

Abstract
TV‐1106 is a human serum albumin genetically fused to recombinant human growth hormone, designed to provide a long‐acting alternative to daily growth hormone (GH) injections in patients with GH deficiency. This study investigated the pharmacokinetics, pharmacodynamics, and safety of single subcutaneous doses of TV‐1106 (7.5, 15, 50, and 100 mg) in Japanese (n = 44) and caucasian (n = 44) healthy subjects. TV‐1106 pharmacokinetics and pharmacodynamics were comparable in Japanese and caucasian populations. TV‐1106 demonstrated relatively slow absorption (median tmax, 10–30 hours) and a mean elimination half‐life of 26–36 hours. Apparent clearance and volume of distribution decreased with increasing TV‐1106 doses in both populations and appeared to increase more than dose proportionality across the tested doses. Insulin‐like growth factor‐1 (IGF‐1) and IGF binding protein‐3 (IGFBP‐3) increased in a dose‐related manner, with maximum responses observed at 33–96 and 42–109 hours, respectively. IGF‐1 and IGFBP‐3 returned to baseline values at 168 hours following 7.5 and 15 mg of TV‐1106, and 336 hours following 50 and 100 mg of TV‐1106. TV‐1106 appeared safe in both populations. There was no evidence of differences in pharmacokinetics, pharmacodynamics, or safety of TV‐1106 between Japanese and caucasian populations. The data also demonstrate long‐acting growth hormone properties of TV‐1106 and support its potential for once‐weekly dosing.
Keywords: growth hormone deficiency (GHD), insulin‐like growth factor (IGF‐1), weekly hGH, TV‐1106, Japanese
Growth hormone (GH) promotes linear growth by regulating production of insulin‐like growth factor 1 (IGF‐1). IGF‐1 is produced by the liver and other target organs such as the epiphyseal growth plates found at the end of the long bones. GH has diverse metabolic actions, which include anabolic, lipolytic, and diabetogenic effects.1
Adult‐onset growth hormone deficiency (AGHD) usually occurs as a result of pituitary or peripituitary tumors and their treatment. More than 90% of people with AGHD have overt pituitary disease. Frequently, this is the result of an expansive pituitary adenoma or therapy aimed at ablating such a tumor, that is, surgery or radiation. AGHD is associated with abnormal body composition, reduced physical performance, altered lipid metabolism, increased cardiovascular disease, skin changes, impaired sense of well‐being and other psychological complaints, and reduced quality of life. The predominant clinical signs and symptoms of pediatric GHD include proportionate short stature, accompanied by decreased growth velocity, delayed bone maturation, truncal obesity, a facial appearance that is younger than expected for chronological age, delayed secondary dentition, and a high‐pitched voice. Puberty may be delayed until the late teens.2
Recombinant human growth hormone (rhGH) is primarily used to treat GHD. Current standard of care for GHD involves daily subcutaneous injections of rhGH, which often results in poor compliance by patients.3, 4 To try to improve patient compliance while maintaining the efficacy and safety profiles of the daily products, there has been a drive to develop long‐acting GH formulations.5 TV‐1106 a human serum albumin (HSA) genetically fused to recombinant human growth hormone, is a GH analogue being developed to provide a long‐acting alternative (frequency once weekly or less) to daily GH injections in patients with GHD.6
Single subcutaneous doses of TV‐1106 have previously been assessed in healthy subjects.6 Doses of 3 to 100 mg of TV‐1106 were found to be safe and well tolerated, and the pharmacokinetics and pharmacodynamics supported once‐weekly administration in patients with GHD. The purpose of this new study was to evaluate the pharmacokinetics, pharmacodynamics, and safety of single subcutaneous doses of TV‐1106 in healthy Japanese subjects and compare the findings with matched healthy caucasian subjects.
Methods
This study was approved by an independent institutional review board (Aspire IRB, LLC Clinical Trials Medical Group, Glendale, California). All subjects had to be willing and able to understand and comply with study procedures and provide written informed consent prior to study entry. The study was conducted at a single center, and in accordance with the principles outlined in the Declaration of Helsinki and Good Clinical Practice.
Subjects
Healthy male and female Japanese and caucasian subjects aged 20 to 50 years with a body mass index (BMI) of 18 to 28 kg/m2 and a serum insulin‐like growth factor 1 (IGF‐1) value within the range of a ‐2.0 and +2.0 age‐adjusted standard deviation score (SDS) were eligible for the study. Japanese subjects were nonnaturalized Japanese citizens, with Japanese parents and grandparents and had been living outside Japan for no more than 10 years. Caucasian subjects had no parents or grandparents of Japanese descent. All female subjects had to be of either nonchildbearing potential or using adequate nonhormonal contraception. Exclusion criteria included previous exposure to recombinant GH or GH supplements prior to the study, known history of severe allergic or anaphylactic reactions, known allergy or hypersensitivity to recombinant GH or HSA, and a history of any clinically significant medical conditions (ie, cardiac, endocrine, hematological, hepatic, renal, metabolic, neurological, or other disease).
Study Design
This was a double‐blind, placebo‐controlled, randomized, parallel‐group study managed by Parexel Early Phase Clinical Unit (Los Angeles, California). Subjects were randomly assigned to one of the following dose groups: 7.5‐, 15‐, 50‐, or 100‐mg TV‐1106, placebo (0.5 mL), or placebo (1.0 mL, to control for the high volume injected at the 100‐mg dose group). All treatments were administered as subcutaneous injections. Each dose group included 8 Japanese and 8 caucasian subjects, except for placebo (1.0 mL), which included 4 Japanese and 4 caucasian subjects. The Japanese and caucasian subjects were matched (at cohort level) on the basis of sex, mean age (±10 years), and BMI (±20%). Each subject received a single dose of either TV‐1106 or placebo on day 1 while resident at the study site. Subjects were discharged from the site on day 3 and then returned daily up to day 8 and then again on day 15 and day 29 (±1) for the final safety assessment.
Treatment Rationale and Administration
The 7.5‐, 15‐, and 50‐mg TV‐1106 doses were selected based on the dose range expected in adults with GHD. In addition, these doses were considered safe and well tolerated and enabled a complete pharmacokinetic/pharmacodynamic comparison to be conducted. A pediatric population is expected to require higher doses per kilogram of body weight than adults (based on experience with daily rhGH), and thus the 100‐mg dose was included to provide safety exposure with the liquid formulation with the highest feasible/expected dose in pediatrics.
No dose adjustment was recommended for Japanese compared with caucasian subjects, as there are no known differences in daily GH exposures and metabolism of GH between these 2 populations. In addition, multiple dosing was not included in this study to avoid or reduce the changes of formation of antidrug antibodies (ADA) in healthy subjects.
TV‐1106 was supplied as single‐use vials, each vial containing 0.25 mL at a concentration of 100 mg/mL in a liquid formulation of 20 mM sodium phosphate, 180 mM mannitol, and 60 mM trehalose dehydrate at pH 6.0. The 7.5‐, 15‐, 50‐, and 100‐mg doses were injected at fixed volumes of 0.075, 0.15, 0.5, and 1.0 mL, respectively. The placebo comprised the TV‐1106 formulation buffer with 4 μg/mL riboflavin (vitamin B2) for color‐matching purposes. Placebo doses were administered as either a 0.5‐ or a 1‐mL injection. Injections were given subcutaneously in the right thigh.
