

Abstract
ARC‐520 Injection, an RNA interference drug for the treatment of hepatitis B that targets cccDNA‐derived viral mRNA transcripts with high specificity, effectively reduces the production of viral proteins and HBV DNA. In this phase 1 randomized, double‐blind, placebo‐controlled study, 54 healthy volunteers (half male, half female) received a single, intravenous dose of 0.01–4.0 mg/kg ARC‐520 Injection (n = 36) or placebo (n = 18). Assessments included safety, tolerability, pharmacokinetics, and pharmacodynamics (cytokines and complement). Pharmacokinetics of the siRNA and peptide excipient components contained in ARC‐520 Injection showed a relatively short half‐life of 3–5 and 8–10 hours, respectively. Dose exposure linearity was demonstrated within the dose range. ARC‐520 Injection was well tolerated, with adverse‐event frequency the same as placebo and no serious adverse events. ARC‐520 Injection was initially found to induce histamine release through mast cell degranulation, resulting in 2 moderate hypersensitivity reactions. However, after initiation of pretreatment with oral antihistamine, no further hypersensitivity reactions occurred. Low‐level, transient complement induction and sporadic, mild, and transient elevations of several cytokines were observed but not associated with any symptoms. ARC‐520 Injection showed a favorable tolerability profile in this single‐dose study in healthy volunteers. Oral antihistamine pretreatment is recommended in the future to offset mast cell degranulation stimulation.
Keywords: viral hepatitis, hepatitis B, treatment, RNA interference, RNAi, pharmacology, pharmacokinetics, safety, tolerability, phase 1, volunteers
With an estimated 240–400 million people worldwide chronically infected with the hepatitis B virus (HBV) and more than 750 000 deaths annually from HBV‐related complications,1, 2, 3, 4, 5 therapies that can induce high rates of hepatitis B surface antigen (HBsAg) seroclearance, an outcome associated with a much improved prognosis, are very desirable. Although current therapies, nucleos(t)ide analog reverse‐transcriptase inhibitors (NUCs) and interferon (IFN), decrease the risk of liver decompensation and improve survival,6, 7, 8 clearance of HBsAg is uncommon. Because discontinuation of NUC therapy results in a high rate of viral rebound and biochemical relapse6, 9, 10, 11, 12 and a long‐term increased risk of hepatocellular carcinoma,13 lifelong NUC therapy is currently recommended for most patients. Innovative therapies that could induce high rates of HBsAg seroclearance, thus reducing off‐treatment disease progression and allowing safe discontinuation of indefinite NUC therapy, are needed.
Therapies based on RNA interference (RNAi) can directly target hepatitis B virus mRNA transcripts with high specificity, profoundly reducing the production of viral proteins, including HBsAg, and potentially allowing restoration of effective host immunity. RNAi uses small, noncoding RNA to regulate the expression of genetic information.14 The RNAi‐based drug ARC‐520 Injection consists of 2 synthetic short interfering RNAs (siRNAs) conjugated to cholesterol, which enhances delivery of the siRNA to hepatocytes. ARC‐520 Injection uses a polymer‐based system, Dynamic PolyConjugates (DPC), for the targeted delivery of siRNA to hepatocyte cytoplasm, where RNAi occurs.15 DPCs use an amphipathic, membrane active peptide, melittin‐like peptide (MLP), which is reversibly masked so that the polymer's membrane activity is only revealed in the acidic environment of the endosome. The masking agent includes the targeting ligand N‐acetylgalactosamine (NAG), causing hepatocyte‐specific delivery via the highly expressed asialoglycoprotein receptor present on the surface of hepatocytes.16 Intravenous coinjection of cholesterol‐conjugated siRNA with the liver‐targeted DPC component results in efficient endosomal escape and cytoplasmic delivery of siRNA to hepatocytes, limiting the toxicity that could result from interaction with nontargeted cells.
The organization of the HBV genome makes it an attractive target for the use of RNAi. HBV persists in the nucleus of hepatocytes as a minichromosome, covalently closed circular DNA (cccDNA), which serves as the template for 5 overlapping viral transcripts including pregenomic RNA.17 Because all viral transcripts expressed from cccDNA share the same termination codon, a single RNAi trigger can target all viral RNA for degradation. The siRNA sequences AD0009 and AD0010 are shown in Table 1. Their selection process and target region within the open‐reading frame of the HBV X protein have been previously described.18 This screening process included a step to eliminate sequences with close similarities to RNA of the human transcriptome. Because ARC‐520 Injection contains 2 siRNAs (AD0009 and AD0010) targeted toward cccDNA‐derived transcripts, it has more extensive genotype coverage than would occur with a single RNAi trigger. AD0009 and AD0010 are an identical match to their target sequence in 96.4% and 92.6%, respectively, of all surveyed HBV genomes; the combination provides coverage of 99.64%18 and reduces the frequency of escape mutants that might arise during treatment because such mutants would need to accumulate at least 2 resistance mutations, one in each target sequence. In addition, the RNAi triggers in ARC‐520 Injection were chemically modified using sugar and backbone analogues to increase nuclease resistance and minimize the potential for induction of an innate immune response.19, 20, 21, 22, 23
Table 1.
Cholesterol‐RNAi Triggers AD0009 and AD0010 Used in the Study
| RNAi Trigger | Strand | Sequence (5’–3’) |
|---|---|---|
| AD0009 | Sense | Chol‐AuCfuGfuAfgGfcAfuAfaAfuUfgGfuAf(invdT) |
| Antisense | dTAfcCfaAfuUfuAfuGfcCfuAfcAfgdTsdT | |
| AD0010 | Sense | Chol‐uAuAfcCfuCfuGfcCfuAfaUfcAfuCfuAf(invdT) |
| Antisense | dTAfgAfuGfaUfuAfgGfcAfgAfgGfudTsdT |
Underline represents hybridization region.
Lower case letter, 2’O‐Me substitution; Nf, 2’F substitution; N, ribose; dN, deoxyribose; invdT, inverted deoxythymidine; s, phosphorothioate bond; Chol, cholesterol.
Using a transiently transgenic pHBV mouse model generated by hydrodynamic tail vein injection of a plasmid harboring the terminally redundant full‐length HBV genome (HBV 1.3),24 it was found that the RNAi triggers were very effective when coinjected with NAG‐MLP, with decreases in HBsAg expression of between 2 and 3 log10 after a single intravenous injection, along with substantial decreases in HBeAg, serum HBV DNA, and liver levels of HBV RNA.18 No knockdown was observed when the RNAi triggers were injected without NAG‐MLP and vice versa, indicating that NAG‐MLP is necessary for effective cytoplasmic delivery and that knockdown is HBV RNAi trigger–dependent. We report here the first clinical assessment of treatment with ARC‐520 Injection in a study designed to evaluate the safety, tolerability, and pharmacokinetics to help determine the dosage range appropriate for study in subsequent clinical trials in patients with chronic HBV infection.
Subjects and Methods
Study Design
This was a single‐center, randomized, double‐blind, placebo‐controlled, sequential, single‐dose‐escalation, first‐in‐human phase 1 study conducted to evaluate the safety, tolerability, and pharmacokinetics of the investigational product ARC‐520 Injection administered intravenously to a healthy volunteer population of adults (the Heparc‐1001 study; Clinicaltrials.gov registration number: NCT01872065). The study was conducted at Nucleus Network, a single center in Melbourne, Australia, with recruitment of subjects starting in July 2013 and database closure in October 2014, with the exception of a follow‐up telephone call for cohort 9, which was completed in November 2014. Eligible subjects were enrolled sequentially into 9 cohorts, with 6 subjects per cohort, and randomized with a 4:2 (active:placebo) ratio. Each subject was assigned to either active (ARC‐520 Injection) or placebo (0.9% normal saline) treatment using a block randomization algorithm. Final confirmation of eligibility was checked on day 1. Because of slight color differences between placebo and ARC‐520 Injection, the syringes were blinded with translucent labels to mask the infusate identity to the blinded staff and participants. Other than pharmacists and staff involved in randomization, dispensing and preparation of the study drug remained blinded. At the completion of the planned initial 6 cohorts through 29 days of follow‐up, the decision was made to unblind the study to assess whether it was safe and appropriate to continue to enroll further cohorts at higher doses. All data were cleaned and queries returned on the first 6 cohorts before database lock. Unblinding each cohort was self‐contained, and it was not believed that unblinding the first 6 cohorts would bias evaluation of subsequent cohorts.
