

Abstract
A recent outbreak of Zika virus (ZIKV) in Brazil is associated with microcephaly in infants born of infected mothers. As this pandemic spreads, rapid scientific investigation is shedding new light on how prenatal infection with ZIKV causes microcephaly. In this analysis we provide an overview of both microcephaly and ZIKV, explore the connection between prenatal ZIKV infection and microcephaly, and highlight recent insights into how prenatal ZIKV infection depletes the pool of neural progenitors in the developing brain. WIREs Dev Biol 2017, 6:e273. doi: 10.1002/wdev.273
For further resources related to this article, please visit the WIREs website.
MICROCEPHALY, BRAIN GROWTH, AND DEVELOPMENT
Microcephaly is a disease that can arise from defects in brain development or from degenerative events following normal brain development. Microcephaly and microencephaly (small brain) are often used synonymously. Microcephaly is characterized by an extremely small head and associated with intellectual disability. Microcephalic individuals typically exhibit head circumferences that are more than two standard deviations below the mean of age‐matched controls with dramatic reductions in size of the cerebral cortex.1, 2, 3, 4 Primary microcephaly, the focus of this primer, describes a group of brain development diseases in which fewer neurons are produced and the resulting smaller brains generally maintain neurotypical gyral patterns.2, 5 Primary microcephaly may arise from a variety of genetic and environmental changes, including maternal viral infections.1, 2, 5 Insights into the cellular and molecular mechanisms governing proliferation of neuronal progenitors and their subsequent differentiation into neurons provide the foundation for understanding the etiology of microcephaly.
The most dramatic loss of neurons in all types of microcephaly occurs in the cerebral cortex, a multilayered collection of neurons and glia in the forebrain that comprises nearly 80% of total brain mass in humans.6 Although all mammals have a cortex, the human cortex is dramatically expanded, and without a fully formed cortex, higher‐order processes such as those required for cognition and sensation are impaired.7 The cortex, like the rest of the brain, originates from the pseudostratified epithelium of the neural tube. In the developing neural tube, progenitor cells initially stretch between the apical (ventricular) and basal (pial) surfaces. These cells undergo three possible modes of division: symmetric proliferative divisions that produce two progenitors, asymmetric divisions that produce one progenitor and one neuron, and symmetric differentiating divisions that produce two neurons. A balance between symmetric proliferative divisions and asymmetric divisions dominates early development. Later, the cell cycle lengthens and progenitors are more likely to undergo symmetric differentiating divisions. Together with programmed cell death, the relative proportions of these modes of division are precisely controlled, ultimately determining the final number of neurons. Early changes in the balance of asymmetric and symmetric divisions impact central nervous system (CNS) development, resulting in pathologies that arise from either too many or too few neurons (reviewed in Ref 8). These basic principles apply to development of all regions of the brain, including the cortex.
During the earliest stages of forebrain development, neuroepithelial cells lining the ventricles divide symmetrically, expanding the pool of neural stem cells (NSCs) that will ultimately generate all neurons and glia. As development proceeds and the cortex begins to emerge, these neuroepithelial cells divide asymmetrically, establishing a pool of NSCs and beginning the production of postmitotic neurons. The NSCs that give rise to the majority of neurons in the cerebral cortex are radial glia (RG). RG are specialized cells that share hallmarks of both glia and neuronal progenitor cells.9, 10, 11, 12, 13 As neuroepithelial cells and RG proliferate, their nuclei migrate along the apicobasal axis through a process termed interkinetic nuclear migration (INM). The nuclear position along the apicobasal axis provides a read‐out for cell cycle progression, with mitotic nuclei positioned apically and G1/S nuclei basally.14 Studies of developing neuroepithelia, primarily in the zebrafish retina (e.g., Refs 15, 16, 17), and also in the mammalian cortex (e.g., Refs 18, 19), suggest that the kinetics of INM and nuclear dwell time near the basal surface may influence whether progenitors produce differentiating daughter cells.
During corticogenesis, which begins around eight weeks postconception in humans, NSCs divide asymmetrically.13, 20 In primates, NSCs are found in two distinct proliferative regions—the ventricular zone (VZ) and the outer subventricular zone (SVZ;). In the VZ, each RG extends a short apical process toward the ventricular surface and a long basally directed process toward the pial surface, providing a migratory track for newly‐born neuronal progeny.21 In the SVZ, transit amplifying non‐RG cells (also known as intermediate progenitors) reside in the inner SVZ while RG‐like cells, termed outer RG (oRG), occupy the outer SVZ.22 Despite retaining only a basal process and no apical process, oRG divide asymmetrically, producing another proliferating oRG progenitor and a differentiating neuron. As they progress through the cell cycle, oRG exhibit mitotic soma translocation, a process somewhat similar to INM,23, 24 that is essential for balancing proliferation and differentiation in the outer SVZ. Once established, the SVZ expands dramatically, especially in humans, and supports extended and extensive cortical neurogenesis.12, 23 Over the next 7 weeks of development, RG and oRG divide asymmetrically and transit amplifying cells divide symmetrically, together generating postmitotic progeny that differentiate into nearly 13 billion excitatory neurons which comprise approximately 80% of all neurons in the cortex (reviewed in Refs 13, 20). The other approximately 20% of neurons in the cortex are inhibitory local circuit neurons that arrive in their final location by migrating radially from the VZ and SVZ or tangentially from the ganglionic eminence of the ventral forebrain.25, 26 By 22 weeks postconception, nearly all RG and oRG have undergone differentiating divisions and cortical neurogenesis slows to undetectable levels, but continued gliogenesis, myelination, and dendrite dynamics support brain growth for years after birth.1, 20 Deviations in these patterns of cell behaviors that support neurogenesis, especially those that negatively influence renewing divisions of RG and oRG, can lead to microcephaly (for example, see Figure 1a).
Figure 1.
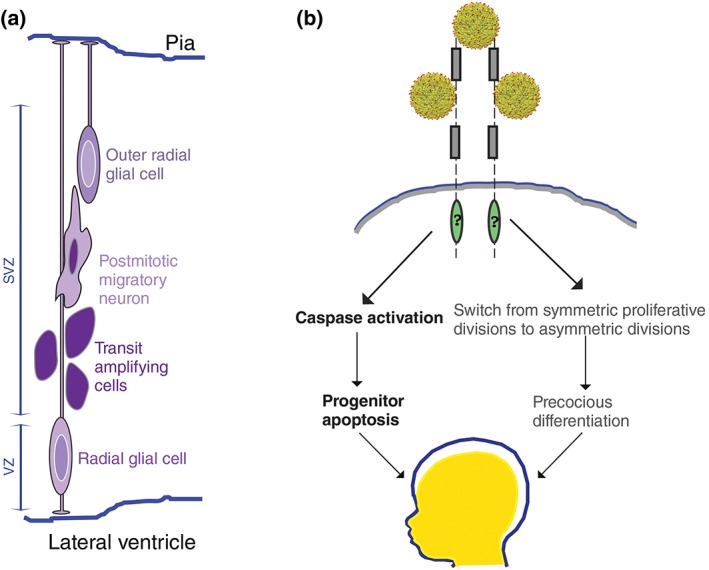
(a) The human cortex develops and grows primarily from two populations of neural stem cells—radial glia [RG; light purple soma with dark purple nucleus positioned near the ventricle in the ventricular zone (VZ)] and outer radial glia [oRG; dark purple soma with light purple nucleus more basally located in the subventricular zone (SVZ)]. These cell types undergo self‐renewing asymmetric divisions, giving rise to either transit amplifying (intermediate progenitor) cells (dark purple) or postmitotic migratory neurons. (b) ZIKV preferentially targets proliferating neural cells including RG, oRG, and neural crest cells. It is likely that a combination of viral‐triggered apoptosis (left arrows) and precocious differentiation (right arrows) contribute to ZIKV‐liked microcephaly. Candidate receptors for Zika virions entry include a number of phosphotidylserine receptors (e.g., AXL, Mer, Tyro3, and TIM1) and are symbolized with a basic domain structure including Ig‐repeats (gray boxes) and kinase domains (green ovals). (Reprinted with permission from 60. Copyright 2016 The American Association for the Advancement of Science)
CELLULAR AND MOLECULAR MECHANISMS OF MICROCEPHALY
Although many features of neural progenitor behavior are conserved among mammals, the human cerebral cortex contains gene expression patterns and cell populations (e.g., oRG) that enable extended proliferation and neurogenesis during development (reviewed in Ref 20). Modeling microcephaly in traditional animal models, therefore, poses a number of challenges. Nonetheless, studies in mice with alterations in genetic loci that have been linked to microcephaly in humans reveal that elevated levels of neuronal and/or neuronal progenitor apoptosis as well as precocious differentiation of neuronal progenitors underlie a number of cortical defects, including microcephaly (reviewed in Refs 20, 27, 28). In addition, recent advances in induced pluripotent stem cell (iPSC) and embryonic stem cell (ESC) technologies enable analyses of neural cells and tissues grown in vitro (e.g., Ref 29). These three‐dimensional culture protocols can generate neurospheres and cerebral organoids that mimic neural growth and development,30 providing a powerful complement to in vivo mouse studies.