Pharmacokinetic Assessments
Blood samples for determination of serum TV‐1106 concentrations were collected prior to dosing on day 1, and 1, 2, 4, 6, 8, 10, 12, 14, 16, 18, 24, 30, 36, 48, 72, 96, 120, 144, 168, 336, and 672 hours postdose, and serum samples were prepared and kept frozen prior to analysis. TV‐1106 serum quantification was performed by Teva Biopharmaceuticals USA Inc. (Rockville, Maryland) using a validated enzyme‐linked immunosorbent assay (ELISA). All scheduled serum samples for all subjects were collected and reported. The method is a sandwich ELISA assay based on the capture of TV‐1106 by anti‐GH antibody coated onto a 96‐well plate and the detection of bound TV‐1106 by horseradish peroxidase (HRP)–labeled anti‐HSA antibody. The method was validated in the concentration range of 4 to 400 ng/mL for TV‐1106 in 100% human serum using a minimum required dilution of 1:20. Accuracy and precision of the method were tested during validation using TV‐1106 spiked at different levels in the serum samples. The recovery of the spiked samples varied between 95% and 113% of the nominal concentrations; the interassay precision varied from 6% to 16% and the intra‐assay from 3% to 6%. For sample testing, 3 levels of quality control (QC) samples spanning the range of quantification were included in each run. The test sample was assayed in duplicate, and the reportable value is the average of the duplicate wells. Only samples from the TV‐1106 dose groups were assayed. Concentrations below the lower limit of quantification (LLOQ; 4 ng/mL) were treated as 0 when calculating summary statistics for serum drug concentrations.
The following pharmacokinetic parameters were calculated by noncompartmental analysis (Phoenix WinNonlin, version 6.2.1; Pharsight Corporation, St Louis, Missouri): maximum observed concentration (Cmax), time of observed maximum concentration (tmax), area under the concentration–time curve from 0 to the time of the last quantifiable concentration (AUC0–t), area under the concentration–time curve from 0 to 168 hours postdose (AUC0–168), area under the concentration–time curve from 0 extrapolated to infinity (AUC0–inf), terminal half‐life (t1/2), apparent total body clearance (CL/F), and the apparent volume of distribution (Vz/F). Dose‐normalized Cmax and AUC parameters were also calculated.
Pharmacodynamic Assessments
Blood samples for determination of IGF‐1 and insulin‐like growth factor binding protein 3 (IGFBP‐3) serum concentrations were collected at screening (IGF‐1 only), day ‐1 (IGF‐1 only), prior to dosing on day 1 and 1, 2, 4, 6, 8, 10, 12, 14, 16, 18, 24, 30, 36, 48, 72, 96, 120, 144, 168, and 336 hours postdose. All scheduled serum samples for all subjects were collected and reported. The measurements of IGF‐1 and IGFBP‐3 were performed by Quest Diagnostics Clinical Trials (Valencia, California) using a validated liquid chromatography–tandem mass spectrometry method (for IGF‐1) and Siemens Immulite 2000 method (for IGFBP‐3). Briefly, IGF‐1 in serum and lyophilized IGF‐1 internal standard (Cerilliant) was first extracted using a mixture of acid and ethanol. The extract was then transferred onto an Onyx guard and Monolithic C18 analytical column (Cohesive Technologies TX‐4 HTLC system). The flow of solvent from the column was diverted to the heated electrospray source of the Agilent TOF system. The analytical measurement range was between 16 and 2000 ng/mL. Precision of the assay was determined during validation. Intra‐assay variation ranged from 1.8% to 2.2% for Bio‐Rad QC pools and from 2.4% to 4.4% for spiked in‐house QCs. Interassay variation for the low, medium, and high Bio‐Rad QC material was 3.3%, 3.1%, and 2.8%, respectively. The overall variation for low, medium, and high in‐house QC material was 5.0%, 5.2%, and 3.5%, respectively. The IGFBP‐3 method was a solid‐phase, enzyme‐labeled chemiluminescent immunometric assay. Anti‐IGFBP‐3 Bead Pack, anti‐IGFBP‐3 reagent wedge, anti‐IGFBP‐3 adjustors, and IGFBP‐3 sample diluent were used according to the manufacturer's procedure (Siemens Healthcare Diagnostic, part number L2KGB2). The range of quantification of the method was 0.5–160.0 mg/L. The inter‐ and intra‐assay precision was less than 6.6% and 3.2%, respectively.
As IGF‐1 and IGFBP‐3 are endogenous substances, baseline‐adjusted values were calculated by subtracting the baseline from all postdose values. For IGF‐1 the baseline was defined as the mean value from screening, day ‐1, and predose day 1 values. For IGFBP‐3, the baseline was the predose day 1 value. The following pharmacodynamic parameters for baseline‐adjusted IGF‐1 and IGFBP‐3 were calculated (Phoenix WinNonlin, version 6.2.1): maximum observed response (Emax), time to achieve Emax (TEmax), area under the effect–time curve from time 0 to the time of the last quantifiable concentration (AUEC0–t) and area under the effect–time curve from time 0 to 168 hours postdose (AUEC0–168).
Safety Assessments
Safety was assessed throughout the study by monitoring of adverse events, vital signs, 12‐lead electrocardiograms (ECGs), clinical laboratory assessments (including fasting blood glucose and insulin), and physical examinations. Fundoscopy (nondilated) was performed at screening and was repeated to rule out intracranial hypertension if headaches associated with vision problems were reported.7
Immunogenicity was also assessed during the study (TV‐1106 dose groups only). Serum samples were collected on day ‐1, day 15, and day 29 (when TV‐1106 was completely washed‐out: concentration < LLOQ 4 ng/mL). Subjects with a positive immunogenicity sample on day 29 postdosing were requested to provide another sample approximately 3 to 5 months after dosing. Analysis for antidrug antibodies (ADAs) was performed by Teva Biopharmaceuticals USA (Rockville, Maryland) using a validated ELISA method. The ADA against TV‐1106 was determined using TV‐1106 as the capture reagent and HRP‐conjugated protein A/G and protein L as the detection reagent. The method was validated for screening, specificity to confirm the presence of specific anti‐TV‐1106 and anti‐GH antibodies, and titration. The method has a sensitivity of 15 ng/mL, a drug tolerance of 2 μg/mL, and intra‐ and interassay variability of less than 7% and 15%, respectively. The positive control for the ADA assay was an affinity‐purified rabbit polyclonal antibody raised against TV‐1106. Samples confirmed as ADA positive were further characterized to evaluate the titer and neutralizing activity of the ADA. The method for analysis of neutralizing antibody is a cell‐based assay, which is based on the ability of anti‐TV‐1106 neutralizing antibodies to block TV‐1106 or hGH‐induced proliferation of Nb2‐11 cells. The neutralizing antibody method has a sensitivity of 495 ng/mL for TV‐1106 or 87 ng/mL for GH when used as an inducer, a drug tolerance of 150 ng/mL, and an overall imprecision of less than 14%.
Statistical Analysis
A total sample size of 8 subjects per dose level per ethnic group was not based on statistical considerations, but was considered sufficient for the assessment of TV‐1106 pharmacokinetics, pharmacodynamics, and safety based on clinical and practical considerations.
Safety data are summarized using descriptive statistics and change from baseline (where appropriate) by dose and ethnic group.
Tabulations with summary descriptive statistics for each TV‐1106 dose and graphs are provided to compare pharmacokinetic and pharmacodynamic parameters and profiles of Japanese and caucasian subjects. Side‐by‐side box plots for dose‐normalized pharmacokinetic (AUC0–t/dose and Cmax/dose) and pharmacodynamic (AUEC0–168) parameters by dose and ethnic group are presented including individual subject parameters.
Results
Subjects
A total of 88 healthy subjects (44 Japanese and 44 caucasian) were randomized and received treatment in the study. One subject (caucasian) who received placebo (0.5 mL) was discontinued from the study after day 15 because of being lost to follow‐up but was included in the pharmacodynamic evaluations. One subject (Japanese) who received 50 mg of TV‐1106 was excluded from the pharmacokinetic and pharmacodynamic analyses because of a dosing error. A flowchart detailing study participation is presented in Supplemental Figure S1.