Cohorts 1–6 and 8–9 were initiated with a sentinel group of 2 subjects (1 randomized to placebo and 1 to ARC‐520 Injection). After review by the Data Safety Monitoring Committee (DSMC; composed of the primary investigator [PI] and medical monitor) of the sentinel group's safety and tolerability data through day 3, the remaining 4 subjects in the cohort were randomized and treated on the same day. For cohort 7, which had the same dose as cohort 6, 6 eligible subjects received ARC‐520 Injection or placebo on the same day, with no sentinel group. The safety data through day 3 for an entire completed cohort of subjects was also reviewed prior to enrollment and dosing of the next cohort. DSMC safety review minutes and recommendations were submitted to the independent ethics committee as a notification prior to dosing of the next cohort. There was no intrasubject dose escalation. Subjects fasted from food for at least 8 hours predose and 4 hours postdose and from water from 1 hour predose to 1 hour postdose. Blood samples were collected predose and at specified times after dosing to determine pharmacokinetic (PK) parameters and pharmacodynamic (PD) end points (cytokines and complement). After 2 participants developed limited rashes (1 mild, 1 moderate) during dose administration, the decision was made that participants in cohorts 7–9 would be pretreated with antihistamine (diphenhydramine 50 mg orally) 2 hours prior to treatment administration.
Subjects were confined at the clinical facility from day 1 to the morning of day 2 and then discharged following completion of safety assessments and blood and urine sampling. Visits to the clinical facility occurred on days 3, 4, 8, 15 ± 1 and 29 ± 1 (end‐of‐study visit). A telephone follow‐up occurred on day 90 ± 5 to verify compliance with contraceptive measures and absence of any known pregnancy. Clinically significant changes including adverse events (AEs) were followed until resolution or grade 1 status was achieved. The study was performed in accordance with the 2008 Declaration of Helsinki25 and good clinical practice guidelines. The study was approved by the Alfred Hospital ethics committee (The Alfred Hospital, Melbourne, Australia). All subjects gave written informed consent before screening.
Participants
Potential subjects recruited from the Melbourne area underwent screening to confirm eligibility and were randomized during the 21 days prior to the scheduled dosing date. Healthy male or female subjects 18 to 55 years old who were nonsmokers were eligible to participate. Exclusion criteria included history of clinically relevant medical illnesses that in the Investigator's opinion might jeopardize subject safety or interfere with participation in the study, including but not limited to hematological, renal, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, psychiatric, neurologic, or allergic disease; acute signs of hepatitis/other infection (eg, moderate fever, jaundice, nausea, vomiting, abdominal pain) evident within 4 weeks of screening and/or at the screening examination; use of any drugs known to induce or inhibit hepatic drug metabolism within 30 days prior to administration of study treatment; being seropositive for human immunodeficiency virus, HBV (HBsAg positive), or hepatitis C virus; current use and/or a history of alcohol and/or drug abuse < 12 months from screening; use of investigational agents or devices within 30 days prior to planned study dosing or current participation in an investigational study; a history of allergy to bee venom, history of hypersensitivity reaction requiring an emergency visit to a physician or hospital and/or requirement for treatment with steroids and/or epinephrine, or a positive reaction to the bee venom allergy skin test performed at screening. The bee venom exclusion criterion was included because the MLP component of the ARC‐520 Injection is similar to melittin, a component of honey bee venom and therefore could theoretically cause an allergic reaction in patients with a history of bee sting allergy.
Participants were free to withdraw from the study at any time for any reason. The PI could withdraw a subject to protect his/her health if the need to take medication could interfere with study measurements; intolerable/unacceptable adverse experiences occurred; major violation or deviation of study protocol procedures occurred; there was noncompliance of subject with protocol; the subject was unwilling to proceed, and/or consent was withdrawn; or if it was deemed in the subject's best interest.
Treatments
Test Formulation
The ARC‐520 Injection was administered as a single intravenous infusion at ascending doses of 0.01, 0.1, 0.3, 0.6, 1.2, 2.0, 3.0, and 4.0 mg/kg. Arrowhead Pharmaceuticals, Inc. (Pasadena, California) supplied the ARC‐520 Injection as 2 sterile 10‐mL vials containing ARC‐520 API (the active pharmaceutical ingredient: siRNAs AD0009 and AD0010) and ARC‐EX1 (the delivery excipient). The API is composed of a 1:1 molar mixture of 2 synthetic, double‐stranded, cholesterol‐conjugated RNA oligonucleotides. ARC‐EX1 is a masked, hepatocyte‐targeted polymeric amine (polymeric amine = l‐melittin‐derived peptide; masking group = carboxydimethyl maleic anhydride N‐acetyl galactosamine [NAG‐MLP]).18 Prior to dosing subjects, a study pharmacist mixed 1 vial of ARC‐EX1 with 1 vial of API to yield the ARC‐520 Injection.
Reference Formulation
Placebo: normal saline (0.9%).
Antihistamine
The oral antihistamine diphenhydramine hydrochloride 50‐mg gel capsules (Unisom Sleep Gel Cap; Petrus Pharmaceuticals, Australia) were supplied to cohorts 7–9; all antihistamine doses were given with 250 mL of water at least 2 hours prior to study drug administration for cohorts 7, 8, and 9 only.
Each subject received a single dose of ARC‐520 Injection or placebo administered intravenously by clinical staff at the infusion rate indicated in Table 2.
Table 2.
Dose and Infusion Rates for Cohorts 1–9
| Cohort | Dose (mg/kg) | Pretreatment | Infusion Rate |
|---|---|---|---|
| Cohort 1 | 0.01 | None | Slow pusha |
| Cohort 2 | 0.10 | None | Slow pusha |
| Cohort 3 | 0.30 | None | 7 mL/min |
| Cohort 4 | 0.60 | None | 3.5 mL/min |
| Cohort 5 | 1.20 | None | 3.5 mL/min |
| Cohort 6 | 2.00 | None | 3.5 mL/min |
| Cohort 7 | 2.00 | Oral antihistamine | 2 mL/minb |
| Cohort 8 | 3.00 | Oral antihistamine | 2 mL/minb |
| Cohort 9 | 4.00 | Oral antihistamine | 2 mL/minb |
Each subject received a single dose of either active drug (ARC‐520 Injection) or placebo (normal saline 0.9%) administered intravenously at the infusion rate indicated.
aInitial volumes for cohorts 1–2 were too small to administer using syringe pump infusion as planned in the protocol; thus, they were given by slow push.
bThe infusion rate for cohorts 7–9 was reduced from 3.5 to 2 mL/min at the overlapping dose of 2 mg/kg to lessen risk of infusion reaction.
Safety Assessments
The safety analysis included all patients who were randomized and received study medication. Safety measures included (1) AEs; (2) physical examinations; (3) vital signs (resting heart rate, semi‐supine systolic/diastolic blood pressure, respiratory rate, and temperature); (4) triplicate electrocardiogram (ECG) measurements at least 2 minutes apart (readings taken after the subject was supine for at least 3 minutes); (5) clinical laboratory tests (hematology, biochemistry, coagulation, urinalysis); (6) use of concomitant medications; and (7) recording reasons for treatment discontinuation because of toxicity. In addition, continuous monitoring by ECG telemetry was used from within 1 hour predose through 24 hours postdose for all enrolled subjects during dosing with ARC‐520 Injection or placebo.
Abnormalities in laboratory findings or other assessments that were deemed clinically significant by the PI and were initially detected during the study or were present at baseline and significantly worsened during the study were reported as AEs, whether or not they were considered drug related. The causality of AEs was determined by the principal investigator. All AEs and serious AEs (SAEs) were followed until resolution, until the condition stabilized, until the event was otherwise explained, or until the subject was lost to follow‐up.
