

Abstract
A placebo‐controlled, randomized, double‐blind, parallel‐group, comparative, multicenter study was conducted to investigate the efficacy and safety of benzoyl peroxide (BPO) gel, administrated once daily for 12 weeks to Japanese patients with acne vulgaris. Efficacy was evaluated by counting all inflammatory and non‐inflammatory lesions. Safety was evaluated based on adverse events, local skin tolerability scores and laboratory test values. All 609 subjects were randomly assigned to receive the study products (2.5% and 5% BPO and placebo), and 607 subjects were included in the full analysis set, 544 in the per protocol set and 609 in the safety analyses. The median rates of reduction from baseline to the last evaluation of the inflammatory lesion counts, the primary end‐point, in the 2.5% and 5% BPO groups were 72.7% and 75.0%, respectively, and were significantly higher than that in the placebo group (41.7%). No deaths or other serious adverse events were observed. The incidences of adverse events in the 2.5% and 5% BPO groups were 56.4% and 58.8%, respectively; a higher incidence than in the placebo group, but there was no obvious difference between the 2.5% and 5% BPO groups. All adverse events were mild or moderate in severity. Most adverse events did not lead to study product discontinuation. The results suggested that both 2.5% and 5% BPO are useful for the treatment of acne vulgaris.
Keywords: acne vulgaris, benzoyl peroxide, comparative study, placebo‐controlled, randomized
Introduction
Acne vulgaris is a chronic inflammatory skin disease that often develops on the face, chest and back in adolescents, starting as comedones in hair follicles. Multiple factors, including increased sebum secretion, endocrine factors (e.g. androgens), dyskeratosis of infundibular hair follicles and inflammation caused by bacterial growth (e.g. Propionibacterium acnes)1 are involved.
Based on its oxidizing property, the active ingredient of the study drug, benzoyl peroxide (BPO), shows sufficient antiseptic activity against P. acnes, a known cause of inflammatory lesions (IL) in acne vulgaris,2 as well as antibiotic‐resistant variants of P. acnes and Staphylococcus epidermidis that develop during long‐term use of antimicrobials.3 It has also been suggested that BPO improves retention hyperkeratosis of infundibular hair follicles.4 Topical medications containing 2.5–10% BPO are widely used for acne vulgaris in Asian countries like Korea, Singapore and Hong Kong, as well as Europe and the USA, where the medical guidelines recognize BPO as a standard of care for acne vulgaris.5, 6
This study was a multicenter, placebo‐controlled, randomized, double‐blind, parallel‐group, comparative study to confirm the efficacy and safety of 2.5% and 5% BPO gel in Japanese patients with acne vulgaris and identify the recommended clinical dose of BPO.
The gels are aqueous gels containing 25 or 50 mg BPO per 1 g of the gel and formulated based on a commercially available BPO product in the USA. The formulation base is composed of conventional additive agents, such as propylene glycol, carboxy vinyl polymer and pH‐adjusting agents. Until the recent approval, BPO was not used as a prescription or over‐the‐counter drug in Japan and there was no clinical experience of BPO in Japanese acne patients. This is the first report that directly compared efficacy and safety of the two formulations with different BPO concentrations to find a better clinical dose of BPO for clinical use in Japanese acne patients, who are considered more susceptible to skin irritation than Caucasian. Although BPO has been used as a standard drug for a long time outside of Japan, there have been few reports that aimed to clarify the relationship between dose and efficacy/safety in BPO products.
This article is based on a study that was first reported in Japanese in the Journal of Clinical Therapeutics and Medicine 2014; 30(8): 651–668, and this secondary publication in English is made with permission of the journal.
Methods
Study design
This phase II/III study was a multicenter (30 centers), randomized, double‐blind, placebo‐controlled, parallel‐group, comparative study that evaluated the efficacy and safety of BPO (2.5% and 5%) relative to the inert vehicle gel alone (placebo) when applied once daily for 12 weeks in Japanese patients with acne vulgaris, and clinically recommended dose was determined. This study was registered at the Japan Pharmaceutical Information Center (JapicCTI‐121784). The study was conducted between February and October 2012. Patients who enrolled in this study were first treated with placebo during the 2‐week observation period, and then randomized (1:1:1) by a computer randomization system (six patients in each block) to receive BPO 2.5%, BPO 5% or placebo. Patients were assessed at baseline (week 0/day 1) and weeks 2, 4, 6, 8, 10 and 12 (Table S1). The final evaluation was at week 12 or discontinuation.
