747–3754. doi: 10.1074/jbc.M607888200. [DOI] [PubMed] [Google Scholar]
- 39.Chi W-J, Chang Y-K, Hong S-K. Agar degradation by microorganisms and agar-degrading enzymes. Appl. Microbiol. Biotechnol. 2012;94:917–930. doi: 10.1007/s00253-012-4023-2. [DOI] [PubMed] [Google Scholar]
- 40.Lee SB, et al. Metabolic pathway of 3, 6-anhydro-L-galactose in agar-degrading microorganisms. Biotechnology and Bioprocess Engineering. 2014;19:866–878. doi: 10.1007/s12257-014-0622-3. [DOI] [Google Scholar]
- 41.Ranea JAG, Buchan DWA, Thornton JM, Orengo CA. Evolution of protein superfamilies and bacterial genome size. J. Mol. Biol. 2004;336:871–887. doi: 10.1016/j.jmb.2003.12.044. [DOI] [PubMed] [Google Scholar]
- 42.Koskiniemi S, Sun S, Berg OG, Andersson DI. Selection-Driven Gene Loss in Bacteria. PLoS Genetics. 2012;8:e1002787–e1002787. doi: 10.1371/journal.pgen.1002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martínezcano DJ, et al. Evolution of small prokaryotic genomes. Frontiers in Microbiology. 2014;5:742. doi: 10.3389/fmicb.2014.00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005;15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Konstantinidis KT, et al. Comparative systems biology across an evolutionary gradient within the Shewanella genus. Proc. Natl. Acad. Sci. USA. 2009;106:15909–15914. doi: 10.1073/pnas.0902000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Todd J, Treangen EPCR. Horizontal Transfer, Not Duplication, Drives the Expansion of Protein Families in Prokaryotes. PLoS Genetics. 2011;7:70–76. doi: 10.1371/journal.pgen.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ravenhall, M., Škunca, N., Lassalle, F. & Dessimoz, C. Inferring Horizontal Gene Transfer. PLoS Computational Biology11, doi:10.1371/journal.pcbi.1004095 (2015). [DOI] [PMC free article] [PubMed]
- 48.Lawrence JG, Ochman H. Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol. 1997;44:383–397. doi: 10.1007/PL00006158. [DOI] [PubMed] [Google Scholar]
- 49.Li RQ, Li YR, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 50.Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. gkw569 (2016). [DOI] [PMC free article] [PubMed]
- 51.Boeckmann B, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:D261–D269. doi: 10.1093/nar/gku1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conesa, A. & Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics2008 (2008). [DOI] [PMC free article] [PubMed]
- 54.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016;428:726–731. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 55.Yin Y, et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexa, A., & Rahnenfuhrer, J. topGO: Enrichment analysis for Gene Ontology. R package version 2.22.0 (2010).
- 57.Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 61.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 63.Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol., msw054 (2016). [DOI] [PMC free article] [PubMed]
- 64.Csűös M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26:1910–1912. doi: 10.1093/bioinformatics/btq315. [DOI] [PubMed] [Google Scholar]
- 65.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:9. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krzywinski M, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.