Demographic and baseline characteristics were comparable between dose groups and between populations groups. Mean ± SD age, weight, and BMI of Japanese subjects were 32.6 ± 8.4 years, 63.4 ± 11.4 kg, and 23.0 ± 2.7 kg/m2, respectively. Mean ± SD age, weight, and BMI of caucasian subjects were 34.2 ± 7.7 years, 71.3 ± 12.9 kg, and 24.1 ± 2.2 kg/m2, respectively. In both populations, the majority of subjects were male (55%).
Pharmacokinetics
In both populations, TV‐1106 serum concentrations increased relatively slowly, with different lag times, and in most cases reached more than a single peak prior to declining in a multiexponential manner (Figure 1). TV‐1106 was quantifiable in all but 3 subjects (1 Japanese and 2 caucasian) at 7.5 mg TV‐1106 up to at least 72 hours. At higher doses (50 and 100 mg), the majority of subjects in each population had quantifiable TV‐1106 concentrations up to 168 hours postdose. TV‐1106 mean serum concentrations increased with increasing dose tested (Table 1). Variability in TV‐1106 exposure was high, with coefficients of variation (CV%) for Cmax and AUC parameters ranging from 25% to 154% across both populations and all doses. Median tmax in Japanese populations was observed 14, 10, 18, and 30 hours following 7.5, 15, 50, and 100 mg, respectively. Similar observations were noted in the caucasian population, with tmax being 12 and 30 hours at 7.5 and 100 mg, respectively. Mean t1/2 ranged from approximately 26 to 36 hours following 15, 50, and 100 mg TV‐1106, in both Japanese and caucasian subjects. Only 3 Japanese subjects (and no caucasian subjects) had evaluable terminal phases at 7.5 mg, and hence as interpretation of the mean t1/2 value (and other dependent variables) at this dose should be treated with caution, the values are not presented. Apparent clearance and volume of distribution decreased with increasing dose (15 to 100 mg) of TV‐1106 in both populations.
Figure 1.
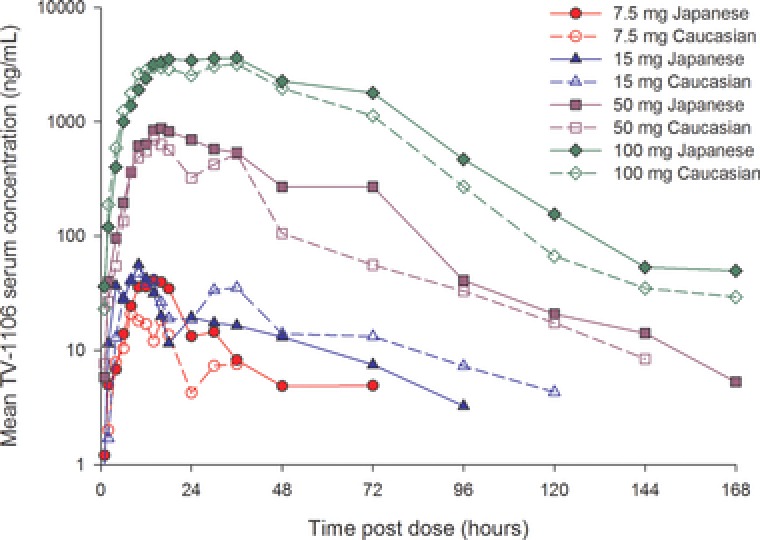
Mean serum TV‐1106 concentration‐versus‐time profiles for Japanese and caucasian subjects.
Table 1.
Summary of Serum Pharmacokinetic Parameters for TV‐1106 in Japanese and Caucasian Subjects
| TV‐1106 Dose | |||||
|---|---|---|---|---|---|
| Population | Parameter (Unit)a | 7.5 mg | 15 mg | 50 mg | 100 mg |
| Japanese | n | 8 | 8 | 7 | 8 |
| Cmax (ng/mL) | 50.06 (67.05) | 68.16 (64.52) | 1031.76 (578.84) | 4516.45 (3438.02) | |
| tmax (h) | 14.00 (4.00, 18.00) | 10.00 (8.00, 48.00) | 18.00 (10.00, 36.00) | 30.00 (16.00, 72.77) | |
| AUC0–t (ng·h/mL) | 938.01 (1445.83) | 1376.15 (1062.82) | 33 950.37 (28 787.01) | 207 742.14 (130 678.73) | |
| AUC0–168 (ng·h/mL)b | —c | 2108.71 (1074.17) | 34 001.52 (28 766.35) | 207 746.35 (130 690.17) | |
| AUC0–inf (ng·h/mL)b | —c | 2222 (114.88) | 34 299.88 (28 873.49) | 210 077.27 (129 234.41) | |
| t1/2 (h)b | —c | 36.02 (8.73) | 30.01 (9.17) | 27.85 (13.13) | |
| CL/F (L/h)b | —c | 7.94 (3.31) | 2.21 (1.21) | 0.66 (0.37) | |
| Vz/F (L)b | —c | 409 (183) | 104 (71.5) | 31.0 (28.4) | |
| Caucasian | n | 8 | 8 | 8 | 8 |
| Cmax (ng/mL) | 25.70 (31.94) | 70.78 (42.87) | 844.73 (748.33) | 3931.05 (3416.77) | |
| tmax (h) | 12.00 (6.00, 36.00) | 10.00 (8.00, 36.00) | 22.00 (10.00, 36.00) | 30.00 (14.00, 72.33) | |
| AUC0–t (ng·h/mL) | 563.47 (566.06) | 1924.16 (899.10) | 20 850.25 (15 691.54) | 168 944.61 (128 206.73) | |
| AUC0–168 (ng·h/mL)d | — | 2281 (805.29) | 20 962 (15 630.57) | 168 363.58 (128 246.44) | |
| AUC0–inf (ng·h/mL)e | — | 2643.29 (660.61) | 21 170.02 (15 666.63) | 169898.21 (125799.41) | |
| t1/2 (h)e | — | 35.39 (8.20) | 25.97 (3.93) | 31.38 (12.65) | |
| CL/F (L/h)e | — | 6.04 (1.81) | 4.25 (3.17) | 1.27 (1.47) | |
| Vz/F (L)e | — | 301 (81) | 158 (120) | 65.8 (82.8) | |
Cmax, maximum observed concentration; tmax, time of observed maximum concentration; AUC0–t, area under the concentration–time curve from 0 to the time of the last quantifiable concentration; AUC0–168, area under the concentration–time curve from 0 to 168 hours postdose; AUC0–inf, area under the concentration–time curve from 0 extrapolated to infinity; t1/2, terminal half‐life; CL/F, apparent total body clearance; Vz/F, apparent volume of distribution.
Parameters are presented as arithmetic mean (standard deviation) except for tmax, which is median (minimum, maximum).
n = 3 for 7.5 mg and n = 4 for 15 mg.
Less than 50% of the population had evaluable parameters at this dose, and hence summary statistics have not been reported.
n = 7 for 15 mg.
n = 6 for 15 mg.
Visual inspection of box plots for dose‐normalized Cmax and AUC0–t versus dose indicates that peak and total exposures of TV‐1106 at the doses studied were comparable between Japanese and caucasian subjects, although given the high variability and small subject numbers, the data should be interpreted with caution (Figure 2). These box plots also demonstrate the lack of dose proportionality. TV‐1106 appears to increase more than dose proportionality across the tested dose range. Lack of dose proportionality over any region of the dose range studied (in both populations) was also supported by exploratory power models fitted to the log‐log transformed data (least‐squares linear regression model; results not shown).
Figure 2.