This study was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice and the ethical principles outlined in the Declaration of Helsinki 2008. The protocol and other relevant study documents were approved by the relevant institutional review boards. Prior to the start of the study, written informed consent was obtained from adult patients and parents or legal guardians of patients under 20 years old.
Patients
Male and female outpatients aged 12–49 years were eligible if they had facial acne (excluding periocular and lip lesions) characterized by 11–40 IL (erythematous papules and pustules), 20–100 non‐IL (open and closed comedones) and two or fewer nodules/cysts at baseline (Table S2).
Before enrollment in the observation period, the following patients were excluded: patients who had complications of other facial acne than acne vulgaris, rosacea or skin diseases that may cause a facial rash (such as atopic dermatitis); female patients who had fluctuation in acne symptoms with the menstrual cycle; female patients who were pregnant, breast‐feeding or hoping to become pregnant; patients who had exhibited hypersensitivity or previous allergic reactions to any component of the study drugs; and patients who had serious complications (including systemic diseases) which precluded participation in the study. After the observation period, patients with low study drug compliance (<70%) or high reduction rate (>30%) in the number of IL during the observation period were excluded from entering the treatment phase. The following patients were also excluded: patients who used systemic or topical retinoids, or 14‐day or longer systemic antibiotics within 12 weeks prior to the beginning of the treatment period; anti‐acne medications (excluding vitamins B2 and B6), steroids, antibiotics or other drugs within 4 weeks (systemic use) or 2 weeks (local application) prior to the beginning of the treatment period, or quasi drugs or cosmetics for the treatment of facial acne; patients who underwent chemical peeling, laser ray treatment or other therapies for acne, which may have an influence on the efficacy of the study drug, within 4 weeks prior to the beginning of the treatment period; or patients who participated in another clinical study within 4 months prior to the beginning of the treatment period.
Treatment regimen
The schedule of the study is shown in Figure 1. Participants applied placebo during the observation period (2 weeks before the treatment started), and either 2.5% or 5% BPO or placebo during the treatment period (12 weeks). Patients were instructed to apply a sufficient quantity of the assigned drug to cover the entire face (except the lips and around the eyes) once daily at night after they washed and dried their faces.
Figure 1.
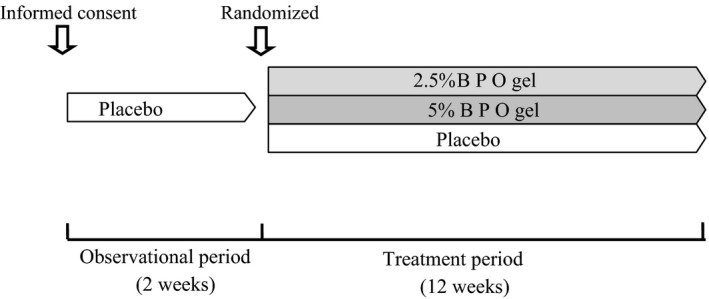
Schedule of the study.
During the treatment period, pharmaceutical agents, quasi‐drugs or cosmetic products (face wash with scrubber, azelaic acid‐containing cosmetic products) for acne, steroids, antimicrobials and others that could affect the evaluation of the study drug were prohibited. Continued administration of vitamins B2 and B6 indicated for acne was allowed, as long as the dose was not changed from the start of observation period to the end of the study or the day when the study was discontinued. Chemical peeling of the face, laser therapy, phototherapy, comedone suction or extraction, other acne treatments, and esthetic facial procedures were prohibited.
Assessments
Facial IL and non‐IL were counted by the investigators at each visit. The primary efficacy end‐point was percentage change in IL count from baseline to the final evaluation (week 12 or discontinuation). Secondary efficacy end‐points were percentage changes in total lesion (TL) and non‐IL counts from baseline to the final evaluation; absolute changes in TL, IL and non‐IL counts from baseline to the final evaluation; and percentage changes in TL, IL and non‐IL counts from baseline to weeks 2, 4, 6, 8, 10 and 12.