Human genetic studies have identified a clear link between mutations in 12 different genes and primary microcephaly (see Table 1). Many of these gene products are enriched in neuronal progenitor cells and impact mitotic cell behaviors that center on the centrosome (e.g., chromosome segregation) and DNA repair (see Table 1 and references therein). Mutations in genes that regulate passage through M‐phase and/or promote differentiation likely underlie many cases of primary microcephaly. Microcephalin (MCPH1) is important for passage through the G2/M checkpoint, influencing both centrosome and DNA repair functions.43 Another microcephaly‐linked gene, CENPJ/CPAP, is important for centrosome integrity and duplication.44, 45 Disruption of this gene is linked to randomization of cell division planes and decreased number of proliferating progenitor cells.36, 46 Another centrosome gene product, CDK5RAP2, is also linked to microcephaly, with decreased/loss of CDK5RAP2 function associated with reduced numbers of neural progenitor cells (NPCs) caused by a switch from symmetric proliferative divisions to asymmetric divisions, most likely by randomizing the cleavage plane.31, 36 Indeed, cerebral organoids made from human cells with truncated CDK5RAP2 were smaller, contained fewer progenitors, and exhibited disruptions cell polarity.29 At least 5 of the 12 genes linked to primary microcephaly are required for centrosome activity, suggesting that the developing brain has a more pronounced need for centrosome function than other tissues.
Table 1.
Genes Disrupted by Both Zika Virus and Microcephaly
| Gene | Proposed Gene Product Function |
|---|---|
| CDK5RAP2 (CDK5 regulatory subunit‐associated protein 2) | Centrosome protein, localizes to centrosome during mitosis31 |
| MCPH1 (MICROCEPHALIN) | G2/M checkpoint regulator32 |
| CASC5 (cancer susceptibility candidate 5) | Formation of kinetochore‐microtubule attachments, chromosome segregation33 |
| WDR62 (WD repeat‐containing protein 62) | Regulation of brain development, localizes to the nucleus and centrosome during mitosis34 |
| ASPM (abnormal spindle‐like microcephaly‐associated protein) | Regulation of mitotic spindle in neuroprogenitor cells35 |
| CENPJ/CPAP (centromere protein J) | Regulation of centrosome integrity and spindle morphology36 |
| STIL (SCL/TAL1‐interrupting locus) | Regulation of mitotic spindle checkpoint37 |
| CEP135 (centrosome protein of 135 kDa) | Maintains structure and organization of the centrosome38 |
| CEP152 (centrosome protein of 152 kDa) | Localizes to centrioles, involved in cell progression through mitosis39 |
| ZNF335 (zinc finger protein 335) | Controls neural progenitor self‐renewal, neurogenesis, and neuronal differentiation40 |
| PHC1 (polyhomeotic‐like protein 1) | Involved in chromatin remodeling/cell cycle regulation41 |
| CDK6 (cyclin‐depenent kinase 6)a | Associates with centrosome during mitosis and regulates centrosome number42 |
The only microcephaly‐linked gene with expression unchanged by ZIKV infection of animals or cerebral organoids.
Environmental factors, especially vertical transmission of viruses from mother to child, have also been associated with microcephaly. Prenatal infection with cytomegalovirus (CMV), herpes simplex (HSV), and rubella have been shown to cause microcephaly and other neurological disorders, with CMV contributing the majority of noninherited cases of primary microcephaly.47 Recent high‐profile examples also link prenatal Zika virus (ZIKV) infection to primary microcephaly.48, 49, 50 Recent surveys of gene expression reveal that 11 microcephaly‐linked genes are downregulated in tissues from ZIKV‐associated microcephaly.51, 52 All of these genes are cell cycle regulated and because ZIKV perturbs cell cycle progression, it is likely decreased expression of these genes is an indirect effect of ZIKV infection.52, 53 Nonetheless, these data raise the possibility that prenatal ZIKV infection may trigger many of the same cellular defects associated with primary microcephaly (Table 1).
ZIKV OVERVIEW
ZIKV is a flavivirus endemic to Africa and Asia. It contains a positive‐strand, RNA genome that encodes a single long open reading frame that is posttranslationally modified to generate structural and nonstructural ZIKV proteins.54, 55, 56 ZIKV can and has evolved rapidly, with distinct strains isolated from numerous locations throughout the world (Refs 57, 58; see also Box 1). Structural studies of ZIKV indicate that its protein coat, comprised primarily of envelope (E) protein, is similar to other flaviviruses.59, 60 There may be ZIKV‐specific and/or strain‐specific changes associated with ten polymorphic amino acids around an asparagine‐linked carbohydrate moiety in the E protein, as this region appears to be subject to selective pressure upon multiple passages in insect cell lines.60, 61
BOX 1. A BRIEF HISTORY OF ZIKV STRAINS AND INFECTIONS.
ZIKV strains can be grouped into two major groups based on sequence similarities—African and Asian.89 ZIKV was first isolated from a rhesus monkey in 1947 in the Zika forest in Uganda, and human cases of ZIKV were documented there and in other areas of East Africa beginning in the 1950s.57 By the mid‐1950s, ZIKV was found scattered throughout East and West Africa, India, and Southeast Asia. A unique strain isolated in Malaysia in 1966 is the most likely parent of currently circulating, microcephaly‐linked Asian strains.57 The first large‐scale human infections were associated with Asian strains in Micronesia and French Polynesia.90, 91, 92 In March 2015, ZIKV was detected in the Americas and reached epidemic levels in Brazil in December 2015, with upwards of 1.3 million reported cases.50 The strains circulating in Brazil are most closely related to an Asian strain isolated in 2013 in French Polynesia. Multiple ZIKV isolates, especially those of the Asian group, exhibit evidence of rapid mutation, including adaptive changes for human infection.57, 89, 93 Analyses of 50 currently circulating Asian strains (isolated from Micronesia as well as South and Central America) exhibit conserved variation when compared to African strains, especially within structural proteins and this may be linked to increased ZIKV virulence over the past 10 years.89, 94, 95 For additional information about the diverging and myriad ZIKV strains, see also Refs 96, 97, 98.
Traditionally, ZIKV was thought to be a mild vector‐borne disease that is transmitted by mosquitoes, primarily Aedes aegypti or Aedes albopictus (both of which are found throughout tropical and subtropical climates, including the contiguous USA). ZIKV can also be sexually transmitted, and high viral titers have been found in semen.50, 62, 63 Animal studies suggest that ZIKV may also be transmitted via ocular secretions including tears.64 ZIKV appears to target cells with an inherent immune privilege such as NPCs,65 retinal cells,66 and testicular cells67; its compact, stable structure enables its survival in relatively harsh physiological conditions including semen, saliva, and urine.59, 63, 68, 69 Most relevant to this primer, a number of recent studies have isolated ZIKV from amniotic fluid, placentae, and tissues of microcephalic infants48, 50, 70, 71, 72, 73 illustrating that ZIKV can pass from the maternal blood stream into the developing CNS in her fetus. For additional details about ZIKV detection and diagnosis, see Box 2.