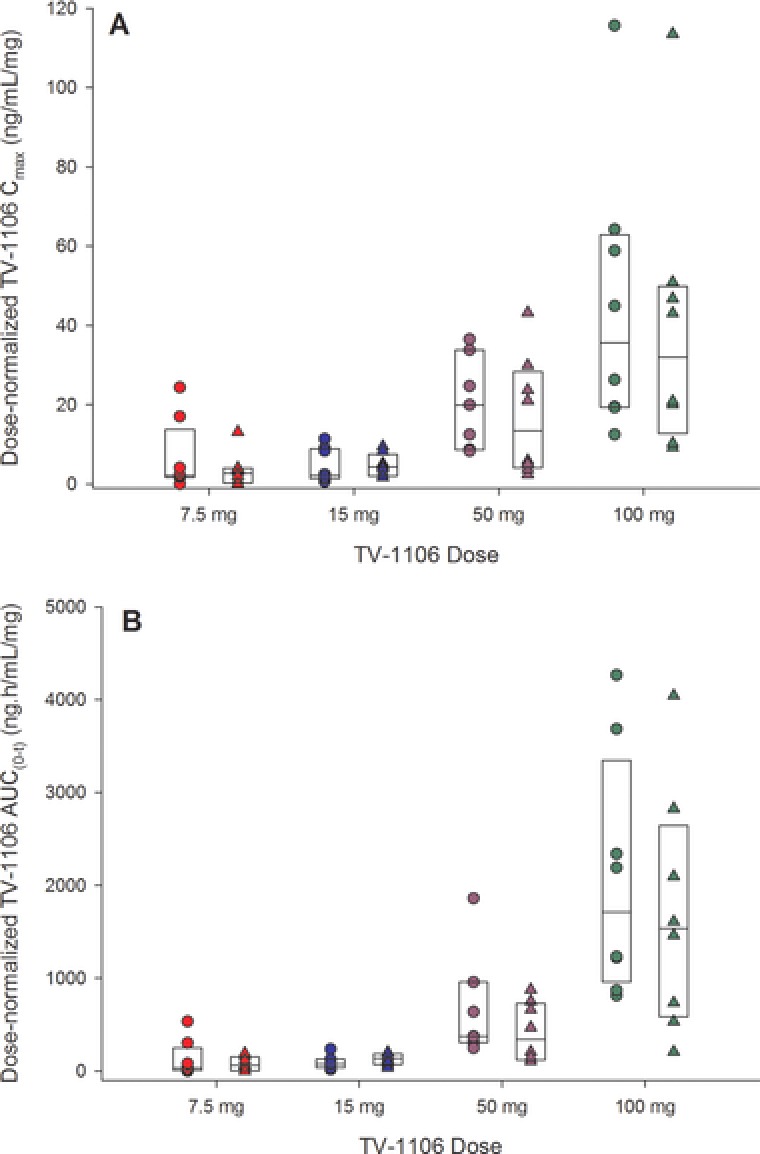
Box plots of (A) dose‐normalized serum TV‐1106 maximum concentrations (Cmax) and (B) area under the serum concentration–time curve from zero to the time of the last quantifiable concentration (AUC0–t) versus dose split by population. The boxes represent the median, 25th, and 75th percentiles. The circles are the individual values for Japanese subjects, and the triangles are individual values for caucasian subjects.
Pharmacodynamics
IGF‐1
Baseline serum concentrations of IGF‐1 were comparable across treatment groups and between populations (Table 2). Mean ± SD baseline values across all dose groups were 173 ± 37 ng/mL (Japanese) and 175 ± 46 ng/mL (caucasian) for IGF‐1.
Table 2.
Summary of Baseline and Baseline‐Adjusted IGF‐1 and IGFBP‐3 Pharmacodynamic Parameters in Japanese and Caucasian Subjects
| Treatment | ||||||||
|---|---|---|---|---|---|---|---|---|
| Population | Parameter (Unit)a | Placebo 0.5 mL | Placebob 1.0 mL | TV‐1106 7.5 mg | TV‐1106 15 mg | TV‐1106 50 mg | TV‐1106 100 mg | |
| IGF‐1 | Japanese | n | 8 | 4 | 8 | 8 | 7 | 8 |
| Baseline (ng/mL) | 176 (27) | 201 (27) | 175 (32) | 171 (53) | 163 (27) | 167 (47) | ||
| Emax (ng/mL) | 60 (28) | 50 (26) | 120 (32) | 147 (47) | 379 (94) | 427 (115) | ||
| TEmax (h) | 21.00 (2.00, 48.00) | 27.00 (6.00, 48.00) | 59.98 (36.00, 96.70) | 48.00 (30.00, 72.73) | 71.88 (30.00, 97.80) | 96.16 (70.63, 144.25) | ||
| AUEC0–t (ng·h/mL) | 2941 (5264) | 1401 (3091) | 11 990 (4897) | 16 278 (8786) | 41 074 (16 068) | 60 835 (14 188) | ||
| AUEC0–168 (ng·h/mL) | 1756 (3184) | 3 (4330) | 10 505 (2510) | 14 384 (6407) | 37 445 (12 000) | 47 522 (11 064) | ||
| Caucasian | n | 8 | 4 | 8 | 8 | 8 | 8 | |
| Baseline (ng/mL) | 186 (47) | 156 (43) | 185 (37) | 165 (49) | 188 (58) | 160 (43) | ||
| Emax (ng/mL) | 42 (22) | 63 (26) | 75 (31) | 135 (73) | 295 (75) | 412 (143) | ||
| TEmax (h) | 30.03 (8.00, 336.67) | 25.00 (0.00, 72.70) | 33.00 (18.00, 71.33) | 36.00 (4.00, 73.23) | 72.56 (48.00, 96.37) | 72.35 (48.00, 119.18) | ||
| AUEC0–t (ng·h/mL) | 1695 (1612) | 4331 (2216) | 6654 (5321) | 13 186 (8427) | 36 277 (12 917) | 53 398 (27 591) | ||
| AUEC0–168 (ng·h/mL) | 347 (1569) | 2815 (2179) | 4973 (3093) | 11 452 (7803) | 29 262 (8151) | 42 974 (16 771) | ||
| IGFBP‐3 | Japanese | n | 8 | 4 | 8 | 8 | 7 | 8 |
| Baseline (mg/L) | 4.46 (0.68) | 4.20 (0.62) | 4.70 (1.05) | 4.73 (0.77) | 4.34 (0.57) | 4.31 (0.38) | ||
| Emax (mg/L) | 0.68 (0.31) | 0.65 (0.55) | 0.90 (0.30) | 0.99 (0.54) | 1.76 (0.33) | 2.26 (0.58) | ||
| TEmax (h) | 24.00 (10.00, 336.67) | 14.00 (0.00, 144.97) | 72.04 (30.00, 167.22) | 59.17 (24.00, 144.43) | 95.20 (71.88, 120.25) | 108.87 (48.00, 167.33) | ||
| AUEC0–t (mg·h/L) | 39.67 (43.98) | 56.03 (86.80) | 89.19 (100.19) | 83.17 (83.33) | 237.57 (109.61) | 284.80 (136.16) | ||
| AUEC0–168 (mg·h/L) | –23.40 (67.90) | 3.75 (68.77) | 71.46 (54.88) | 67.45 (66.94) | 175.00 (46.63) | 219.38 (57.85) | ||
| Caucasian | n | 8 | 4 | 8 | 8 | 8 | 8 | |
| Baseline (mg/L) | 3.94 (0.62) | 4.55 (0.71) | 4.36 (1.13) | 4.24 (0.64) | 4.58 (1.13) | 3.81 (0.96) | ||
| Emax (mg/L) | 0.90 (0.69) | 0.33 (0.05) | 0.88 (0.41) | 1.04 (0.34) | 1.80 (0.85) | 2.11 (0.65) | ||
| TEmax (h) | 84.23 (24.00, 336.67) | 22.00 (4.00, 36.00) | 42.00 (10.00, 96.33) | 72.66 (8.13, 119.58) | 72.18 (2.00, 144.40) | 106.82 (72.25, 143.90) | ||
| AUEC0–t (mg·h/L) | 128.82 (186.89) | 2.35 (9.03) | 64.16 (72.33) | 99.96 (89.57) | 192.61 (169.75) | 328.69 (133.34) | ||
| AUEC0–168 (mg·h/L) | 60.50 (104.99) | –54.89 (39.03) | 33.21 (33.58) | 76.83 (58.68) | 136.83 (100.51) | 209.45 (86.24) | ||
Emax, maximum observed response; TEmax, time to achieve maximum observed response; AUEC0–t, area under the effect–time curve from time 0 to the time of the last quantifiable concentration; AUEC0–168, area under the effect–time curve from time 0 to 168 hours postdose.