Safety was assessed based on adverse events (AE), local skin tolerability scores (scaling and erythema) and laboratory tests (hematology, blood chemistry and urine analysis). The local skin tolerability scores were evaluated on a four‐grade scale (0 = none, 1 = mild, 2 = moderate, 3 = severe; Table S3) by the investigator at each visit. An increase in the score over baseline level was recorded as an AE.
Statistical analysis
Assuming a difference in percentage change in IL count at week 12 of 15% (standard deviation of 44) between the BPO 2.5% and placebo groups, a sample size of 182 or more per group was calculated to provide a more than 90% power to demonstrate the superiority of BPO 2.5% over placebo at the 0.05 level of significance (two‐sided). Taking dropout rate into consideration, a sample size of 200 subjects per group (600 in total) was set. The main analysis population for efficacy was the full analysis set (FAS), which included patients who received the study drugs at least once and were evaluated for efficacy at least once. Non‐parametric Wilcoxon two‐sample test was used for testing superiority of BPO over placebo at the 0.05 two‐sided significance level. Assuming that percentage reductions in IL count from baseline to week 12 were equivalent in the 2.5% and 5% BPO groups, a close testing procedure was used for group comparison in ascending order: if it was significant between BPO 5% and placebo, then BPO 2.5% gel was compared with placebo. No multiplicity adjustments were made for the P‐value for the secondary end‐points. To ensure the robustness of the statistical tests, we applied the same tests to the per protocol set (PPS), which consisted of patients in FAS excluding those who did not satisfy the inclusion criteria or conflicted with the exclusion criteria, patients with low compliance rate (<70%) for one or more evaluation term(s), patients who received a prohibited concomitant drug/therapy, patients who underwent final evaluation for efficacy less than 81 days after the start of the treatment, except for patients who discontinued prematurely because the principal investigator confirmed that all of their IL lesions had completely resolved.
Patients who received the study drugs at least once and were evaluated for safety at least once were included in the safety assessment. All AE were coded using the Medical Dictionary for Regulatory Activities (MedDRA version 15.0) and summary statistics were calculated for each treatment group by the System Organ Class and Preferred Term and by causal relationship with the study treatments.
Results
Disposition of enrolled patients and their profiles
The disposition of enrolled patients is shown in Figure 2. In this study, 679 patients participated in the observation period, 70 discontinued and 609 proceeded to the treatment period, after which they were randomized and administrated the study drugs. During the treatment period, 31 participants withdrew and 578 completed the study. The number of subjects who discontinued and the reasons for withdrawal were similar in each group (Table S4). Although emergency codes were broken for three participants who became pregnant during the study, blinding of the other patients was maintained throughout the trial.
Figure 2.
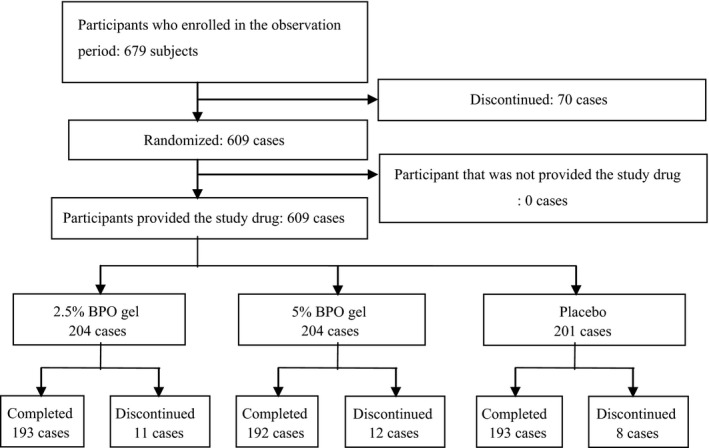
Disposition of participants.
Excluding two subjects who were not evaluated for safety, FAS included 607 subjects who applied the study drug at least once (2.5% BP, n = 203; 5% BPO, n = 203; placebo, n = 201).
Baseline demographics and disease characteristics of patients included in the FAS are shown in Table 1. The number of female patients was greater than that of male patients in all groups. Age distribution was similar among the groups, namely, most patients were aged 16–20 years, followed by 12–15 years and then 21–25 years. The total number of subjects in these three age categories accounted for more than 80% of all participants. No bias between the groups regarding history of hypersensitivity, concomitant drug use or combination therapy, or the number of each lesion type at baseline was observed. The PPS consisted of 554 patients (2.5% BPO, n = 184, 5% BPO, n = 179; placebo, n = 181).