BOX 2. ZIKV DETECTION AND DIAGNOSIS.
Because ZIKV contains a positive‐strand RNA genome, which can simply be translated by infected host cells, the incubation period is short and the mutation rate can be rapid. Symptoms of ZIKV infection are thought to be minor in adults, including a transient rash and arthralgia, and are similar to symptoms associated with infection by other flaviviruses such as Dengue (DENV) or Chikungunya.91 ZIKV is usually detected from blood or urine samples with RT‐PCR, which identifies the presence of viral RNA, or MAC‐ELISA, which detects antibodies to viral components. RT‐PCR only works as long as patients are actively infected with the virus, typically 3–5 days after infection and MAC‐ELISA can yield false positives due to cross‐reactivity with other flaviviruses like DENV.117, 118 ELISA testing of neonatal sera and cerebrospinal fluid for ZIKV‐specific IgM has confirmed congenital ZIKV infections in newborns with microcephaly.119 Most recently, a reverse transcription loop mediated isothermal amplification method for detecting ZIKV has been reported as a more rapid and field‐safe test for ZIKV infection.120
HOW DOES ZIKV ENTER CELLS?
Despite its initial isolation in the mid‐20th century, the mechanisms of ZIKV infection are poorly understood. How ZIKV enters human cells is an active area of investigation. Like other flaviruses, ZIKV infects cells when it interacts with cell surface receptors and is then internalized via endocytosis. Potential candidate receptors for ZIKV on human cells include receptor tyrosine kinases (RTKs) in the TYRO, AXL, and MER (TAM) family. Members of the TAM family typically interact with ligands that bind phosphotidylserine, a lipid found predominantly on the surfaces of apoptotic cells and enveloped viruses. The TAM RTKs were originally thought to only regulate homeostasis of mature tissues,74, 75 but additional evidence shows that TAM receptors are also found on proliferating progenitor cells in developing neural tissues.76, 77 For example, one member of the TAM family, AXL, is expressed on proliferating neuroepithelial cells in the brain and retina but not on mature neurons. AXL receptors appear to be enriched in neuroepithelial VZ areas in mice, ferrets, and human cerebral organoids,77 and may serve as one entry route for a number of flaviviruses.78 Recent studies have demonstrated that depletion of AXL reduces ZIKV infection of cultured fibroblasts79and astrocytes.80 Although these data raise the possibility that AXL may provide a route for ZIKV entry, recent work shows that ZIKV infection rate and cell viability were the same in iPSC‐derived neural progenitors and cerebral organoids with or without AXL.81 Moreover, depletion of AXL in an immunodeficient mouse model did not inhibit eye or brain infection by ZIKV.82 Alternative candidate ZIKV entry receptors include other TAM RTKs—TYRO3 and MER—as well as other receptors known to facilitate entry of enveloped viruses such as those in the T‐cell and immunoglobulin and mucin domain family (e.g., TIM1). These receptors can promote infection by other flaviviruses including Dengue (DENV) and West Nile Virus (WNV)78, 83 and have been found to facilitate ZIKV infection in cultured cells (e.g., Refs 79, 81, 84). Together, these data suggest that Zika virions likely use multiple methods for entry dependent on cell type and context of infection.
Current evidence demonstrates that ZIKV induces microcephaly in humans when passed vertically from mother to fetus, particularly when the mother is infected during the first trimester of pregnancy. The mechanism by which ZIKV attaches to and crosses the placenta remains unclear, and the likelihood of viral infection may change based on placental age. One study exposed a variety of human placental cells to Zika virions, finding that placental trophoblastic cells (PTCs) from full‐term placentae were resistant to ZIKV infection, protected by the type III interferon they constitutively secrete. However, placental cell lines were sensitive to ZIKV infection, but were protected by conditioned media from noninfected PTCs. ZIKV infection of primary placental cells and chorionic villus explants is strongly linked to the enveloped virus receptor, TIM‐1.85 Consistent with this finding, administration of duramycin, a compound that prevents binding between TIM1 protein and enveloped viruses, was highly effective in decreasing ZIKV infection in cell culture.84 Whether levels of TIM1 decrease as type III interferon secretion increases in PTCs has not yet been determined.
HOW DOES ZIKV CAUSE MICROCEPHALY?
Reports from Brazil in 2016 first correlated maternal ZIKV infection with an increased likelihood of congenital defects, especially microcephaly. It has since become clear that ZIKV exhibits tropism for NPCs and that prenatal infection with this virus induces cell cycle arrest, apoptosis, and differentiation defects in the developing nervous system.51, 53, 86, 87 To cross from the maternal blood stream into the developing fetal brain, ZIKV must cross the placental barrier and the developing blood–brain barrier (BBB). Studies that model the BBB in culture (hBMEC) or investigate ZIKV infection in adult mice suggest that ZIKV can infect BBB cells and cross the mature BBB, but it remains unclear exactly how.87, 88
Primary microcephaly is established during the first trimester of pregnancy when cortical neurogenesis is most pronounced. The same appears to be true for ZIKV‐linked microcephaly, with the most severe cases of ZIKV‐associated microcephaly correlating with maternal infection during the first trimester.99, 100 Embryonic and fetal neural progenitors in the developing forebrain appear to be especially susceptible to adverse effects of ZIKV infection. A number of reports correlate maternal ZIKV infection with microcephaly as well as developmental ocular abnormalities.50, 101, 102, 103 Unlike related flaviviruses, such as WNV and DENV, which can infect neural cells but either target mature neurons (WNV) or elicit a less cytotoxic response (DENV), ZIKV exhibits clear tropism for proliferative neural cells, often with cytotoxic effects.53, 104 ZIKV also productively and detrimentally infects cranial neural crest cells (CNCC), which give rise to cranial bones and can exert paracrine effects on the developing brain.105
Once Zika virions enter neurogenic regions in the brain, they reduce the number of mitotic progenitor cells by promoting cell cycle arrest and apoptotic cell death,51, 52, 86, 87, 106, 107 autophagy,55, 88 or possibly by precocious differentiation (Figure 1b). ZIKV is not the only flavivirus that triggers apoptosis and autophagy. For example, both DENV and WNV can trigger caspase activation to induce apoptosis.108, 109 In addition, a number of flaviviruses have been shown to increase autophagy, presumably in an effort to evade the host immune system and furthering their own replication.110, 111, 112, 113 Thus, ZIKV appears to cause microcephaly not because it elicits ZIKV‐specific responses but because it triggers cell behaviors that perturb proliferation and survival of NPCs during critical periods of brain development. ZIKV may also nonautonomously promote microcephaly by promoting precocious cell cycle exit and differentiation of cortical progenitors, as infection of CNCC causes secretion of proteins that promote neuronal differentiation.105
ZIKV infection after the first trimester can also detrimentally impact neural development. Some infants, who were not classified as microcephalic at birth and who may have been infected during the second or even third trimester of development, have been shown to develop microcephaly and other neurological disorders. In fact, of 12 infants diagnosed with microcephaly within months after birth, only three showed prenatal ultrasound abnormalities consistent with ZIKV infection.114 In addition, ZIKV infection has been associated with an increased likelihood of Guillain–Barré Syndrome,115, 116 an autoimmune disorder that targets the peripheral nervous system in children and adults.