Parameters are presented as mean (standard deviation) except for TEmax, which is median (minimum, maximum).
The 1.0‐mL placebo group was included to control for the high volume injected in the 100‐mg TV‐1106 dose group.
Dose‐related increases in baseline‐adjusted serum IGF‐1 concentrations were observed following TV‐1106 in both Japanese and caucasian subjects (Figure 3), with the changes at all doses being greater than those observed in the placebo groups. Serum concentrations of IGF‐1 increased rapidly after dosing with maximum observed responses ultimately occurring at approximately 48 to 96 hours (Japanese subjects) and 33 to 73 hours (caucasian subjects). After reaching a peak, IGF‐1 concentrations then slowly decreased, returning to baseline values at approximately 168 hours for the lower doses (7.5 and 15 mg) and by 336 hours for the higher doses (50 and 100 mg).
Figure 3.
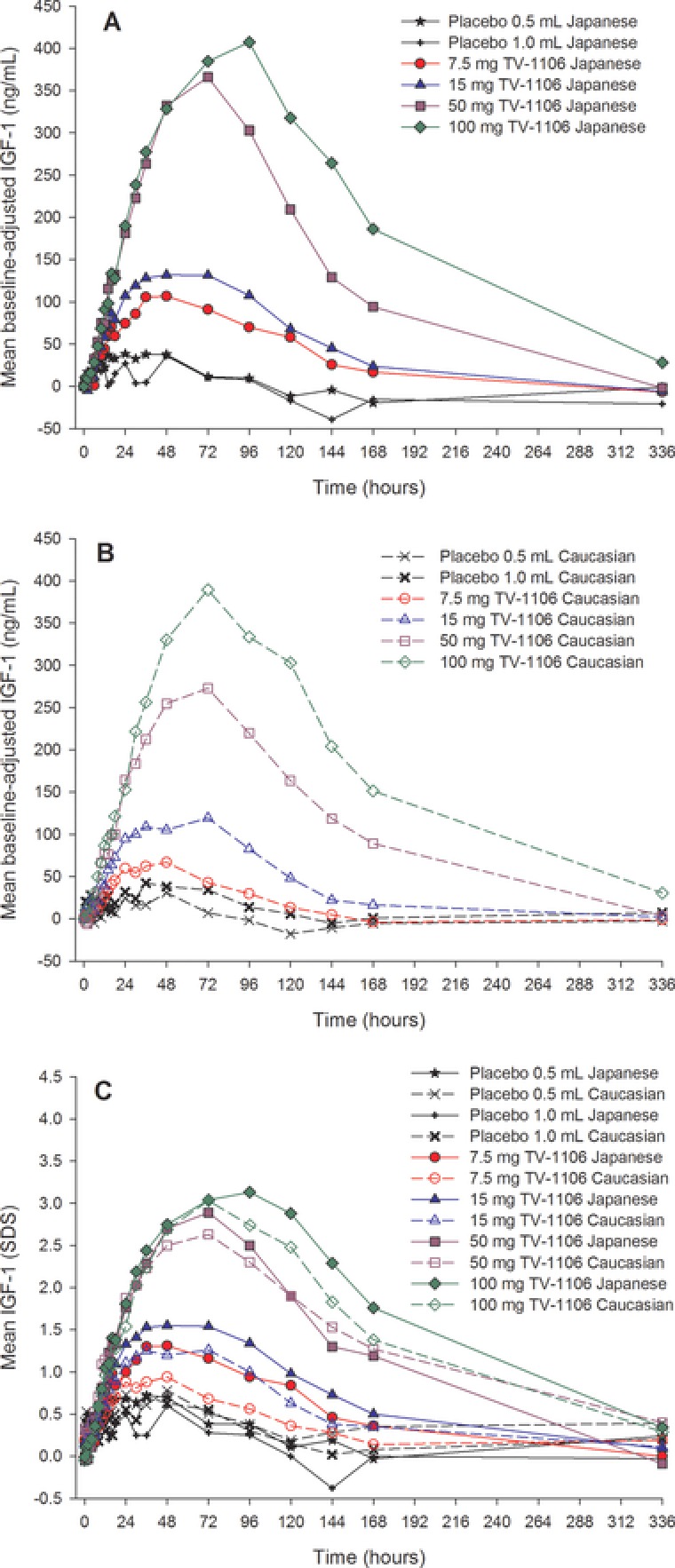
Mean serum concentration‐versus‐time profiles for baseline‐adjusted insulin‐like growth factor 1 (IGF‐1; ng/mL) in Japanese (A) and caucasian (B) subjects and for unadjusted IGF‐1 (SDS) in Japanese and caucasian subjects (C).
IGF‐1 was also presented in SDS units by taking into account the age of the subject (Figure 3C). The findings were similar to those discussed above with comparable observations in both Japanese and caucasian populations; the difference between the populations was approximately ±0.5 SDS (by observing IGF‐1 concentrations over time). Combined data from both populations showed peak mean unadjusted IGF‐1 serum concentrations were approximately 1.1, 1.4, 2.8, and 3.0 SDS for the 7.5‐, 15‐, 50‐, and 100‐mg TV‐1106 groups, respectively. Peak mean unadjusted IGF‐1 concentrations of approximately 0.7 and 0.6 SDS were observed in the 0.5‐ and 1.0‐mL placebo groups, respectively.
In both populations, serums IGF‐1 baseline‐adjusted mean pharmacodynamic parameters AUEC0–t, AUEC0–168, and Emax increased with an increase in dose, with the maximum observed mean Emax and AUEC occurring with the 100‐mg dose (Table 2). Variability in baseline‐adjusted IGF‐1 pharmacodynamic parameters was high, although the variability generally decreased with increasing dose of TV‐1106. Visual inspection of box plots showed comparable baseline‐adjusted IGF‐1 AUEC0–168 split by treatment and population (Figure 4) in Japanese and caucasian subjects with respect to the effect of TV‐1106 on IGF‐1.
Figure 4.
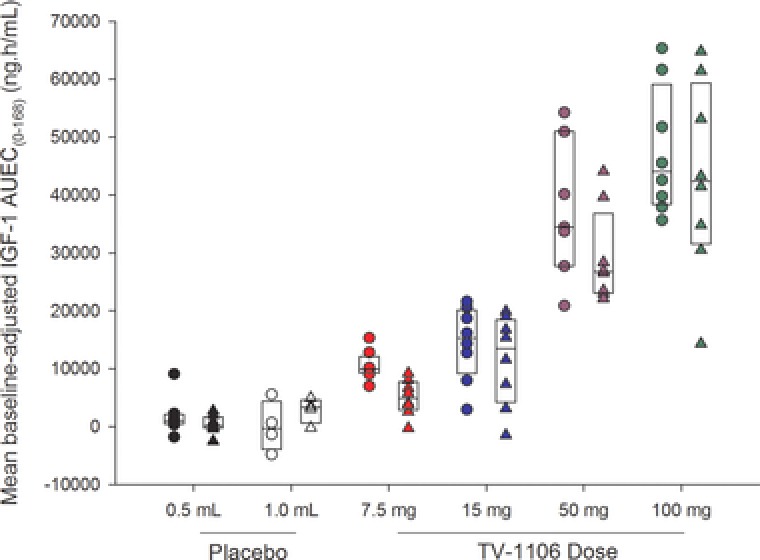
Box plots of baseline‐adjusted serum insulin‐like growth factor 1 (IGF‐1) area under the effect–time curve from 0 to 168 hours postdose (AUEC0–168) versus dose split by population. The boxes represent the median, 25th, and 75th percentiles. The circles are the individual values for Japanese subjects, and the triangles are individual values for caucasian subjects.