Pharmacokinetic Assessments
Eight blood samples were collected from each subject during the study for pharmacokinetic analysis. Approximately 4 mL of whole blood was collected using ethylenediaminetetraacetic acid (EDTA) as an anticoagulant. Samples were centrifuged at 4°C, and plasma was stored at ‐20°C until transfer to the bioanalytical laboratory. Plasma samples were assayed by a validated liquid chromatography with fluorescence detection method for the ARC‐520 siRNAs.26 The sequences for the siRNAs AD0009 and AD0010 are shown in Table 1. In this method, siRNAs are first hybridized with a fluorescently labeled peptide nucleic acid (PNA)27 probe complementary to the antisense strand, and the resulting complex is analyzed by high‐pressure liquid chromatography (HPLC) with fluorescence detection. Briefly, thawed plasma samples were resolubilized in lysis buffer containing 50 μg/mL proteinase K. Sequence‐specific PNA probes were generated for RNAi triggers AD0009 (Atto425‐OO‐CTGTAGGCATAAATT) and AD0010 (Atto425‐OO‐ACCTCTGCCTAATCA), (Panagene, Daejeon, Korea), which bound to the antisense strand of each RNAi trigger. These contained the fluorescent Atto425 label at the N‐terminus, separated from the PNA chain by a diethylene glycol linker. One PNA probe was added to each supernatant sample. Hybridization of the probe to the antisense siRNA strand was performed in 96‐well conical‐bottom plates. Plates were sealed and incubated at 95°C for 15 minutes in a thermal cycler. Plates were rapidly cooled to 20°C by incubating in an ice bath. HPLC analysis was carried out using a Shimadzu HPLC system with a DNAPac PA‐100 4 × 250‐mm analytical column (Fisher Scientific, Pittsburgh, Pennsylvania). Analysis was carried out at a flow rate of 1 mL/min with a column oven temperature of 60°C. A gradient elution from 9% to 13% mobile phase B over 5 minutes was used with mobile phase A (1 mM EDTA in 25 mM Tris‐HCl [pH 8.0], 50% acetonitrile) and mobile phase B (1 mM EDTA and 1.6 M NaClO4 in 25 mM Tris‐HCl [pH 8.0], 50% acetonitrile). Fluorescence detection was set to 436‐nm excitation and 484‐nm emission. The range of the method was 1.0 to 100 ng/mL for both AD0009 and AD0010, using a 25‐μL aliquot of human plasma. For AD0009, intraday precision and accuracy were between 1.8% and 10.9% coefficient of variation (CV) and between 89.9% and 110.0% of theoretical, respectively. Interday precision and accuracy were between 4.0% and 9.2% CV and between 97.5% and 103.6% of theoretical, respectively. For AD0010, intraday precision and accuracy were between 2.3% and 12.2% CV and between 93.2% and 108.0% of theoretical, respectively. Interday precision and accuracy were between 3.7% and 7.9% CV and between 98.4% and 101.6% of theoretical, respectively.
A validated liquid chromatography–tandem mass spectrometry method was used for the excipient peptide (MLP). This method initially converts NAG‐MLP into unmasked MLP via acid hydrolysis. MLP with the sequence LIGAILKVLATGLPTLISWIKNKRKQ and its internal standard, a synthetic MLP peptide with 5 heavy isotope‐labeled leucine amino acids (Leu: U13C6, 15N), were then extracted from human plasma using solid‐phase extraction (Oasis HLB 1 cc; Waters). The analytes were separated by HPLC on a Zorbax 300SB C18 reverse‐phase column (2.1 × 150‐mm, Agilent), using a gradient of 34% acetonitrile, 0.1 TFA in water to 62% acetonitrile, 0.1% TFA in water at a flow rate of 0.3 mL/min over 5 minutes at 50°C. The eluates are monitored by an API 4000 tandem mass spectrometry detector (Sciex) in positive MRM mode. The multiple charged Q1/Q3 transition was 719.8/884.0 mass to charge (m/z) for MLP and 728.7/893.2 m/z for the internal standard. The data are acquired and processed by the data acquisition system Analyst (AB Sciex). The range of the method is 10.0 to 10 000 ng/mL, using a 100‐μL aliquot of human plasma. Intraday precision and accuracy were between 1.4% and 4.6% CV and between 98.5% to 101.7% of theoretical, respectively. Interday precision and accuracy were between 3.2% and 11.3% CV and between 100.7% and 108.0% of theoretical, respectively. Plasma concentrations of ARC‐520 API (siRNAs) and MLP (excipient peptide) were determined at different doses. Plasma concentrations of ARC‐520 components were used to calculate single‐dose pharmacokinetic parameters. Samples of all subjects were analyzed.
Pharmacodynamic Assessments
Whole blood was collected at specified times for analysis of cytokines and complement.
Cytokines
Whole‐blood samples were processed for serum. Serum was stored at ‐20°C until transfer to the bioanalytical laboratory and was analyzed by Luminex assay for the following cytokine panel: IL‐6, MCP‐1, TNF‐α, IL‐8, IL‐1β, IFN‐α, and MIP‐1α. Initially the 0‐ (pre‐) and 6‐hour postdose samples were analyzed, with the remaining samples analyzed if a change from baseline was observed at 6 hours. Cytokines were chosen based on literature data on innate immune responses and cytokines induced by double‐stranded RNA and phosphorothioate oligonucleotides.28, 29
Complement
Venous blood samples were collected and processed to produce serum (for complement CH50 analysis) or plasma (for split products–Bb analysis). Initially the 0‐ (pre‐) and 0.5‐hour postdose samples were analyzed, with the remaining samples analyzed if a change from baseline was observed at 0.5 hours.
Statistical Analysis
All study subjects receiving any test article were included in the safety analyses. Treatment‐emergent AEs were summarized using the latest version of MedDRA by system organ class (SOC) and preferred term (PT). Laboratory results were classified according to National Cancer Institute Common Terminology Criteria for Adverse Events and summarized by treatment group. Vital sign measurements were summarized at each scheduled point using descriptive statistics. ECG parameter changes overall, changes from baseline, and qualitative assessments were summarized. Pharmacokinetic data were presented for all subjects who were administered ARC‐520 Injection. Plasma concentrations less than the limit of quantitation of the assay were designated a value of zero for pharmacokinetic analysis and included in the calculation of the means. The pharmacokinetic parameters were determined using noncompartmental method(s). Descriptive statistics of pharmacokinetic parameters included mean, standard deviation (SD), and CV, minimum and maximum. Dose‐related trends in pharmacokinetic parameters were assessed. Phoenix WinNonlin version 6.3 (1998–2012; Certara L.P., distributed by Pharsight Corporation) was used for pharmacokinetic analysis. SAS 9.2 (SAS Institute Inc., Cary, North Carolina) was used for statistical analysis, including generating data listings and summary tables.
In Vitro Mast Cell Degranulation Studies
Rat and human mast cell degranulation assays were performed as previously described.30 Briefly, freshly isolated rat peritoneal mast cells were resuspended in RPMI Medium 1640 containing 1% fetal bovine serum (FBS) and incubated with ARC‐EX1, ARC‐520 API, the combination of the two, or positive controls (IgE‐DNP or Compound 48/80) at increasing concentrations up to 250 μg/mL for ARC‐EX1 or 1000 μg/mL for ARC‐520 API. Incubation was at 37°C for 10 or 30 minutes, after which supernatants were analyzed for histamine and chymase release as a measure of degranulation and lactate dehydrogenase (LDH) release as a measure of viability.
The human mast cell line HMC‐1 was cultured in Iscove's media containing 2% FBS and incubated with ARC‐EX1, ARC‐520 API, the combination of the two, or positive controls (combination of phorbol myristate acetate and calcium ionophore A23187) at increasing concentrations up to 250 μg/mL for ARC‐EX1 and ARC‐520 API. Incubation was at 37°C for 30 minutes, after which supernatants were analyzed for histamine release and β‐hexosaminidase release as a measure of degranulation and LDH release as a measure of viability.
In Vitro Transporter Assays
The inhibition of membrane transporters OCT‐2, OCT‐3, OAT‐3, MATE1, and MATE2‐K or bile acid transporters ASBT, MRP1, MRP2, MRP3, NTCP, and BSEP was evaluated in vitro at drug concentrations up to 240 μg/mL by measuring the inhibition of transporter‐specific substrate transport across cell membranes in cell lines stably expressing the respective transporter as previously described.31, 32, 33, 34
Results
All 9 cohorts (54 persons, half male and half female) were enrolled and received the planned doses (Figure 1); 35 of 36 persons in the ARC‐520 Injection treatment group and 17 of 18 in the placebo group continued through their day 90 follow‐up call. One person in the treatment group withdrew for personal reasons and 1 in the placebo group because of travel conflicts; there were no discontinuations for AEs. Most participants were white; 4 were of Asian and 1 of African descent. Age ranged from 18 to 53 years (mean, 26.85 years). ARC‐520 Injection doses evaluated were 0.01, 0.1, 0.3, 0.6, 1.2, 2.0, 3.0, and 4.0 mg/kg (see Table 2 for specific dose and infusion rates for all cohorts).