INSIGHTS FROM MODELING ZIKV‐LINKED MICROCEPHALY
Given the neurotropic tendency of the virus and the particularly devastating effects of embryonic and early fetal ZIKV infection, in vitro studies examining NPCs are particularly necessary. To determine whether and how prenatal ZIKV infection causes microcephaly, researchers have employed a number of cell culture and animal models. Neural cell culture experiments have provided insight into ZIKV infection, and recently yielded an extensive list of genes dysregulated upon ZIKV infection. A study comparing the transcriptomes of human neural progenitors infected with a microcephaly‐associated Asian ZIKV strain with other closely related flaviviruses (an African ZIKV and DENV) highlights the importance of apoptotic regulators and viral response genes.121 For instance, the tumor suppressor and pro‐apoptotic gene, tp53 is elevated only in NPCs infected with the microcephaly‐associated strain. Infection with this same strain of ZIKV also alters expression of immune system genes, including elevation of the Toll‐like receptor 3 (TLR3) and its targets. TLR3 is present in neurons and NPCs and has been shown to modulate proliferation of NPCs and promote apoptosis (e.g., Refs 122, 123).
A number of studies using three‐dimensional culture protocols to generate neurospheres and cerebral organoids have yielded important insight into the unique pathogenicity of ZIKV. For example, forebrain mini‐organoids were infected with ZIKV and observed at 4 and 14 days postinfection (dpi) to simulate prenatal ZIKV infection during the first trimester.124 At 4 dpi, a negligible number of neurons were infected but many neural progenitors showed signs of active ZIKV infection. By 14 dpi, many more cells were infected, indicative of a productive viral infection. The 14‐dpi forebrain organoids contained fewer proliferating cells and increased numbers of apoptotic progenitors and neurons. ZIKV infection of older forebrain organoids, resembling second trimester fetal brains with oRG in the SVZ, showed ZIKV‐positive RG and oRG, consistent with ZIKV tropism for NSCs.124 To examine the early consequences of ZIKV infection, another study exposed NSCs, neurospheres, and cerebral organoids to ZIKV or a closely related flavivirus, DENV.107 Both viruses were detected in NSCs 24 hours postinfection, but only ZIKV‐containing cells exhibited hallmarks of apoptosis. Neurospheres cultured from ZIKV‐infected NSCs failed to thrive, containing numerous Zika virions and apoptotic cell bodies. Similarly, brain organoids derived from human iPSCs and then exposed to ZIKV manifested signs of defective growth and neurogenesis, contained the remnants of apoptotic cells, and were 40% smaller than mock‐treated controls. Because none of these defects were observed when cells were exposed to DENV, the authors concluded that ZIKV, not flaviviruses in general, promote aberrant proliferation, differentiation, and death in neural stem and progenitor cells.107 Coupled with analyses of human microcephaly patients, these two foundational studies indicate that neuronal progenitors are especially susceptible to ZIKV infection, with data supporting the hypothesis that loss of neural progenitors, especially during early development, underlies dramatic changes in brain size.
Despite the power of organoids and cell culture, animal models provide a vital tool for fully understanding ZIKV and developing methods that will combat infection. Studies have shown that laboratory mice respond to ZIKV infection differently depending on mouse strain and mode of infection. For instance, wild‐type C57BL/6 mice can be infected by Zika (e.g., Ref 93), but the virus has only been detected in pups of mothers infected by vaginal swab.125 A number of studies, however, provide evidence that C57BL/6 mice become more susceptible to ZIKV infection upon suppression of interferon signaling. Specifically, disrupting the function of interferon α/β either through genetic deletion87, 125, 126 or administration of an interferon‐blocking antibody during early development126 confers susceptibility to ZIKV. When female mice lacking the receptor for interferon α/β, IFNAR, were crossed to wild‐type male mice, these Ifnar−/− pregnant mothers showed signs of placental damage and their Ifnar+/− pups exhibited extensive spread of Zika virions and brain defects consistent with microcephaly.127 Moreover, direct vaginal infection with ZIKV in Ifnar−/− mothers can be vertically transmitted to developing pups that often die in utero due to extensive viral‐induced morphogenetic defects.125 Further supporting a protective role for interferon signaling, mice in which the receptors for both interferon α/β and γ are genetically deleted (AG129 mice), are even more susceptible to ZIKV infection.128 When adult AG129 mice are injected with Asian strains of ZIKV, they die a week after infection from viral‐induced pathologies (including extensive cell death) in the brain and muscle. These mice were also susceptible to ZIKV transmitted from eye secretions and homogenates.64 To further investigate the role of interferon signaling in the context of ZIKV, adult mice lacking interferon regulatory factor were infected with ZIKV. In these mice, ZIKV infection negatively affected neurogenic regions, the anterior SVZ of the forebrain and the subgranular zone (SGZ) of the hippocampal dentate gyrus.87 Cells in these regions showed elevated caspase activation and reduced proliferative potential. Creating a susceptible mouse model by disrupting key components of the immune system may obfuscate the exact mechanisms by which ZIKV infects the host. These models do, however, allow for recapitulation of the ZIKV‐associated phenotypes, and provide data suggestive of a protective role for interferon signaling, at least in mice.
Other inbred mouse strains have been tested for susceptibility to ZIKV‐linked neural developmental defects. ICR mice, which are often used as a model for carcinogen‐induced colon cancer, are also providing insight into how ZIKV causes microcephaly.51 Consistent with in vitro findings, ZIKV injection into the brain of ICR mice induced activation of the apoptotic caspase and immune response pathways, cell cycle arrest, and increased expression of candidate flavivirus entry receptors. Specifically, ZIKV infection of RG and oRG triggered apoptosis of all classes of cells examined, including cycling NPCs, immature, and mature neurons.51 Another mouse strain, SJL, which has functional interferon signaling and is used to model spontaneous myopathies, may be most useful for studying vertical transmission of ZIKV as pups born to ZIKV‐infected mothers have Zika virons throughout their bodies and concentrated in their brains.93 These pups exhibit brain and eye phenotypes similar to ZIKV‐infected fetuses, including cortical malformations, fewer cells, and apoptotic remnants throughout the CNS.
In addition to murine models, nonhuman primates such as rhesus macaque monkeys are particularly important for studying primate‐specific consequences of ZIKV infection and for vaccine development. Several recent studies revealed that monkeys could be infected with ZIKV, highlighting the multiorgan repercussions of ZIKV infection (e.g., Ref 129). Two other studies provide additional insight into ZIKV infection in primates and offer evidence for protective immunity from previous infection with different130 or the same131 ZIKV strains. Nonhuman primate models are also important for understanding how the primate immune system responds to infection with multiple flaviviruses, especially in the context of vaccine development (see final section for more details).
In all of the animal models used to date, various routes of ZIKV infection have been used throughout these models, including vaginal swab,125 transmission via ocular secretions,64 and injections into multiple sites including the footpad,128 intraperitoneal space,126 retroorbital space,87 or cerebellar ventricles (e.g., Ref 51). While the injection into the cerebral ventricles ensures that virions reach neural tissues of interest, peripheral injections align more closely with the mosquito–human route of administration. Vaginal swab mimics sexual transmission. Existing mouse and primate models will likely further our understanding of ZIKV pathology and identify targets for the development of vaccines and prophylactic treatments.
OPEN QUESTIONS AND FUTURE DIRECTIONS
Current evidence demonstrates that ZIKV induces microcephaly in humans when passed vertically from mother to fetus, particularly when this transfer occurs during the first trimester of pregnancy. Although a flurry of recent experimentation has begun to identify potential mechanisms by which ZIKV‐induced microcephaly emerges, much work remains. Additional research is needed to understand the molecular properties and infection mechanisms of ZIKV. In particular, the genes that are dysregulated by ZIKV infection should be more closely examined. While studies have reported a host of genes that are differentially expressed after Zika infection (e.g., Refs 52, 93, 104, 121), it remains unclear how these catalogued changes effect ZIKV pathogenesis. In particular, it will be interesting to learn how virus‐induced gene expression changes influence how ZIKV gains access to immune privileged areas, whether by crossing placental and BBBs or by unique transmission routes. Moreover, identifying the cell surface receptor(s) for ZIKV will provide valuable information for halting ZIKV infection, especially in particularly vulnerable tissues.