IGFBP‐3
Baseline serum concentrations of IGFBP‐3 were comparable across treatment groups and between populations (Table 2). Mean baseline values across all dose groups were 4.5 mg/L (Japanese) and 4.2 mg/L (caucasian) for IGFBP‐3.
Increases in baseline‐adjusted serum IGFBP‐3 concentrations were observed following TV‐1106 in both Japanese and caucasian subjects, although the difference between the lower doses was less clear than for IGF‐1 (Figure 5).
Figure 5.
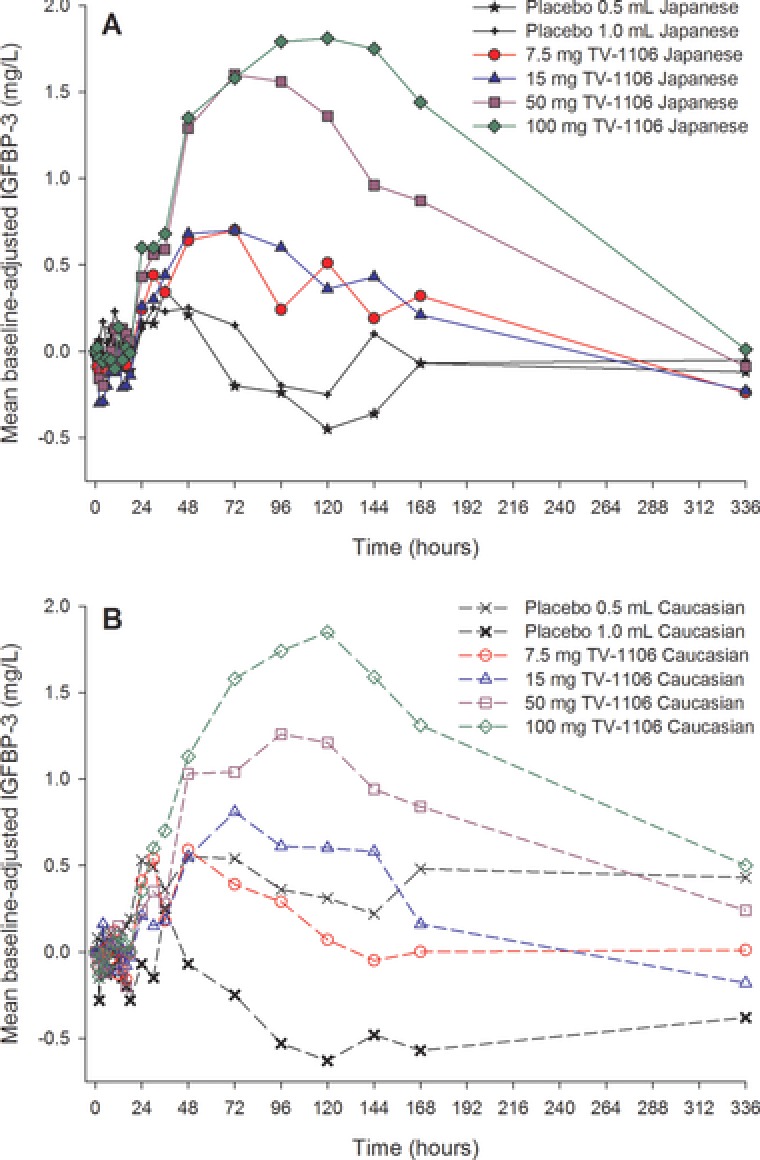
Mean baseline‐adjusted serum insulin‐like growth factor binding protein 3 (IGFBP‐3; mg/L) concentration versus time for Japanese (A) and caucasian (B) subjects.
The maximum observed mean Emax and AUEC parameters for baseline‐adjusted IGFBP‐3 occurred following 100 mg TV‐1106 in both Japanese and caucasian subjects (Table 2). The mean pharmacodynamic parameters were higher following all doses of TV‐1106 compared with placebo in Japanese subjects, although the responses to 7.5 and 15 mg TV‐1106 were similar. In the caucasian subjects, mean Emax and AUEC parameters increased with increasing TV‐1106 dose (7.5 to 100 mg); however, the values at 7.5 mg TV‐1106 were lower than those in the 0.5‐mL placebo group. In general, mean baseline‐adjusted IGFBP‐3 pharmacodynamic parameters varied between the placebo groups; in particular, the caucasian 0.5‐mL placebo group had notably higher Emax and AUEC parameters than the caucasian 1.0‐mL placebo group and both the Japanese placebo groups (Table 2). The higher mean baseline‐adjusted parameters in the caucasian 0.5‐mL placebo group may be partly because of 1 subject who had a low baseline value (2.6 mg/mL), whereas the majority of his later values (from 14 to 336 hours) were in the 4 to 5 mg/mL range. Subjects in the 3 other placebo groups had baseline values of 3.5 to 6 mg/mL. Variability in baseline‐adjusted IGFBP‐3 pharmacodynamic parameters was high in both Japanese and caucasian subjects, although the variability generally decreased with increasing dose of TV‐1106. Visual inspection of box plots showed comparable baseline‐adjusted IGFBP‐3 AUEC0–168 split by treatment and population (Figure 6) in Japanese and caucasian subjects.
Figure 6.
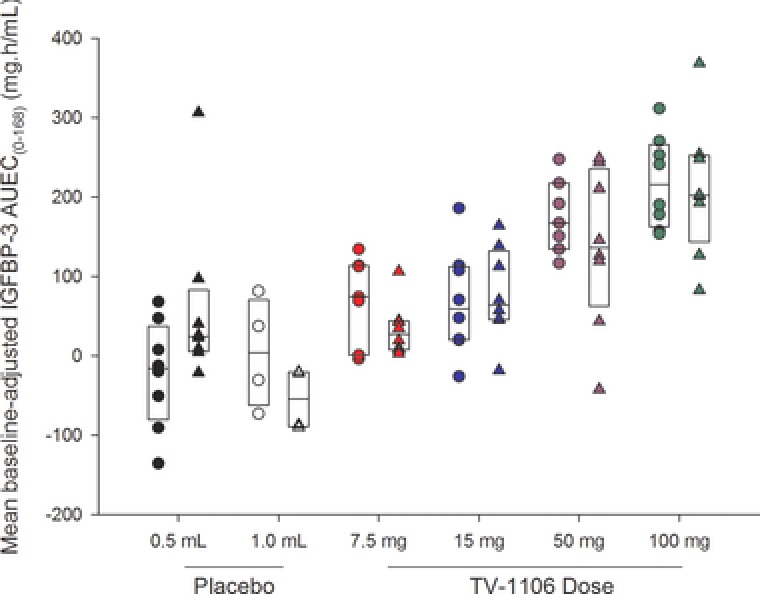
Box plots of baseline‐adjusted serum insulin‐like growth factor binding protein 3 (IGFBP‐3) area under the effect–time curve from 0 to 168 hours postdose (AUEC0–168) versus dose split by population. The boxes represent the median, 25th, and 75th percentiles. The circles are the individual values for Japanese subjects, and the triangles are individual values for caucasian subjects.
Safety
Overall, 29 of the total 88 subjects (33%) reported adverse events. A total of 24 of the 64 subjects who received TV‐1106 (38%) reported adverse events, compared with 5 of the 22 subjects who received placebo (23%).