Figure 1.
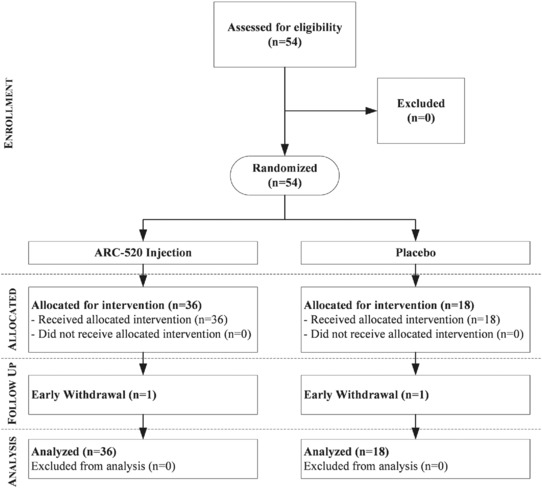
Study flow diagram. A total of 54 healthy adults (half were male and half were female) were enrolled and received the planned doses; 35 of 36 persons in the ARC‐520 Injection treatment group and 17 of 18 in the placebo group continued through their day 90 follow‐up call. One person in the treatment group withdrew for personal reasons and 1 in the placebo group because of travel conflicts; there were no discontinuations for adverse events.
Safety and Tolerability
Following single intravenous doses of ARC‐520 Injection there were no clinically significant changes in ECG, transaminases, troponin, hematology, electrolytes, or urinalysis. No participant developed sensitivity to bee venom based on changes in hymenoptera‐specific IgE. Vital signs were within normal limits in all participants but 1, in whom there were no clinical symptoms, but blood pressure as measured with an automated device was shown to be decreased (58/24 mm Hg) 5 minutes after receiving 0.3 mg/kg ARC‐520 Injection. However, the device used stopped working, and a manual reading within 1 minute was normal (115/75 mm Hg; heart rate, 75 bpm), as were subsequent pressures measured with a different automated device; although recorded as an AE, it is likely that this was machine failure.
One subject in cohort 3 (0.3 mg/kg ARC‐520 Injection) was observed during overnight telemetry while asleep (≈16 hours after dosing) to have a 7.9‐second pause associated with atrioventricular (AV) block; ∼ 1 hour later an episode of Mobitz type I AV block was noted; brief telemetry data available predosing revealed Mobitz type I AV block prior to study dosing. There were no associated ECG or vital sign abnormalities; creatine kinase (CK) and troponin values were normal. Twenty‐four‐hour Holter monitoring conducted 9 days after dosing revealed a Wenckebach (Mobitz type I) rhythm and a 2.5‐second pause. The patient reported a history of syncope prior to enrolling in the study. A consulting cardiologist diagnosed hypervagal syndrome, and there were no further medical interventions.
Few changes in serum chemistry were reported in subjects receiving ARC‐520 Injection and placebo. With the few laboratory abnormalities noted postdosing, no pattern was apparent regarding time of occurrence or increasing incidence with increasing dose or study treatment.
Complement
An increase in individual Bb levels (a measure of alternative pathway activation) was detected beginning in cohort 3 (1 subject) and cohort 4 (1 subject). A pattern of Bb increase was seen in the cohort average for groups 6, 7, 8, and 9. Increases were mild, with no subject having a 4‐fold or greater increase over baseline, which would be considered clinically significant.35 In all cases, the increase in Bb occurred at 0.5 and 2 hours postdose. In addition, in cohort 9, there was a pattern toward a decrease in CH50 at 2 hours postdose in 4 subjects (3 treated with ARC‐520 Injection, 1 treated with placebo). All changes had resolved by 6 hours after dosing.
Cytokines
ARC‐520 Injection administration led to a transient increase in MCP‐1 at 2 hours postdose. The magnitude of the increase in MCP‐1 (generally around 10 times predose level) was similar at ARC‐520 Injection doses of 2.0 mg/kg and above, and this increase was not seen for placebo‐treated subjects. A total of 12 subjects had a similar pattern of MCP‐1 increase, of whom 1 had symptoms of generalized flushing, and 1 had symptoms of an urticarial rash. A pattern of a lesser increase was seen for IL‐8 and MIP‐1α. The level of increase for these 2 analytes was slight (2.5 to 5 times predose levels). All increases were transient, decreasing substantially by 6 hours postdose, reaching levels comparable to predose levels by 24 hours postdose. Finally, although the occurrence of these modest increases appeared to be dose dependent, the intensity did not increase with dose. None of the other cytokines, including IFN‐α2 (IFN‐α), IL‐1β, IL‐6, IL‐12p40, IL‐12p70, and TNF‐α, showed any increases posttreatment.
Adverse Events
There were no SAEs or AEs leading to premature discontinuation of therapy or study discontinuation. Table 3 summarizes AEs by cohort and in the placebo group. The incidence of all reported adverse events was the same in the placebo and treatment groups: 12 of 18 (67%) in the placebo group and 24 of 36 (67%) in the ARC‐520 Injection treatment group. There was no clear pattern of an increased rate or severity of AEs with dose escalation. The most commonly reported AEs across all subjects were (in decreasing order of frequency) headache, upper respiratory infection (URI), somnolence, lethargy, and creatinine elevation. Headaches, sore throats, and URIs were a common occurrence, as the study was conducted during the Australian winter. Dizziness was reported in 2 subjects in the 2 mg/kg group, was mild in both subjects, and was not accompanied by any vital sign changes. One incident of dizziness was reported to resolve in less than a minute; the other lasted 47 minutes. These were not associated with ECG or telemetry changes. Mild drowsiness was noted in 4 subjects and was not thought to be related to study treatment but rather early‐morning predose administration of diphenhydramine. In 3 subjects there were transient creatinine elevations. One subject, a 2‐ year‐old black man who received placebo, had a predose creatinine of 0.86 mg/dL and the following postdose levels; 0.814 mg/dL, 4 hours; 1.33 mg/dL, 12 hours; 0.916 mg/dL, 24 hours; .927 mg/dL, 72 hours. One subject, a 31‐year‐old white woman who received ARC‐520 Injection at 4 mg/kg, had a predose creatinine of 0.77 mg/dL and the following postdose levels: 0.77 mg/dL, 4 hours; 1.78 mg/dL, 12 hours; 0.848 mg/dL, 24 hours; 0.825 mg/dL, 48 hours; 0.814 mg/dL, 72 hours. The third subject, a 20‐year‐old white woman who also received ARC‐520 Injection at 4 mg/kg, had a predose creatinine of 1.188 mg/dL and the following postdose levels; 1.4 mg/dL, 4 hours; 2.29 mg/dL, 12 hours; 1.17 mg/dL, 24 hours; 1.09 mg/dL, 48 hours; 1.05 mg/dL, 72 hours. There were no associated adverse changes in blood urea nitrogren, electrolytes, or CK. Urinalysis for all 3 subjects were within normal limits at 4, 12, and 24 hours without hematuria, proteinuria, or tubular casts. To evaluate if creatinine increases were related to inhibition of renal transporters, in vitro studies were performed. ARC‐520 Injection did not inhibit any of 5 evaluated renal transporters (OCT‐2, OCT‐3, OAT‐3, MATE1, and MATE2‐K).
Table 3.