Another crucial goal of on‐going work will be to develop treatments and prophylactic measures to halt the spread of ZIKV. Based on the protective capabilities of interferons (as discussed earlier), it will be important to examine whether boosting interferon signaling can reduce the susceptibility of embryos to Zika infection. In addition, several ZIKV vaccines have been tested in mice and rhesus monkeys and appear to provide protection from microcephaly‐associated ZIKV strains,132, 133, 134 and Phase I clinical trials are beginning to evaluate the safety and efficacy of a ZIKV vaccine.135 Despite this progress, immunogenic crosstalk between ZIKV and DENV complicate the development of effective vaccines for each virus. The amino acid sequence of ZIKV E protein, which is associated with viral entry and host defense, is more similar to that of DENV than any other flavivirus.136 Thus, monoclonal antibodies produced in response to DENV bind weakly to Zika virions at levels that cause antibody‐dependent enhancement (ADE) rather than virus neutralization.137 Epidemiological analyses will be necessary to determine whether infection with any of the four DENV serotypes raises the risk of ZIKV infection. One recent study suggested that exposure to another flavivirus, HSV‐2, raises susceptibility to infection by ZIKV.138 Given this risk, and the fact that immunity to one DENV serotype does not protect against other serotypes and may increase risk of infection, it is therefore paramount to develop a vaccine that is cross‐protective, with one potential target being the fusion loop of the E protein.136
Scientists are also identifying pharmacological approaches to treat Zika infection. One large‐scale screen identified 20 potential candidate FDA‐approved drugs that may stymie ZIKV infection.139 Another large‐scale screen identified azithromycin, a macrolide antibiotic, as capable of reducing both viral proliferation and ZIKV‐induced cytopathy in developing human brain tissues.80 In addition, chloroquine, a drug commonly used to prevent malarial infection, can reduce ZIKV infection in several cell lines regardless of ZIKV strain.88 Finally, the mechanism by which ZIKV neutralizing antibodies work is also being actively investigated.140, 141 Additional studies are needed to understand how these various treatments work and whether they will be efficacious in the clinic.
From all this recent work, most of it within a short 16‐month period, it is clear that basic research into the molecular mechanisms of Zika infection will pave the way for the development of effective treatments and protective measures.
References
FURTHER READINGS
REFERENCES
- 1. Woods CG. Human microcephaly. Curr Opin Neurobiol 2004, 14:112–117. [DOI] [PubMed] [Google Scholar]
- 2. Faheem M, Naseer MI, Rasool M, Chaudhary AG, Kumosani TA, Ilyas AM, Pushparaj P, Ahmed F, Algahtani HA, Al‐Qahtani MH. Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics 2015, 8(suppl 1):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashwal S, Michelson D, Plawner L, Dobyns WB. Practice parameter: evaluation of the child with microcephaly (an evidence‐based review). Report of the quality standards subcommittee of the American academy of neurology and the practice committee of the child neurology society. Neurology 2009, 73:887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adachi Y, Mochida G, Walsh C, Barkovich J. Posterior fossa in primary microcephaly: relationships between forebrain and mid‐hindbrain size in 110 patients. Neuropediatrics 2014, 45:93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gilmore EC, Walsh CA. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip Rev Dev Biol 2013, 2:461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, Jacob Filho W, Lent R, Herculano‐Houzel S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled‐up primate brain. J Comp Neurol 2009, 513:532–541. [DOI] [PubMed] [Google Scholar]
- 7. Geschwind DH, Rakic P. Cortical evolution: judge the brain by its cover. Neuron 2013, 80:633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taverna E, Gotz M, Huttner WB. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol 2014, 30:465–502. [DOI] [PubMed] [Google Scholar]
- 9. Malatesta P, Hartfuss E, Gotz M. Isolation of radial glial cells by fluorescent‐activated cell sorting reveals a neuronal lineage. Development 2000, 127:5253–5263. [DOI] [PubMed] [Google Scholar]
- 10. Miyata T, Kawaguchi A, Okano H, Ogawa M. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron 2001, 31:727–741. [DOI] [PubMed] [Google Scholar]
- 11. Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature 2001, 409:714–720. [DOI] [PubMed] [Google Scholar]
- 12. Rakic P. Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci 2009, 10:724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kriegstein A, Alvarez‐Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 2009, 32:149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sauer FC. Mitosis in the neural tube. J Comp Neurol 1935, 62:377–405. [Google Scholar]
- 15. Strzyz PJ, Lee HO, Sidhaye J, Weber IP, Leung LC, Norden C. Interkinetic nuclear migration is centrosome independent and ensures apical cell division to maintain tissue integrity. Dev Cell 2015, 32:203–219. [DOI] [PubMed] [Google Scholar]
- 16. Del Bene F, Wehman AM, Link BA, Baier H. Regulation of neurogenesis by interkinetic nuclear migration through an apical‐basal notch gradient. Cell 2008, 134:1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baye LM, Link BA. Interkinetic nuclear migration and the selection of neurogenic cell divisions during vertebrate retinogenesis. J Neurosci 2007, 27:10143–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang YT, Wang CL, Van Aelst L. DOCK7 interacts with TACC3 to regulate interkinetic nuclear migration and cortical neurogenesis. Nat Neurosci 2012, 15:1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 2009, 5:320–331. [DOI] [PubMed] [Google Scholar]
- 20. Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron 2016, 89:248–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun T, Hevner RF. Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat Rev Neurosci 2014, 15:217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010, 464:554–561. [DOI] [PubMed] [Google Scholar]
- 23. LaMonica BE, Lui JH, Wang X, Kriegstein AR. OSVZ progenitors in the human cortex: an updated perspective on neurodevelopmental disease. Curr Opin Neurobiol 2012, 22:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ostrem B, Di Lullo E, Kriegstein A. oRGs and mitotic somal translocation—a role in development and disease. Curr Opin Neurobiol 2016, 42:61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron 2011, 72:231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Letinic K, Zoncu R, Rakic P. Origin of GABAergic neurons in the human neocortex. Nature 2002, 417:645–649. [DOI] [PubMed] [Google Scholar]
- 27. Homem CC, Repic M, Knoblich JA. Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci 2015, 16:647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barbelanne M, Tsang WY. Molecular and cellular basis of autosomal recessive primary microcephaly. Biomed Res Int 2014, 547986:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Camp JG, Farhath B, Marta F, Sabina K, Tobias G, Michaela W‐B, Eric L, Alex S, Wulf H, Madeline L, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA 2015, 112:15672–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lizarraga SB, Margossian SP, Harris MH, Campagna DR, Han AP, Blevins S, Mudbhary R, Barker JE, Walsh CA, Fleming MD. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 2010, 137:1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alderton GK, Galbiati L, Griffith E, Surinya KH, Neitzel H, Jackson AP, Jeggo PA, O'Driscoll M. Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat Cell Biol 2006, 8:725–733. [DOI] [PubMed] [Google Scholar]