One caucasian subject had a serious adverse event (spontaneous abortion) following 7.5‐mg TV‐1106 dosing, which was considered treatment‐related and severe. The subject had had a negative serum pregnancy test at screening and a negative urine test on day ‐1, but had a positive urine test on day 15. As the event occurred after dosing, the investigator assessed the event as related. No action was taken and the event resolved. The subject was allowed to remain in the study. No other serious or severe adverse events were reported, and no subjects were withdrawn from the study because of adverse events.
Overall, 10 of 88 subjects (11%) reported at least 1 treatment‐related adverse event (TRAE). Two subjects after TV‐1106 7.5 mg (13%) reported TRAEs, 3 subjects after 15 mg TV‐1106 (19%), 1 subject after TV‐1106 50 mg (6%), and 3 subjects after TV‐1106 100 mg (19%). One subject after placebo 0.5 mL (6%) and no subjects after placebo 1.0 mL reported TRAEs. The number of subjects who reported treatment‐related adverse events was similar between the Japanese and caucasian subjects (4 subjects [9%] and 6 subjects [14%], respectively). There were no dose‐related trends for adverse events (all causes or treatment‐related) for either Japanese or caucasian subjects.
The most commonly reported adverse event was headache, which was reported by 4 subjects (5%; 1 in the 50‐mg TV‐1106 group and 3 in the 100‐mg TV‐1106 group). The only other adverse events reported by more than 1 subject were vomiting, injection‐site pain, upper respiratory tract infection, and dizziness (2 subjects each). The majority of adverse events were reported as mild.
There were no clinically significant findings regarding clinical laboratory assessments, ECGs, vital signs, or other safety parameters. There was evidence of higher mean pulse rates from 30 to 144 hours postdose compared with the initial 24‐hour postdose period for the 50‐ and 100‐mg TV‐1106 groups. The mean maximum change from baseline was 10.8 and 19.0 bpm in the 50‐ and 100‐mg groups, respectively. The changes were similar in Japanese and caucasian populations.
There was a trend toward increases in mean fasting blood glucose and insulin levels after TV‐1106 dosing that was not deemed clinically meaningful, and there was no evidence of a difference between Japanese and caucasian subjects.
Three of the 64 TV‐1106‐treated subjects (4.7%) demonstrated treatment‐emergent anti‐TV‐1106 antibody response, with titers ranging from 3 to 83 (linear scale); however, all ADA‐positive samples tested negative for neutralizing antibody activity. Follow‐up samples were collected from 2 patients approximately 3 months after dosing. One patient tested negative; the other showed a titer decrease from 83 to 18. No neutralizing activity was observed for the ADA‐positive follow‐up sample.
Discussion
TV‐1106 is a long‐acting rhGH being developed as a subcutaneously administered long‐acting therapy to provide a safe and effective alternative for hormone replacement therapy in adult and pediatric patients with GHD. Presumably, a long‐acting nature and predicted less frequent administration regimen will lead to improved patient compliance and an enhanced quality of life for patients requiring GH replacement therapy. The primary aim of this study was to evaluate the pharmacokinetics and pharmacodynamics of TV‐1106 in healthy Japanese and caucasian subjects. Safety and tolerability of TV‐1106 were also assessed in both populations.
The pharmacokinetics of TV‐1106 were generally comparable between the caucasian and Japanese populations and were characterized by relatively slow increases in systemic exposure, with delayed peak serum concentrations. Multiple peaks were commonly observed in both populations, particularly in the first 24–48 hours. The reason for the multiple peaks is not known, but it is considered likely to be because of fluctuations in absorption of the liquid formulation from the subcutaneous injection site.
Serum TV‐1106 concentrations were detectable for approximately 3 to 6 days at doses up to 15 mg, and for up to 1 week following doses of 50 to 100 mg. Dose proportionality was not observed over any region of the dose range studied in both populations. TV‐1106 exposure appeared to increase more than dose proportionality, and this observation was also supported by exploratory power models. It should be noted, though, that the study was not powered for this analysis. The supraproportionality has also been observed with other long‐acting rhGHs.8 Apparent clearance and volume of distribution decreased with increasing dose of TV‐1106 in both populations. The TV‐1106 population PK model (unpublished data) with nonlinear elimination was found to describe the data adequately. Thus, the model suggests that the dose‐dependent changes in exposures are linked to saturable elimination, but the mechanism for this observation is still unknown. The clinical and therapeutic implications of the non–dose proportionality of TV‐1106 PK should be taken into account from a safety point of view; the current recommendations in clinical practice for GH replacement (may apply to TV‐1106 treatment) is that a lower starting dose should be employed and that IGF‐1 SDS level is used as the biomarker to guide dose titration for patients with GHD receiving GH replacement.
The long‐circulating concentrations of TV‐1106 indicate that a reduced frequency of injections (compared with daily injections) may be feasible in GHD patients. Indeed, the terminal t1/2 was generally in the range of 26 to 36 hours at 15 to 100 mg. These t1/2 values are consistent with those observed with other long‐acting rhGHs designed for once‐weekly dosing.8, 9, 10, 11, 12
The pharmacodynamic properties of TV‐1106 were assessed by investigation of IGF‐1 and IGFBP‐3, which are accepted pharmacodynamic markers of GH activity.13, 14, 15 IGF‐1 mediates the effect of endogenous GH, and it is primarily bound to IGFBP‐3, which plays an important role in controlling the bioavailability and action of IGF‐1.16
IGF‐1 serum concentrations increased rapidly in both Japanese and caucasian populations, with maximum concentrations observed 1.5 to 4 days after TV‐1106 dosing. The IGF‐1 response then decreased slowly in both populations, returning to baseline values within approximately 1 week for the lower doses and within 2 weeks for the higher doses. A similar profile was observed when IGF‐1 was expressed as SDS units, with comparable changes observed between the Japanese and caucasian populations. The difference between the Japanese and caucasian populations (±0.5 SDS) was similar to the magnitude of changes seen in the placebo groups (0.6 to 0.7 SDS), suggesting that any differences may be because of the inherent variability of the endogenous compound. In addition, the variability is consistent with the day‐to‐day intrapatient variability observed in week 12 in IGF‐1 SDS data in patients treated with daily Genotropin (Pfizer) in a phase 2 study in adults with GHD. In this phase 2 study, patients with confirmed GHD and experienced users of rhGH were titrated to an IGF‐1 SDS level of ±0.5 of their individual prewashout value over a period of 12 weeks, during which 1 of the treatment arms was daily Genotropin (data on file, Teva Pharmaceutical Industries).
In general, IGFBP‐3 also showed changes similar to IGF‐1, with maximum baseline‐adjusted concentrations observed 2 to 4.5 days after TV‐1106 dosing. As for IGF‐1, the IGFBP‐3 response decreased slowly in both populations, returning to baseline values within approximately 1 week for the lower doses and 2 weeks for the higher doses. The IGFBP‐3 response was comparable between the Japanese and caucasian populations.
For both IGF‐1 and IGFBP‐3, the data were variable across all TV‐1106 dose groups and populations and within the placebo groups. The changes observed in IGF‐1 and IGFBP‐3 in the placebo groups may suggest inherent endogenous variability, or there may be some degree of placebo effect.6, 17 Despite the variability, overall dose‐related increases in both IGF‐1 and IGFBP‐3 were observed following TV‐1106 dosing, with the greatest effects observed at the highest dose (100 mg) with mean IGF‐1 concentrations up to +3.1 SDS. It should be noted that the IGF‐1 levels at the higher doses (50 and 100 mg) were beyond the desired normal IGF‐1 level (+2.0 SDS). The high levels of IGF‐1 were detected in healthy subjects with normal physiologic baseline levels of GH who were administered exogenous GH and are therefore not likely to be relevant to GH‐deficient patients when the therapeutic goal is to restore a normal physiologic GH level (‐2.0 to +2.0 SDS).