Incidence of Adverse Events in the Heparc‐1001 Study
| PBO | 0.01 mg/kg | 0.1 mg/kg | 0.3 mg/kg | 0.6 mg/kg | 1.2 mg/kg | 2.0 mg/kg | 2.0 mg/kg + DPH | 3.0 mg/kg + DPH | 4.0 mg/kg + DPH | |
|---|---|---|---|---|---|---|---|---|---|---|
| n = 18 | n = 4 | n = 4 | n = 4 | n = 4 | n = 4 | n = 4 | n = 4 | n = 4 | n = 4 | |
| Adverse event | Number of subjects with adverse events related (not related) to study drug | |||||||||
| Upper respiratory tract infection | (5) | (1) | (3) | (1) | 1 (1) | |||||
| Influenza | (1) | |||||||||
| Pharyngitis | (1) | |||||||||
| Hypersensitivity | 1 | |||||||||
| Decreased appetite | 1 | |||||||||
| Dizziness | 2 | |||||||||
| Dysgeusia | 1 | |||||||||
| Headache | 3 (2) | 1 (1) | (1) | 1 | ||||||
| Lethargy | 1 | (1) | 1 | |||||||
| Somnolence | (1) | |||||||||
| Ocular hyperemia | (1) | |||||||||
| Atrioventricular block complete | 1 | |||||||||
| Palpitations | (1) | |||||||||
| Flushing | 1 | |||||||||
| Hypotension | (1) | |||||||||
| Oropharyngeal pain | (1) | (1) | ||||||||
| Dry throat | (1) | |||||||||
| Abdominal discomfort | (1) | 1 | ||||||||
| Nausea | 1 | |||||||||
| Paresthesia oral | 1 | |||||||||
| Myalgia | (1) | 1 | ||||||||
| Pain in extremity | (1) | |||||||||
| Catheter site bruise | (1) | |||||||||
| Contusion | (1) | (1) | ||||||||
| Laceration | (1) | |||||||||
| Creatinine increase | 1 | 2 | ||||||||
| Relationship | Number of subjects with moderate or severe adverse events | |||||||||
| Severe All | ||||||||||
| Moderate all | 5 (28%) | 1 (25%) | 2 (50%) | 4 (100%) | 1 (25%) | 1 (25%) | 1 (25%) | |||
| Moderate related | 1 (6%) | 1 (25%) | 2 (50%) | 1 (25%) | ||||||
PBO, placebo; DPH, diphenhydramine; n, number of subjects; blank cell, no AE reported.
Several ARC‐520 Injection–treated subjects showed mild, transient bilirubin elevations 4 hours postadministration, an effect that was not observed in placebo subjects. An in vitro evaluation of bile acid transporters showed no inhibition of ASBT, MRP1, MRP2, or MRP3, partial inhibition of NTCP at high assay concentrations (close to the Cmax in plasma), and potent inhibition of BSEP. However, because of the redundant nature of bile acid transport, it is not clear that bile acid transporter inhibition was the cause of the observed bilirubin increases.
Suspected hypersensitivity reactions were observed in 2 ARC‐520 Injection–treated subjects (1 generalized flushing at 0.3 mg/kg and 1 urticarial rash at 2.0 mg/kg) who were not pretreated with an antihistamine. The urticarial rash was treated with intravenous promethazine but was noted to be resolving before the treatment was administered. No further hypersensitivity reactions were observed after patients were pretreated with an oral antihistamine (cohorts 7–9) despite progressing to higher doses.
Mast Cell Degranulation Studies
After 2 subjects developed hypersensitivity reactions, 1 in cohort 3 and 1 in cohort 6, in vitro studies with rat and human mast cells were performed. These studies indicated that ARC‐520 Injection induces dose‐dependent degranulation and histamine release, for which the major trigger is the ARC‐EX1 component, with a lower level of degranulation induced by the API component (Figure 2). Histamine release equivalent to that produced by the positive control, or nearly so, occurred at concentrations ≥ 2 ug/mL ARC‐EX1 injection. This is below the Cmax for MLP in patients in cohorts 3–9. The effects observed were specific for degranulation activity and not cellular cytotoxicity.
Figure 2.
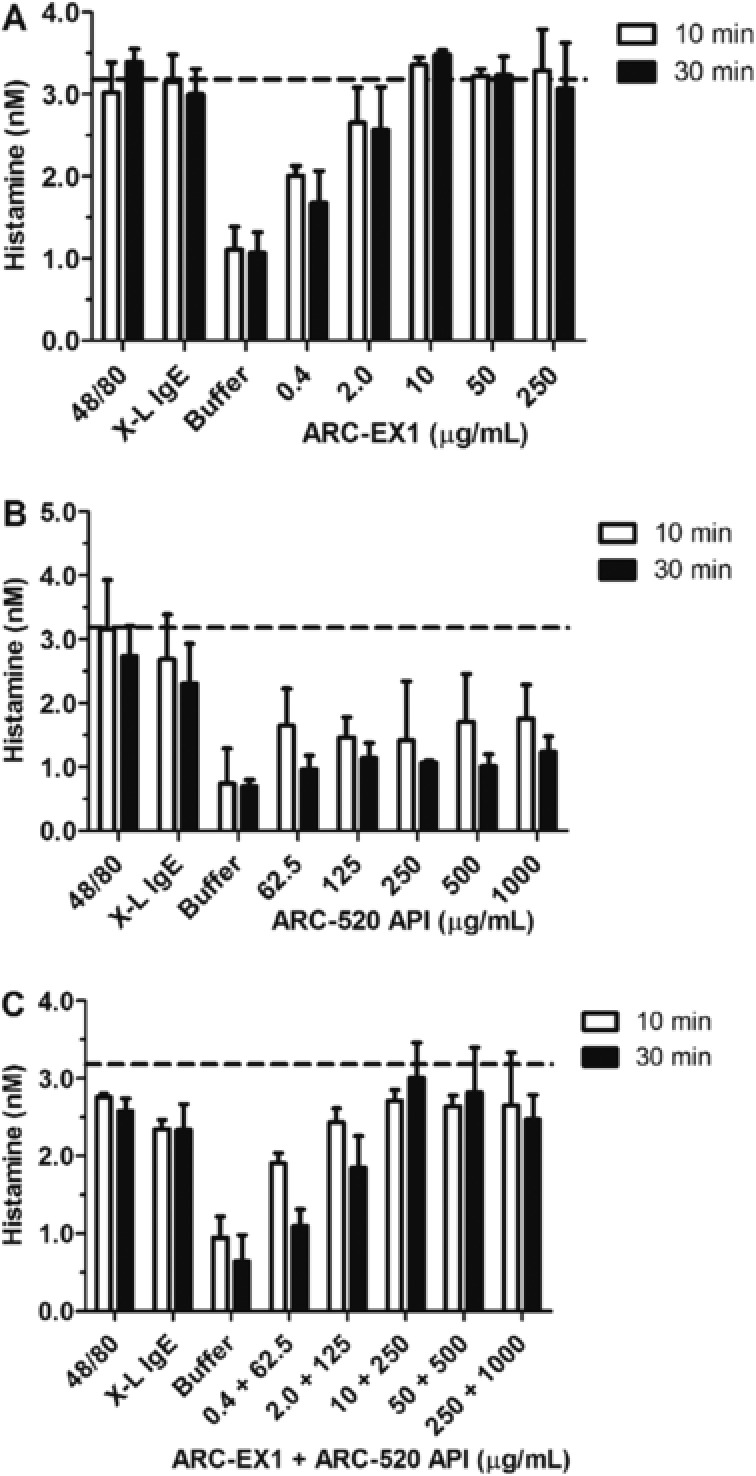
Induction of histamine release from rat peritoneal mast cells in vitro. Histamine concentrations (nM) ± SD in the supernatant of cells is shown for various treatments at 10 and 30 minutes of incubation. Both ARC‐520 Injection (C) and ARC‐EX1 (A) but not ARC‐520 API (B) induced mast cell degranulation and histamine release in a dose‐dependent manner. Positive controls included were immunoglobulin E–dinitrophenol complex (IgE‐DNP) and compound 48/80. ARC‐520 API was evaluated at higher doses than ARC‐EX1, and maximum histamine release was observed at ARC‐EX1 concentrations of 10 μg/mL and greater. As a control for total cellular content, mast cells treated with Triton X‐100 led to release of 3.18 ± 0.26 nM histamine (dotted lines); this process induces cell lysis and should not be considered an activation event.
Pharmacokinetics
Pharmacokinetic results indicate that the siRNA components (AD0009 and AD0010) circulated with a short half‐life of 3–5 hours (Table 4, Figure 3). The MLP excipient component had a somewhat longer half‐life of 8–10 hours. Dose proportionality was evaluated using a log‐log regression model. Exposure to the ARC‐520 components MLP, AD0009, and AD0010 increased proportionally with dose (Table 5). Ninty‐five percent confidence intervals for PK parameters for this study were extremely narrow, indicating minimal interindividual variability. As a consequence, the slope often did not include 1.0 in the confidence interval. However, with point estimates varying between 1.0 and 1.08, this deviation from linearity was considered minimal.