- 33. Kiyomitsu T, Obuse C, Yanagida M. Human blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev Cell 2007, 13:663–676. [DOI] [PubMed] [Google Scholar]
- 34. Nicholas AK, Khurshid M, Desir J, Carvalho OP, Cox JJ, Thornton G, Kausar R, Ansar M, Ahmad W, Verloes A, et al. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet 2010, 42:1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kouprina N, Pavlicek A, Collins NK, Nakano M, Noskov VN, Ohzeki J, Mochida GH, Risinger JI, Goldsmith P, Gunsior M, et al. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum Mol Genet 2005, 14:2155–2165. [DOI] [PubMed] [Google Scholar]
- 36. Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 2005, 37:353–355. [DOI] [PubMed] [Google Scholar]
- 37. Pfaff KL, Straub CT, Chiang K, Bear DM, Zhou Y, Zon LI. The zebra fish cassiopeia mutant reveals that SIL is required for mitotic spindle organization. Mol Cell Biol 2007, 27:5887–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ohta T, Essner R, Ryu JH, Palazzo RE, Uetake Y, Kuriyama R. Characterization of Cep135, a novel coiled‐coil centrosomal protein involved in microtubule organization in mammalian cells. J Cell Biol 2002, 156:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, Perry S, Babineau‐Sturk T, Beis J, Dumas N, Evans SC, et al. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet 2010, 87:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall‐Walker CA, Barry BJ, Murn J, et al. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 2012, 151:1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Awad S, Al‐Dosari MS, Al‐Yacoub N, Colak D, Salih MA, Alkuraya FS, Poizat C. Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis. Hum Mol Genet 2013, 22:2200–2213. [DOI] [PubMed] [Google Scholar]
- 42. Hussain MS, Baig SM, Neumann S, Peche VS, Szczepanski S, Nurnberg G, Tariq M, Jameel M, Khan TN, Fatima A, et al. CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly. Hum Mol Genet 2013, 22:5199–5214. [DOI] [PubMed] [Google Scholar]
- 43. Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1‐Cdc25 pathway. Nat Cell Biol 2011, 13:1325–1334. [DOI] [PubMed] [Google Scholar]
- 44. Cottee MA, Muschalik N, Wong YL, Johnson CM, Johnson S, Andreeva A, Oegema K, Lea SM, Raff JW, van Breugel M. Crystal structures of the CPAP/STIL complex reveal its role in centriole assembly and human microcephaly. Elife 2013, 2:e01071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vulprecht J, David A, Tibelius A, Castiel A, Konotop G, Liu F, Bestvater F, Raab MS, Zentgraf H, Izraeli S, et al. STIL is required for centriole duplication in human cells. J Cell Sci 2012, 125:1353–1362. [DOI] [PubMed] [Google Scholar]
- 46. Garcez PP, Diaz‐Alonso J, Crespo‐Enriquez I, Castro D, Bell D, Guillemot F. Cenpj/CPAP regulates progenitor divisions and neuronal migration in the cerebral cortex downstream of Ascl1. Nat Commun 2015, 6:6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Coyne CB, Lazear HM. Zika virus—reigniting the TORCH. Nat Rev Microbiol 2016, 14:707–715. [DOI] [PubMed] [Google Scholar]
- 48. Driggers RW, Ho CY, Korhonen EM, Kuivanen S, Jaaskelainen AJ, Smura T, Rosenberg A, Hill DA, DeBiasi RL, Vezina G, et al. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med 2016, 374:2142–2151. [DOI] [PubMed] [Google Scholar]
- 49. Basarab M, Bowman C, Aarons EJ, Cropley I. Zika virus. BMJ 2016, 352:i1049. [DOI] [PubMed] [Google Scholar]
- 50. Petersen LR, Jamieson DJ, Powers AM, Honein MA. Zika virus. N Engl J Med 2016, 374:1552–1563. [DOI] [PubMed] [Google Scholar]
- 51. Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, Zhang N, Shi L, Qin C‐F, Xu Z. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 2016, 19:120–126. [DOI] [PubMed] [Google Scholar]
- 52. Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brault JB, Khou C, Basset J, Coquand L, Fraisier V, Frenkiel MP, Goud B, Manuguerra JC, Pardigon N, Baffet AD. Comparative analysis between flaviviruses reveals specific neural stem cell tropism for zika virus in the mouse developing neocortex. EBioMedicine 2016, 10:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cunha MS, Alves EDL, Maria RI, Yurika MA, Silva VFG, Silva NJ, Pereira DSR, Akemi S, Marcelo A‐C, Lourdes B‐CM, et al. First complete genome sequence of zika virus (Flaviviridae, Flavivirus) from an autochthonous transmission in Brazil. Genome Announc 2016, 4:2015–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, Ge J, Wang S, Goldman SA, Zlokovic BV, et al. Zika virus NS4A and NS4B proteins deregulate Akt‐mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 2016, 19:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saiz JC, Vazquez‐Calvo A, Blazquez AB, Merino‐Ramos T, Escribano‐Romero E, Martin‐Acebes MA. Zika virus: the latest newcomer. Front Microbiol 2016, 7:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kindhauser MK, Allen T, Frank V, Santhana RS, Dye C. Zika: the origin and spread of a mosquito‐borne virus. Bull World Health Organ 2016, 94:675C–686C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang H, Liu S, Zhang B, Wei W. Analysis of synonymous codon usage bias of zika virus and its adaption to the hosts. PLoS One 2016, 11:e0166260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kostyuchenko VA, Lim EX, Zhang S, Fibriansah G, Ng TS, Ooi JS, Shi J, Lok SM. Structure of the thermally stable Zika virus. Nature 2016, 533:425–428. [DOI] [PubMed] [Google Scholar]
- 60. Sirohi D, Zhenguo C, Lei S, Thomas K, Pierson TC, Rossmann MG, Kuhn RJ. The 3.8 Å resolution cryo‐EM structure of Zika virus. Science 2016, 5316:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ekins S, Liebler J, Neves BJ, Lewis WG, Coffee M, Bienstock R, Southan C, Andrade CH. Illustrating and homology modeling the proteins of the Zika virus. F1000Res 2016, 5:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Foy BD, Kobylinski KC, Foy JLC, Blitvich BJ, da Rosa AT, Haddow AD, Lanciotti RS, Tesh RB. Probable non‐vector‐borne transmission of Zika virus, Colorado, USA. Emerg Infect Dis 2011, 17:880–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mansuy JM, Dutertre M, Mengelle C, Fourcade C, Marchou B, Delobel P, Izopet J, Martin‐Blondel G. Zika virus: high infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect Dis 2016, 16:405. [DOI] [PubMed] [Google Scholar]
- 64. Miner JJ, Sene A, Richner JM, Smith AM, Santeford A, Ban N, Weger‐Lucarelli J, Manzella F, Ruckert C, Govero J, et al. Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep 2016, 16:3208–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hori J, Ng TF, Shatos M, Klassen H, Streilein JW, Young MJ. Neural progenitor cells lack immunogenicity and resist destruction as allografts. Stem Cells 2003, 21:405–416. [DOI] [PubMed] [Google Scholar]
- 66. Zhou R, Caspi RR. Ocular immune privilege. F1000 Biol Rep 2010, 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fijak M, Meinhardt A. The testis in immune privilege. Immunol Rev 2006, 213:66–81. [DOI] [PubMed] [Google Scholar]
- 68. Barzon L, Pacenti M, Berto A, Sinigaglia A, Franchin E, Lavezzo E, Brugnaro P, Palu G. Isolation of infectious Zika virus from saliva and prolonged viral RNA shedding in a traveller returning from the Dominican Republic to Italy, January 2016. Euro Surveill 2016, 21:pii30159. [DOI] [PubMed] [Google Scholar]
- 69. Gourinat AC, O'Connor O, Calvez E, Goarant C, Dupont‐Rouzeyrol M. Detection of Zika virus in urine. Emerg Infect Dis 2015, 21:84–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Calvet G, Aguiar RS, Melo AS, Sampaio SA, de Filippis I, Fabri A, Araujo ES, de Sequeira PC, de Mendonca MC, de Oliveira L, et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 2016, 16:653–660. [DOI] [PubMed] [Google Scholar]