In both populations it was noted that the pharmacodynamic effects peaked later than the serum TV‐1106 concentrations and that the decline in the pharmacodynamic response was slower than the decline in TV‐1106 concentrations. In addition, the multiple peaks observed in the TV‐1106 serum concentrations were not observed in the pharmacodynamic parameters. These data suggest that there is a delayed effect between TV‐1106 and IGF‐1. A delayed effect between TV‐1106 and IGF‐1 levels has also been reported following the first week of dosing of TV‐1106 in monkeys during a 4‐week study of once‐weekly dosing. TV‐1106 peak concentrations were approximately 24 hours postdosing, whereas IGF‐1 levels peaked on day 5 after the 5 mg/kg dose of TV‐1106 and on day 8 after the 10 and 20 mg/kg doses (data on file, Teva Pharmaceutical Industries).
TV‐1106 appeared to be safe in both populations. The only notable safety findings were mild changes in heart rate at the higher TV‐1106 doses and mild increases in fasting blood glucose and insulin values, none of which were considered clinically relevant. Changes in glucose and insulin have also been noted with other rhGH compounds.9, 18 The changes in heart rate at the higher doses (50 and 100 mg) are likely attributable to increasing the circulating levels of IGF‐1, which in turn increases heart rate. A similar observation was detected in a phase 1 single ascending‐dose (SAD) study with 3‐ to 100‐mg doses of TV‐1106. A dedicated exploratory analysis assessing the relationship between heart rate and IGF‐1 level suggested that higher doses of TV‐1106 may indirectly affect heart rate in healthy subjects by increasing the circulating levels of IGF‐1, which in turn increases heart rate.6, 7
The immunogenic response to TV‐1106 was investigated. Similar to the phase 1 SAD study conducted with TV‐1106,6 3 of the 64 TV‐1106‐treated subjects demonstrated treatment‐emergent TV‐1106 antibody response, with no observed neutralizing activity. ADAs have also been observed with other rhGH compounds.18
Conclusions
Overall, the data from this study suggest that there is no evidence of a difference between Japanese and caucasian populations in the pharmacokinetics or pharmacodynamics of TV‐1106. In addition, the data further demonstrate the long‐acting GH properties of TV‐1106 and support its potential for once‐weekly treatment of patients with GH deficiency.
Declaration of Conflicting Interests
O.C.B., H.B., M.R., K.B., K.Y., M.B., and O.S. are employees of Teva Pharmaceuticals. E.Y. is an employee of Parexel, which was contracted by Teva Pharmaceuticals to conduct the study of this report.
Funding
The study described in this report was funded by Teva Pharmaceutical Industries Research and Development, Petach Tiqva, Israel.
Supporting information
Supporting Information
Acknowledgments
The authors acknowledge the assistance of Samantha Abel (Valley Writing Solutions Ltd, Canterbury, United Kingdom) and Pippa Loupe, PhD (Teva Pharmaceuticals, Overland Park, Kansas) for help with the preparation of this article.
References
- 1. Eledrisi MS. Overview on Growth Hormone Deficiency. Emedicine [homepage on the Internet]. http://emedicine.medscape.com/article/120767-overview#a0104. Accessed January 9, 2015.
- 2. Gharib H, Cook DM, Saenger PH, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for growth hormone use in adults and children–2003 update. Endocr Pract. 2003;9:64–76. [DOI] [PubMed] [Google Scholar]
- 3. Rosenfeld R, Bakker B Compliance and persistence in pediatric and adult patients receiving growth hormone therapy. Endocr Pract. 2008;14:143–154. [DOI] [PubMed] [Google Scholar]
- 4. Osterberg L, Blaschke T. Adherence to medication. N Engl J Med. 2005;353:487–497. [DOI] [PubMed] [Google Scholar]
- 5. Høybye C, Cohen P, Hoffman AR, Ross R, Biller BMK, Christiansen JS. Status of long‐acting‐growth hormone preparations — 2015. Growth Horm IGF Res. 2015. https://doi.org/10.1016/j.ghir.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 6. Cohen‐Barak O, Sakov A, Rasamoelisolo M, et al. Safety, pharmacokinetic and pharmacodynamic properties of TV‐1106, a long‐acting growth hormone treatment for growth hormone deficiency. Eur J Endocrinol. 2015;173:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crock PA, McKenzie JD, Nicoll AM, et al. Benign intracranial hypertension and recombinant growth hormone therapy in Australia and New Zealand. Acta Paediatr. 1998;87:381–386. [DOI] [PubMed] [Google Scholar]
- 8. Rasmussen MH, Bysted BV, Anderson TW, Klitgaard T, Madsen J. Pegylated long‐acting human growth hormone is well‐tolerated in healthy subjects and possesses a potential once weekly pharmacokinetic and pharmacodynamic treatment profile. J Clin Endocrinol Metab. 2010;95:3411–3417. [DOI] [PubMed] [Google Scholar]
- 9. Sondargaard E, Klose M, Hansen M, et al. Pegylated long‐acting human growth hormone possesses a promising once‐weekly treatment profile, and multiple dosing is well tolerated in adult patients with growth hormone deficiency. J Clin Endocrinol Metab. 2011;96:681–688. [DOI] [PubMed] [Google Scholar]
- 10. Peter F, Savoy C, Ji HJ, Juhasz M, Bidlingmaier M, Saenger P. Pharmacokinetic and pharmacodynamic profile of a new sustained‐release GH formulation, LB03002, in children with GH deficiency. Eur J Endocrinol. 2009;160:349–355. [DOI] [PubMed] [Google Scholar]
- 11. Kemp SF, Fielder PJ, Attie KM, et al. Pharmacokinetic and pharmacodynamic characteristics of a long‐acting growth hormone (GH) preparation (nutropin depot) in GH‐deficient children. J Clin Endocrinol Metab. 2004;89:3234–3240. [DOI] [PubMed] [Google Scholar]
- 12. Zadika Z, Rosenfeld R, Radziuk K, et al. Top line results of once‐weekly, CTP‐modified human GH (MOD‐4023): phase 2 dose finding study in children with GH deficiency. Horm Res Paediatr. 2014;82(suppl 1):63 [Google Scholar]
- 13. Ooi GT, Boisclair YR. Molecular biology of the IGF binding proteins In: Rosenfeld R, Roberts CT, eds. The IGF System. Totowa, New Jersey: Humana Press Inc; 1999:111–139. [Google Scholar]
- 14. Fuhr U, Tuculanu D, Berghout A, Balser S, Schwebig A, Saenger P. Bioequivalence between novel ready‐to‐use liquid formulations of the recombinant human GH Omnitrope and the original lyophilized formulations for reconstitution of Omnitrope and Genotropin. Eur J Endocrinol. 2010;162:1051–1058. [DOI] [PubMed] [Google Scholar]
- 15. Pawlikowska‐Haddal, Cohen P , Cook DM. How useful are serum IGF‐1 measurements for managing GH replacement therapy in adults and children? Pituitary. 2012;15:126–134. [DOI] [PubMed] [Google Scholar]
- 16. Delafontaine P, Song YH, Li Y. Expression, regulation, and function of IGF‐1, IGF‐1R, and IGF‐1 binding proteins in blood vessels. Arterioscler Thromb Vasc Biol. 2004;24:435–444. [DOI] [PubMed] [Google Scholar]
- 17. Miller FG, Colloca L, Kaptchuk TJ. The placebo effect: illness and interpersonal healing. Perspect Biol Med. 2009;52:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Schepper J, Rasmussen MH, Gucev Z, Eliakim A, Battelino T. Long‐acting pegylated human GH in childrenwith GH deficiency: a single‐dose, dose‐escalation trial investigating safety, tolerability, pharmacokinetics and pharmacodynamics. Eur J Endocrinology. 2011;165:401–409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information