Table 4.
Summary of Pharmacokinetic Parameters Following Administration of ARC‐520 Injection
| Treatment Group (n = 4) | Analyte | AUC0–inf (μg·h/mL) | Cmax (μg/mL) | T1/2 (h) |
|---|---|---|---|---|
| Cohort 1 | MLP | 2.5 (0.4, 2.0–2.9) | 0.2 (0.03, 0.2–0.3) | 8.70 (2.1, 6.16–10.80) |
| 0.01 mg/kg | AD0009 | 0.5 (0.05, 0.4–0.5) | 0.1 (0.02, 0.1–0.1) | 2.68 (0.4, 2.32–3.25) |
| AD0010 | 0.7 (0.2, 0.5–0.9) | 0.1 (0.02, 0.1–0.2) | 3.48 (0.7, 2.79–4.40) | |
| Cohort 2 | MLP | 21.2 (2.7, 18.4–23.7) | 2.3 (0.4, 2.0–3.0) | 7.03 (0.8, 6.26–7.92) |
| 0.10 mg/kg | AD0009 | 6.4 (1.5, 4.6–8.0) | 1.1 (0.2, 0.9–1.5) | 3.41 (0.5, 2.63–3.71) |
| AD0010 | 8.4 (2.1, 5.8–10.6) | 1.2 (0.2, 1.0–1.5) | 4.33 (0.8, 3.20–5.14) | |
| Cohort 3 | MLP | 70.9 (11.5, 59.5–86.5) | 6.4 (0.6, 5.9–7.1) | 8.32 (2.2, 6.36–11.60) |
| 0.30 mg/kg | AD0009 | 18.0 (2.2, 15.7–21.0) | 3.1 (0.2, 2.9–3.4) | 3.48 (0.6, 2.93–4.29) |
| AD0010 | 23.2 (2.8, 20.2–27.0) | 3.3 (0.4, 2.9–3.8) | 4.39 (0.6, 3.62–5.11) | |
| Cohort 4 | MLP | 157.8 (49.6, 127.8–231.5) | 13.2 (2.7, 11.1–16.9) | 9.26 (2.8, 7.00–13.30) |
| 0.60 mg/kg | AD0009 | 34.0 (5.8, 27.5–39.8) | 6.1 (1.4, 5.0–8.0) | 3.62 (0.5, 2.91–3.92) |
| AD0010 | 42.6 (7.7, 33.6–51.3) | 6.5 (1.2, 5.4–8.0) | 4.15 (0.2, 3.93–4.34) | |
| Cohort 5 | MLP | 297.3 (66.3, 231.9–386.9) | 25.4 (4.4, 21.4–31.6) | 7.45 (1.1, 6.40–8.73) |
| 1.20 mg/kg | AD0009 | 77.5 (15.2, 66.5–99.7) | 11.4 (1.9, 8.8–13.3) | 4.00 (0.2, 3.83–4.13) |
| AD0010 | 108.3 (26.1, 89.8–146.7) | 13.3 (2.2, 10.4–15.4) | 4.40 (0.2, 4.22–4.72) | |
| Cohort 6 | MLP | 634.9 (131.7, 553.2–831.8) | 50.3 (3.8, 46.6–55.4) | 8.85 (1.8, 6.70–10.7) |
| 2.00 mg/kg | AD0009 | 125.4 (37.8, 100.5–181.3) | 21.0 (1.8, 19.2–23.2) | 3.82 (0.6, 3.46–4.72) |
| AD0010 | 190.4 (46.0, 162.3–258.5) | 25.5 (1.1, 24.3–26.7) | 4.19 (1.0, 3.49–5.53) | |
| Cohort 7 | MLP | 528.1 (62.6, 452.1–590.9) | 47.8 (5.5, 40.2–52.2) | 7.68 (1.5, 5.48–8.81) |
| 2.00 mg/kg + DPH1 | AD0009 | 138.6 (18.2, 117.1–161.4) | 21.9 (2.3, 20.1–25.0) | 3.86 (0.1, 3.72–3.99) |
| AD0010 | 196.2 (21.8, 171.9–223.7) | 26.1 (2.9, 22.6–29.6) | 4.19 (0.2, 3.95–4.37) | |
| Cohort 8 | MLP | 968.7 (116.8, 858.2–1102.4) | 65.5 (3.3, 61.2–69.3) | 9.84 (1.7, 7.44–11.40) |
| 3.00 mg/kg + DPH1 | AD0009 | 221.8 (22.9, 198.0–252.2) | 34.4 (1.5, 33.0–35.8) | 3.73 (0.2, 3.61–3.94) |
| AD0010 | 292.5 (23.6, 280.1–327.8) | 35.6 (1.3, 34.5–37.1) | 4.23 (0.2, 4.06–4.54) | |
| Cohort 9 | MLP | 1236.4 (207.1, 969.7–1457.6) | 77.8 (4.1, 72.4–82.1) | 10.3 (2.0, 8.65–13.20) |
| 4.00 mg/kg + DPH1 | AD0009 | 374.3 (29.7, 340.3–408.2) | 48.8 (3.8, 46.2–54.3) | 3.93 (0.3, 3.68–4.60) |
| AD0010 | 490.9 (146.4, 389.5–706.9) | 44.3 (1.3, 42.7–45.6) | 4.68 (0.5, 4.04–5.23) |
Arithmetic mean (SD, min–max) values for select parameters of the excipient MLP and siRNAs AD0009 and AD0010 contained in ARC‐520 Injection are listed.
n, number of subjects per PK cohort; DPH, diphenhydramine; MLP, melittin‐like peptide, AD0009, AD0010: siRNAs targeted against HBV. The ratio (on a weight basis) of MLP:AD0009:AD0010 in ARC‐520 Injection is 2:1:1, dose level is in total API (sum of siRNAs) administered.
Figure 3.
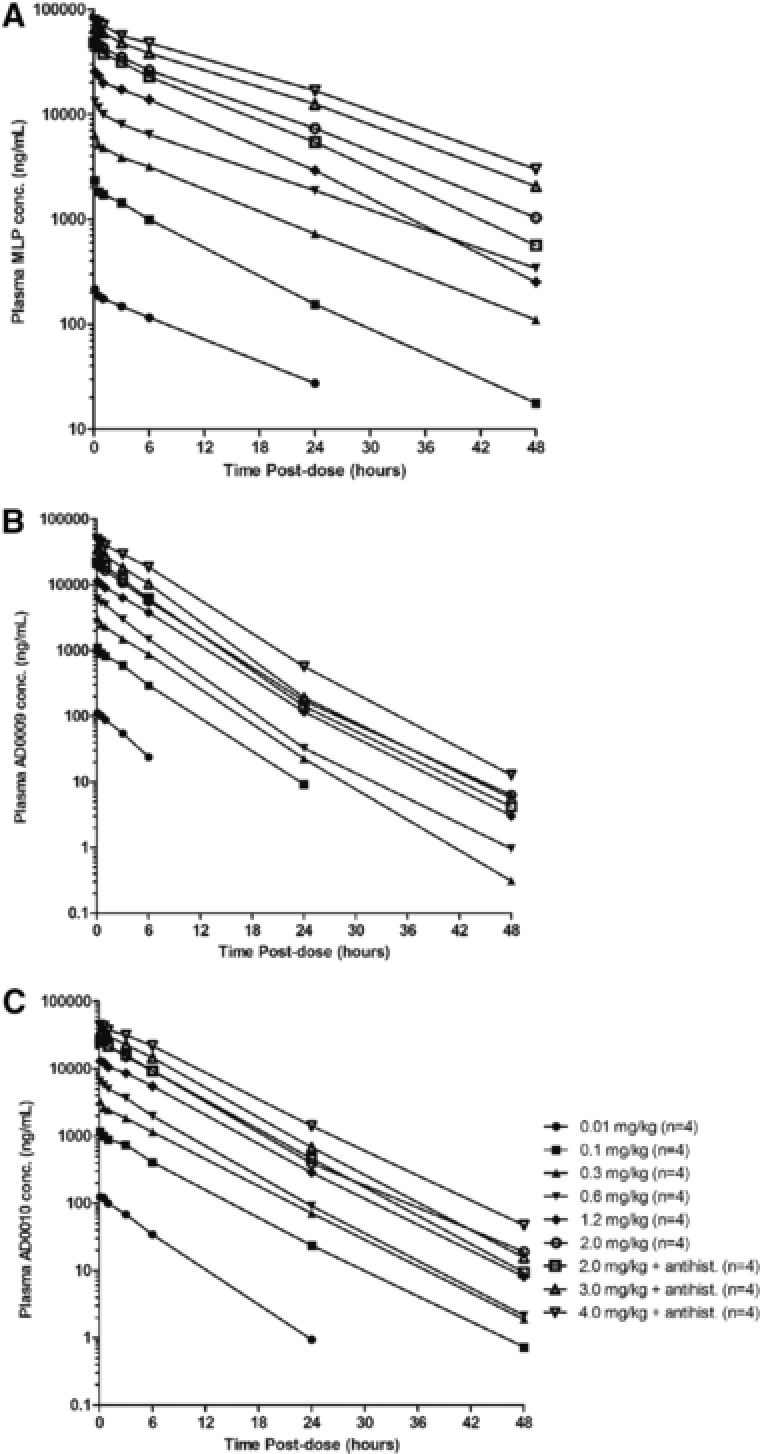
Mean plasma concentration‐versus‐time profiles for ARC‐520 delivery component melittin‐like peptide (MLP) (A) and siRNAs AD0009 (B), and AD0010 (C). Plasma concentrations less than the limit of quantitation (LOQ) of the assay were designated a value of zero for pharmacokinetic analysis and included in the calculation of arithmetic means. All components showed a relatively short half‐life of 3–5 hours for siRNAs and 8–10 hours for MLP, respectively. Exposure increased approximately linearly with dose. Given the anticipated dosing frequency of once every 4 to 6 weeks, the risk of drug accumulation is minimal.