- 71. Culjat M, Darling SE, Nerurkar VR, Ching N, Kumar M, Min SK, Wong R, Grant L, Melish ME. Clinical and imaging findings in an infant with Zika embryopathy. Clin Infect Dis 2016, 63:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jurado KA, Simoni MK, Tang Z, Uraki R, Hwang J, Householder S, Wu M, Lindenbach BD, Abrahams VM, Guller S, et al. Zika virus productively infects primary human placenta‐specific macrophages. JCI Insight 2016, 1:e88461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mlakar J, Korva M, Tul N, Popovic M, Poljsak‐Prijatelj M, Mraz J, Kolenc M. Zika virus associated with microcephaly. N Engl J Med 2016, 374:951–958. [DOI] [PubMed] [Google Scholar]
- 74. Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol 2013, 5:a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Deng T, Chen Q, Han D. The roles of TAM receptor tyrosine kinases in the mammalian testis and immunoprivileged sites. Front Biosci (Landmark Ed) 2016, 21:316–327. [DOI] [PubMed] [Google Scholar]
- 76. Ji R, Meng L, Jiang X, Cvm NK, Ding J, Li Q, Lu Q. TAM receptors support neural stem cell survival, proliferation and neuronal differentiation. PLoS One 2014, 9:e115140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nowakowski TJ, Pollen AA, Di Lullo E, Sandoval‐Espinosa C, Bershteyn M, Kriegstein AR. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 2016, 18:591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Perera‐Lecoin M, Meertens L, Carnec X, Amara A. Flavivirus entry receptors: an update. Viruses 2013, 6:69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera‐Lecoin M, Surasombatpattana P, Talignani L, Thomas F, et al. Biology of Zika virus infection in human skin cells. J Virol 2015, 89:8880–8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Retallack H, Di Lullo E, Arias C, Knopp KA, Laurie MT, Sandoval‐Espinosa C, Mancia Leon WR, Krencik R, Ullian EM, Spatazza J, et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc Natl Acad Sci USA 2016, 113:14408–14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wells MF, Salick MR, Wiskow O, Ho DJ, Worringer KA, Ihry RJ, Kommineni S, Bilican B, Klim JR, Hill EJ, et al. Genetic ablation of AXL does not protect human neural progenitor cells and cerebral organoids from Zika virus infection. Cell Stem Cell 2016, 19:703–708. [DOI] [PubMed] [Google Scholar]
- 82. Miner JJ, Diamond MS. Understanding how Zika virus enters and infects neural target cells. Cell Stem Cell 2016, 18:559–560. [DOI] [PubMed] [Google Scholar]
- 83. Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel‐Benhassine F, Lew E, Lemke G, Schwartz O, Amara A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12:544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tabata T, Petitt M, Puerta‐Guardo H, Michlmayr D, Wang C, Fang‐Hoover J, Harris E, Pereira L. Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe 2016, 20:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tabata T, Petitt M, Puerta‐Guardo H, Michlmayr D, Wang C, Fang‐Hoover J, Harris E, Pereira L. Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe 2016, 20:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wu KY, Zuo GL, Li XF, Ye Q, Deng YQ, Huang XY, Cao WC, Qin CF, Luo ZG. Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res 2016, 26:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li H, Saucedo‐Cuevas L, Regla‐Nava JA, Chai G, Sheets N, Tang W, Terskikh AV, Shresta S, Gleeson JG. Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell 2016, 19:593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Delvecchio R, Higa LM, Pezzuto P, Valadao AL, Garcez PP, Monteiro FL, Loiola EC, Dias AA, Silva FJ, Aliota MT, et al. Chloroquine, an endocytosis blocking agent, inhibits Zika virus infection in different cell models. Viruses 2016, 8, 322. doi:10.3390/v8120322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Shrinet J, Agrawal A, Bhatnagar RK, Sunil S. Analysis of the genetic divergence in Asian strains of ZIKA virus with reference to 2015–2016 outbreaks. Bull World Health Organ (Epub ahead of print; April 22, 2016). [Google Scholar]
- 90. Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 2009, 360:2536–2543. [DOI] [PubMed] [Google Scholar]
- 91. Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, Stanfield SM, Duffy MR. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis 2008, 14:1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cao‐Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP, Sall AA, Musso D. Zika virus, French polynesia, South Pacific, 2013. Emerg Infect Dis 2014, 20:1085–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP, Benazzato C, Almeida N, Pignatari GC, Romero S, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016, 534:267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhang Z, Li Y, Loh YR, Phoo WW, Hung AW, Kang C, Luo D. Crystal structure of unlinked NS2B‐NS3 protease from Zika virus. Science 2016, 354:1597–1600. [DOI] [PubMed] [Google Scholar]
- 95. Zhang C, Feng T, Cheng J, Li Y, Yin X, Zeng W, Jin X, Li Y, Guo F, Jin T. Structure of the NS5 methyltransferase from Zika virus and implications in inhibitor design. Biochem Biophys Res Commun (Epub ahead of print; 17 November 17, 2016). [DOI] [PubMed] [Google Scholar]
- 96. Ye Q, Liu ZY, Han JF, Jiang T, Li XF, Qin CF. Genomic characterization and phylogenetic analysis of Zika virus circulating in the Americas. Infect Genet Evol 2016, 43:43–49. [DOI] [PubMed] [Google Scholar]
- 97. Haddow AD, Schuh AJ, Yasuda CY, Kasper MR, Heang V, Huy R, Guzman H, Tesh RB, Weaver SC. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl Trop Dis 2012, 6:e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Haddow AD, Nasar F, Guzman H, Ponlawat A, Jarman RG, Tesh RB, Weaver SC. Genetic characterization of Spondweni and Zika viruses and susceptibility of geographically distinct strains of Aedes aegypti, Aedes albopictus and Culex quinquefasciatus (Diptera: Culicidae) to Spondweni virus. PLoS Negl Trop Dis 2016, 10:e0005083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schuler‐Faccini L, Ribeiro EM, Feitosa IM, Al E. Possible association between Zika virus infection and microcephaly‐Brazil, 2015. Morb Mortal Wkly Rep 2016, 65:59–62. [DOI] [PubMed] [Google Scholar]
- 100. Johansson MA, Mier‐y‐Teran‐Romero L, Reefhuis J, Gilboa SM, Hills SL. Zika and the risk of microcephaly. N Engl J Med 2016, 375:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. de Paula FB, de Oliveira Dias JRJ, Prazeres J, Sacramento GAG, Ko AI, Maia MM, Belfort RJ, Belfort R Jr. Ocular findings in infants with microcephaly associated with presumed zika virus congenital. JAMA Ophthalmol 2016, 134:529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ventura CV, Maia M, Ventura BV, Linden VV, Araujo EB, Ramos RC, Rocha MA, Carvalho MD, Belfort R Jr, Ventura LO. Ophthalmological findings in infants with microcephaly and presumable intra‐uterus Zika virus infection. Arq Bras Oftalmol 2016, 79:1–3. [DOI] [PubMed] [Google Scholar]
- 103. Ventura CV, Maia M, Dias N, Ventura LO, Belfort R Jr. Zika: neurological and ocular findings in infant without microcephaly. Lancet 2016, 387:2502. [DOI] [PubMed] [Google Scholar]
- 104. Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535:164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bayless NL, Greenberg RS, Swigut T, Wysocka J, Blish CA. Zika virus infection induces cranial neural crest cells to produce cytokines at levels detrimental for neurogenesis. Cell Host Microbe 2016, 20:423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hanners NW, Eitson JL, Usui N, Richardson RB, Wexler EM, Konopka G, Schoggins JW. Western Zika virus in human fetal neural progenitors persists long term with partial cytopathic and limited immunogenic effects. Cell Rep 2016, 15:2315–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352:816–818. [DOI] [PubMed] [Google Scholar]