Table 5.
Dose Proportionality of ARC‐520 Injection Using Log‐Log Regression
| Analyte | PK Parameter | r² | Estimate of β | Standard Error | 95% Confidence Interval |
|---|---|---|---|---|---|
| MLP | Cmax | 0.995 | 1.00 | 0.01 | 0.98–1.03 |
| MLP | AUCinf | 0.990 | 1.05 | 0.02 | 1.01–1.08 |
| AD0009 | Cmax | 0.994 | 1.00 | 0.01 | 0.98–1.03 |
| AD0009 | AUCinf | 0.991 | 1.08 | 0.02 | 1.05–1.12 |
| AD0010 | Cmax | 0.995 | 1.00 | 0.01 | 0.97–1.02 |
| AD0010 | AUCinf | 0.990 | 1.08 | 0.02 | 1.04–1.11 |
Discussion
Because, although effective at suppressing viral DNA and reducing liver inflammation, existing HBV therapies rarely result in sustained functional cures off therapy, new therapies are needed that can lead to a high percentage of functional cures (HBsAg clearance). ARC‐520 Injection is the first RNAi therapeutic targeting HBV to enter clinical testing. It is designed to reduce all RNA transcripts derived from viral cccDNA, leading to a reduction in viral antigens, as well as HBV DNA. Viral antigens, especially HBsAg, have been implicated in the suppression of the immune system, leading to immune exhaustion and persistence of chronicity. RNAi therapy leading to reductions in viral antigens may allow for immune reconstitution and functional cure, defined as sustained loss of HBsAg with or without seroconversion.
In this first‐in‐human phase 1 study that assessed the safety, tolerability, and pharmacokinetics of intravenously administered ARC‐520 Injection in healthy volunteers, ARC‐520 Injection was well tolerated with an acceptable safety profile up to a dose of 4 mg/kg. This is supported by the finding that the incidence and severity of AEs were the same in placebo‐ and ARC‐520 Injection–treated subjects. ARC‐520 Injection, and more specifically its peptide component, was found to cause mast cell degranulation and histamine release in vitro. Although 2 subjects treated with ARC‐520 Injection exhibited mild or moderate hypersensitivity reactions, no further hypersensitivity reactions were observed following initiation of oral antihistamine pretreatment, supporting this hypothesis and also indicating that sequelae are easily prevented.
Although transient increases in creatinine with no other changes in laboratory parameters were observed in 3 study participants, including 1 who received placebo, the rapid creatinine decrease and return to baseline within 24 hours lead to the conclusion that it is unlikely to be related to renal tubule injury. Although temporal inhibition of tubular creatinine secretion or an effect of ARC‐520 Injection on glomerular hemodynamics is a possibility, the notable elevation in 1 placebo participant makes it more likely that a cooked protein‐containing meal after fasting contributed to the transient elevation in creatinine. A similar effect was seen in a study in healthy volunteers consuming cooked meat after fasting.36 In addition, in vitro studies showed that ARC‐520 Injection did not inhibit any of 5 evaluated renal transporters, supporting this assessment. Although the mild bilirubin elevations observed in ARC‐520 Injection–treated participants 4 hours postadministration were not seen in those given placebo, the clinical significance of these observations is considered minimal based on the mildness and transient nature of the increases and given that this drug is expected to be delivered once monthly or less. Laboratory signs of innate immune activation were assessed through measurement of complement and cytokine activation in plasma. Low‐level complement increases were all transient and minimal, indicating that their clinical consequence would be questionable.35 The same is true of the asymptomatic low‐level cytokine increase in a few ARC‐520 Injection–treated subjects. Taken together, this indicates that ARC‐520 Injection has a low potential to activate the innate immune system.
Pharmacokinetics of the siRNA and MLP excipient components contained in ARC‐520 Injection showed a relatively short half‐life of 3–5 and 8–10 hours, respectively. Exposure increased approximately linearly with dose. A long duration of activity of more than 4 weeks has been demonstrated after a single dose of ARC‐520 Injection in preclinical animal studies18 and in patients with chronic HBV.37 The persistence of activity significantly beyond the period of plasma exposure is because of the unique RNAi mechanism, in which a small amount of siRNA guide strand can persist and be active within the RNA induced silencing complex (RISC) in the cytoplasm of target cells for extended periods. Given that ARC‐520 Injection is expected to be dosed infrequently, systemic exposure to the components of ARC‐520 Injection can be considered episodic and brief in clinical practice, with a minimal risk of accumulation under multiple‐dose conditions.
In summary, ARC‐520 Injection showed an acceptable tolerability profile in this single‐dose study in healthy volunteers. Pretreatment with an oral antihistamine is recommended for future studies based on the findings of this study and supporting in vitro data indicating direct stimulation of mast cell degranulation. Single‐ and multidose studies in patients with chronic HBV are currently ongoing.
Declaration of Conflicting Interests
Drs. Schluep, Hamilton, Lewis, and Given are employed by Arrowhead Pharmaceuticals, Inc. Dr. Lai is on the advisory committees or review panels of Bristol‐Myers Squibb and Gilead Sciences. Dr. Lai does consulting for Arrowhead Pharmaceuticals, Inc., Bristol‐Myers Squibb, and Gilead Sciences. Dr. Lai is on the speakers’ bureau for Bristol‐Myers Squibb and Gilead Sciences. Dr. Lau is on advisory committees or review panels of Arrowhead Pharmaceuticals, Inc. Dr. Locarnini is employed by Melbourne Health. Dr. Locarnini does consulting for Gilead Sciences and Arrowhead Pharmaceuticals, Inc. Dr. Gish has had grants/research support from Gilead Sciences and Merck & Co. Dr. Gish has performed as consultant and/or adviser to Akshaya Pharmaceuticals, Arbutus Biopharma Corporation, Arrowhead Pharmaceuticals, Inc., Bristol‐Myers Squibb, ContraVir Pharmaceuticals, Enyo Pharma, Gilead Sciences, HumAbs BioMed, Ionis Pharmaceuticals, Merck & Co., Nanogen Biopharmaceutical, and Novira Therapeutics. Dr. Gish has current activity with the scientific or clinical advisory boards of Arrowhead Pharmaceuticals, Inc., Merck & Co., ContraVir Pharmaceuticals, Gilead Sciences, Isis Pharmaceuticals, Enyo Pharma, HumAbs BioMed, and Nanogen Biopharmaceutical. Dr. Gish is a member of the speakers’ bureau for Bristol‐Myers Squibb, Gilead Sciences, and Merck & Co. Dr. Gish has stock options with Arrowhead Pharmaceuticals, Inc. Dr. Given is a stock shareholder of ICON.
Funding
This study was funded in full by Arrowhead Pharmaceuticals, Inc.
Author Contributions
Jason Lickliter performed the research; Thomas Schluep, James Hamilton, David L. Lewis, Ching‐Lung Lai, Johnson Y.N. Lau, Stephen A. Locarnini, and Bruce D. Given analyzed the data; Thomas Schluep, James Hamilton, Robert G. Gish, and Bruce D. Given designed the study; Thomas Schluep, James Hamilton, Robert G. Gish, and Bruce D. Given drafted the article; David L. Lewis, Ching‐Lung Lai, Johnson Y. N. Lau, and Stephen A. Locarnini critically revised the article. All authors approved the article including the author list.
Acknowledgments
Writing and editing support was provided by independent medical editor Lark Lands, PhD, and funded by Arrowhead Pharmaceuticals, Inc. The authors express their gratitude for her assistance in preparing the article for publication.
AUTHORSHIP
Guarantor of the article: Thomas Schluep
References
- 1. Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age‐specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30(12):2212–2219. [DOI] [PubMed] [Google Scholar]
- 2. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127(5 suppl. 1):S35–S50. [DOI] [PubMed] [Google Scholar]
- 4. Marcellin P, Castelnau C, Martinot‐Peignoux M, Boyer N. Natural history of hepatitis B. Minerva Gastroenterol Dietol. 2005;51(1):63–75. [PubMed] [Google Scholar]
- 5. European Association For The Study Of The Liver . EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J Hepatol. 2012;57(1):167–185. [DOI] [PubMed] [Google Scholar]
- 6. Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology. 2009;50(3):661–662. [DOI] [PubMed] [Google Scholar]
- 7. Rijckborst V, ter Borg MJ, Cakaloglu Y, et al. A randomized trial of peginterferon alpha‐2a with or without ribavirin for HBeAg‐negative chronic hepatitis B. Am J Gastroenterol. 2010;105(8):1762–1769. [DOI] [PubMed] [Google Scholar]
- 8. Sung JJ, Tsoi KK, Wong VW, Li KC, Chan HL. Meta‐analysis: treatment of hepatitis B infection reduces risk of hepatocellular carcinoma. Aliment Pharmacol Ther. 2008;28(9):1067–1077. [DOI] [PubMed] [Google Scholar]
- 9. Seto WK, Hui AJ, Wong VW, et al. Treatment cessation of entecavir in Asian patients with hepatitis B e antigen negative chronic hepatitis B: a multicentre prospective study. Gut. 2015;64(4):667–672. [DOI] [PubMed] [Google Scholar]
- 10. Jafri SM, Lok AS. Antiviral therapy for chronic hepatitis B. Clin Liver Dis. 2010;14(3):425–438. [DOI] [PubMed] [Google Scholar]
- 11. Frenette CT, Gish RG. To “be” or not to “be”: that is the question. Am J Gastroenterol. 2009;104(8):1948–1952. [DOI] [PubMed] [Google Scholar]
- 12. Fung J, Lai CL, Tanaka Y, et al. The duration of lamivudine therapy for chronic hepatitis B: cessation vs. continuation of treatment after HBeAg seroconversion. Am J Gastroenterol. 2009;104(8):1940–1946; quiz 1947. [DOI] [PubMed] [Google Scholar]
- 13. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Long‐term therapy with adefovir dipivoxil for HBeAg‐negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131(6):1743–1751. [DOI] [PubMed] [Google Scholar]
- 14. Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rozema DB, Lewis DL, Wakefield DH, et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci U S A. 2007;104(32):12982–12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu J, Nantz MH, Zern MA. Targeting hepatocytes for drug and gene delivery: emerging novel approaches and applications. Front Biosci. 2002;7:d717–d725. [DOI] [PubMed] [Google Scholar]
- 17. Gish RG, Yuen MF, Chan HL, et al. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antiviral Res. 2015;121:97–108. [DOI] [PubMed] [Google Scholar]
- 18. Wooddell CI, Rozema DB, Hossbach M, et al. Hepatocyte‐targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21(5):973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Allerson CR, Sioufi N, Jarres R, et al. Fully 2ʹ‐modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J Med Chem. 2005;48(4):901–904. [DOI] [PubMed] [Google Scholar]
- 20. Czauderna F, Fechtner M, Dames S, et al. Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003;31(11):2705–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Volkov AA, Kruglova NS, Meschaninova MI, et al. Selective protection of nuclease‐sensitive sites in siRNA prolongs silencing effect. Oligonucleotides. 2009;19(2):191–202. [DOI] [PubMed] [Google Scholar]
- 22. Choung S, Kim YJ, Kim S, Park HO, Choi YC. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem Biophys Res Commun. 2006;342(3):919–927. [DOI] [PubMed] [Google Scholar]
- 23. Judge AD, Bola G, Lee AC, MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther. 2006;13(3):494–505. [DOI] [PubMed] [Google Scholar]
- 24. Yang PL, Althage A, Chung J, Chisari FV. Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc Natl Acad Sci U S A. 2002;99(21):13825–13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. World Medical Association . World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. Ferney‐Voltaire, France: World Medical Association; 2008. [Google Scholar]
- 26. Roehl I, Schuster M, Seiffert S. Oligonucleotide detection method. Patent: U.S. 20110201006. 2011. http://www.google.com/patents/US20110201006.
- 27. Uhlmann E, Peyman A, Breipohl G, Will DW. PNA: synthetic polyamide nucleic acids with unusual binding properties. Angew Chem Int Ed. 1998;37:2796–2823. [DOI] [PubMed] [Google Scholar]
- 28. Robbins M, Judge A, MacLachlan I. siRNA and innate immunity. Oligonucleotides. 2009;19(2):89–102. [DOI] [PubMed] [Google Scholar]
- 29. Frazier KS, Sobry C, Derr V, et al. Species‐specific inflammatory responses as a primary component for the development of glomerular lesions in mice and monkeys following chronic administration of a second‐generation antisense oligonucleotide. Toxicol Pathol. 2014;42(5):923–935. [DOI] [PubMed] [Google Scholar]
- 30. Gillespie E, Levine RJ, Malawista SE. Histamine release from rat peritoneal mast cells: inhibition by colchicine and potentiation by deuterium oxide. J Pharmacol Exp Ther. 1968;164(1):158–165. [PubMed] [Google Scholar]
- 31. International Transporter Consortium; Giacomini KM, Huang SM, Tweedie DJ, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Craddock AL, Love MW, Daniel RW, et al. Expression and transport properties of the human ileal and renal sodium‐dependent bile acid transporter. Am J Physiol. 1998;274(1 Pt 1):G157–G169. [DOI] [PubMed] [Google Scholar]
- 33. Weiss J, Theile D, Ketabi‐Kiyanvash N, Lindenmaier H, Haefeli WE. Inhibition of MRP1/ABCC1, MRP2/ABCC2, and MRP3/ABCC3 by nucleoside, nucleotide, and non‐nucleoside reverse transcriptase inhibitors. Drug Metab Dispos. 2007;35(3):340–344. [DOI] [PubMed] [Google Scholar]
- 34. Pedersen JM, Matsson P, Bergstrom CA, et al. Early identification of clinically relevant drug interactions with the human bile salt export pump (BSEP/ABCB11). Toxicol Sci. 2013;136(2):328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burns VE, Edwards KM, Ring C, Drayson M, Carroll D. Complement cascade activation after an acute psychological stress task. Psychosom Med. 2008;70(4):387–396. [DOI] [PubMed] [Google Scholar]
- 36. Jacobsen FK, Christensen CK, Mogensen CE, Andreasen F, Heilskov NS. Postprandial serum creatinine increase in normal subjects after eating cooked meat. Proc Eur Dial Transplant Assoc. 1979;16:506–512. [PubMed] [Google Scholar]
- 37. Yuen M, Chan H, Given B, et al. Phase II, dose ranging study of ARC‐520, a siRNA‐based therapeutic, in patients with chronic hepatitis B virus infection. Hepatology. 2014;60(suppl. 1):LB‐21. [Google Scholar]