- 108. Melian EB, Edmonds JH, Nagasaki TK, Hinzman E, Floden N, Khromykh AA. West Nile virus NS2A protein facilitates virus‐induced apoptosis independently of interferon response. J Gen Virol 2013, 94:308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu Y, Liu H, Zou J, Zhang B, Yuan Z. Dengue virus subgenomic RNA induces apoptosis through the Bcl‐2‐mediated PI3k/Akt signaling pathway. Virology 2014, 448:15–25. [DOI] [PubMed] [Google Scholar]
- 110. MacLean B, Hess RF, Bonvillain E, Kamate J, Dao D, Cosimano A, Hoy S. Seroprevalence of hepatitis B surface antigen among pregnant women attending the Hospital for Women & Children in Koutiala, Mali. S Afr Med J 2011, 102:47–49. [PubMed] [Google Scholar]
- 111. Heaton NS, Randall G. Dengue virus‐induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Blazquez AB, Martin‐Acebes MA, Saiz JC. Amino acid substitutions in the non‐structural proteins 4A or 4B modulate the induction of autophagy in West Nile virus infected cells independently of the activation of the unfolded protein response. Front Microbiol 2014, 5:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Blazquez AB, Escribano‐Romero E, Merino‐Ramos T, Saiz JC, Martin‐Acebes MA. Stress responses in flavivirus‐infected cells: activation of unfolded protein response and autophagy. Front Microbiol 2014, 5:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. van der Linden V, Pessoa A, Dobyns W, Barkovich AJ, Junior HV, Filho EL, Ribeiro EM, Leal MC, Coimbra PP, Aragao MF, et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth—Brazil. MMWR Morb Mortal Wkly Rep 2016, 65:1343–1348. [DOI] [PubMed] [Google Scholar]
- 115. Cao‐Lormeau VM, Blake A, Mons S, Lastère S, Roche C, Vanhomwegen J, Dub T, Baudouin L, Teissier A, Larre P, et al. Guillain‐Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case–control study. Lancet 2016, 387:1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Parra B, Lizarazo J, Jimenez‐Arango JA, Zea‐Vera AF, Gonzalez‐Manrique G, Vargas J, Angarita JA, Zuniga G, Lopez‐Gonzalez R, Beltran CL, et al. Guillain‐Barre Syndrome associated with Zika virus infection in Colombia. N Engl J Med 2016, 375:1513–1523. [DOI] [PubMed] [Google Scholar]
- 117. Waggoner JJ, Pinsky BA. Zika virus: diagnostics for an emerging pandemic threat. J Clin Microbiol 2016, 54:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Faye O, Faye O, Diallo D, Diallo M, Weidmann M, Sall AA. Quantitative real‐time PCR detection of Zika virus and evaluation with field‐caught mosquitoes. Virol J 2013, 10:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cordeiro MT, Brito CA, Pena LJ, Castanha PM, Gil LH, Lopes KG, Dhalia R, Meneses JA, Ishigami AC, Mello LM, et al. Results of a Zika virus (ZIKV) immunoglobulin M‐specific diagnostic assay are highly correlated with detection of neutralizing anti‐ZIKV antibodies in neonates with congenital disease. J Infect Dis 2016, 214:1897–1904. [DOI] [PubMed] [Google Scholar]
- 120. Wang X, Yin F, Bi Y, Cheng G, Li J, Hou L, Li Y, Yang B, Liu W, Yang L. Rapid and sensitive detection of Zika virus by reverse transcription loop‐mediated isothermal amplification. J Virol Methods 2016, 238:86–93. [DOI] [PubMed] [Google Scholar]
- 121. Zhang F, Hammack C, Ogden SC, Cheng Y, Lee EM, Wen Z, Qian X, Nguyen HN, Li Y, Yao B, et al. Molecular signatures associated with ZIKV exposure in human cortical neural progenitors. Nucleic Acids Res 2016, 44:8610–8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lathia JD, Okun E, Tang SC, Griffioen K, Cheng A, Mughal MR, Laryea G, Selvaraj PK, Ffrench‐Constant C, Magnus T, et al. Toll‐like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. J Neurosci 2008, 28:13978–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, Rana TM. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 2016, 19:258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. Brain‐region‐specific organoids using mini‐bioreactors for modeling ZIKV exposure. Cell 2016, 165:1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yockey LJ, Varela L, Rakib T, Khoury‐Hanold W, Fink SL, Stutz B, Szigeti‐Buck K, Van den Pol A, Lindenbach BD, Horvath TL, et al. Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 2016, 166:1247–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. A mouse model of Zika virus pathogenesis. Cell Host Microbe 2016, 19:720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 2016, 165:1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Aliota MT, Caine EA, Walker EC, Larkin KE, Camacho E, Osorio JE. Characterization of lethal Zika virus infection in AG129 mice. PLoS Negl Trop Dis 2016, 10:e0004682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Li XF, Dong HL, Huang XY, Qiu YF, Wang HJ, Deng YQ, Zhang NN, Ye Q, Zhao H, Liu ZY, et al. Characterization of a 2016 clinical isolate of Zika virus in non‐human primates. EBioMedicine 2016, 12:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Aliota MT, Dudley DM, Newman CM, Mohr EL, Gellerup DD, Breitbach ME, Buechler CR, Rasheed MN, Mohns MS, Weiler AM, et al. Heterologous protection against Asian Zika virus challenge in rhesus macaques. PLoS Negl Trop Dis 2016, 10:e0005168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dudley DM, Aliota MT, Mohr EL, Weiler AM, Lehrer‐Brey G, Weisgrau KL, Mohns MS, Breitbach ME, Rasheed MN, Newman CM, et al. A rhesus macaque model of Asian‐lineage Zika virus infection. Nat Commun 2016, 7:12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Larocca RA, Abbink P, Peron JP, Zanotto PM, Iampietro MJ, Badamchi‐Zadeh A, Boyd M, Ng'ang'a D, Kirilova M, Nityanandam R, et al. Vaccine protection against Zika virus from Brazil. Nature 2016, 536:474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Dowd KA, Ko SY, Morabito KM, Yang ES, Pelc RS, DeMaso CR, Castilho LR, Abbink P, Boyd M, Nityanandam R, et al. Rapid development of a DNA vaccine for Zika virus. Science 2016, 354:237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Abbink P, Larocca RA, De La Barrera RA, Bricault CA, Moseley ET, Boyd M, Kirilova M, Li Z, Ng'ang'a D, Nanayakkara O, et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 2016, 353:1129–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Routh J NIH begins testing investigational Zika Vaccine in humans. Available at: https://www.niaid.nih.gov/news‐events/nih‐begins‐testing‐investigational‐zika‐vaccine‐humans. (Accessed March 7, 2017).
- 136. Harrison SC. Immunogenic cross‐talk between dengue and Zika viruses. Nat Immunol 2016, 17:1010–1012. [DOI] [PubMed] [Google Scholar]
- 137. Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba‐Spaeth G, Duangchinda T, Sakuntabhai A, Cao‐Lormeau VM, Malasit P, Rey FA, et al. Dengue virus sero‐cross‐reactivity drives antibody‐dependent enhancement of infection with zika virus. Nat Immunol 2016, 17:1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Aldo P, You Y, Szigeti K, Horvath TL, Lindenbach B, Mor G. HSV‐2 enhances ZIKV infection of the placenta and induces apoptosis in first‐trimester trophoblast cells. Am J Reprod Immunol 2016, 76:348–357. [DOI] [PubMed] [Google Scholar]
- 139. Barrows NJ, Campos RK, Powell ST, Prasanth KR, Schott‐Lerner G, Soto‐Acosta R, Galarza‐Mun˜oz G, McGrath EL, Urrabaz‐Garza R, Gao J, et al. A screen of FDA‐approved drugs for inhibitors of Zika virus infection. Cell Host Microbe 2016, 20:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhao H, Fernandez E, Dowd KA, Speer SD, Platt DJ, Gorman MJ, Govero J, Nelson CA, Pierson TC, Diamond MS, et al. Structural basis of Zika virus‐specific antibody protection. Cell 2016, 166:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Zhang S, Kostyuchenko VA, Ng TS, Lim XN, Ooi JS, Lambert S, Tan TY, Widman DG, Shi J, Baric RS, et al. Neutralization mechanism of a highly potent antibody against Zika virus. Nat Commun 2016, 7:13679. [DOI] [PMC free article] [PubMed] [Google Scholar]