Table 1.
Baseline demographic and disease characteristics of participants (FAS)
| 2.5% BPO gel | 5% BPO gel | Placebo | |
|---|---|---|---|
| No. of cases (%), n = 203 | No. of cases (%), n = 203 | No. of cases (%), n = 201 | |
| Sex | |||
| Male | 84 (41.4) | 79 (38.9) | 91 (45.3) |
| Female | 119 (58.6) | 124 (61.1) | 110 (54.7) |
| Age (years) | |||
| 12–15 | 52 (25.6) | 46 (22.7) | 57 (28.4) |
| 16–20 | 84 (41.4) | 80 (39.4) | 85 (42.3) |
| 21–25 | 36 (17.7) | 44 (21.7) | 33 (16.4) |
| 26–30 | 18 (8.9) | 21 (10.3) | 15 (7.5) |
| 31–35 | 11 (5.4) | 8 (3.9) | 6 (3.0) |
| 36–40 | 1 (0.5) | 4 (2.0) | 3 (1.5) |
| 41–45 | 1 (0.5) | 0 | 2 (1.0) |
| 46–49 | 0 | 0 | 0 |
| Average | 19.5 | 20.0 | 19.2 |
| SD | 5.7 | 5.6 | 5.5 |
| History of hypersensitivity | |||
| Yes | 15 (7.4) | 12 (5.9) | 14 (7.0) |
| No | 188 (92.6) | 191 (94.1) | 187 (93.0) |
| Concomitant drugs | |||
| Yes | 88 (43.3) | 110 (54.2) | 96 (47.8) |
| No | 115 (56.7) | 93 (45.8) | 105 (52.2) |
| Concomitant therapies | |||
| Yes | 10 (4.9) | 9 (4.4) | 14 (7.0) |
| No | 193 (95.1) | 194 (95.6) | 187 (93.0) |
| No. of IL at baseline | |||
| Median | 18 | 18 | 18 |
| Range | 11–40 | 11–40 | 11–40 |
| No. of TL at baseline | |||
| Median | 50 | 51 | 51 |
| Range | 31–125 | 31–134 | 31–140 |
| No. of non‐IL at baseline | |||
| Median | 29 | 30 | 30 |
| Range | 20–90 | 20–96 | 20–100 |
| No. of nodules/cysts | |||
| Median | 0 | 0 | 0 |
| At baseline | |||
| Range | 0–2 | 0–2 | 0–2 |
BPO, benzoyl peroxide; FAS, full analysis set; IL, inflammatory lesions; SD, standard deviation; TL, total lesions.
Compliance rate
For FAS, patients with less than 70% compliance rate for treatment from the start to the completion of the study were three of 203 for 2.5% BPO, five of 203 for 5% BPO and none for placebo (0/201). Specifically, the number of such patients for any evaluation term was 4 or less. Overall, the compliance rate with treatment in each group was high.
Efficacy
Primary efficacy end‐point
The percentage reduction in the number of IL at the end of study for FAS is shown in Table 2. Median percentage reductions for 2.5% BPO and 5% BPO were similar, namely, 72.7% and 75.0%, respectively, while that for placebo was 41.7%. Compared with placebo, the percentage reduction of IL with 5% BPO was significantly different (Wilcoxon two‐sample test, P < 0.001). Then, the 2.5% BPO was compared with the placebo group, and the difference between the two groups was also significant (Wilcoxon two‐sample test, P < 0.001). Similar results were obtained for the PPS.
Table 2.
Absolute number and percentage reduction in inflammatory lesions (FAS)
| Treatment group | Baseline, no. of IL | End of study, no. of IL | Absolute reduction, no. of IL | Percentage reduction, % |
|---|---|---|---|---|
| 2.5% BPO gel (203 cases) | 18 (14–26) | 5 (2–11) | 12*** (8–18) | 72.7*** (46.2–87.5) |
| Difference from placebo | – | −5 | 5 (4–7) | 25.7 (19.2–32.9) |
| 5% BPO gel (203 cases) | 18 (14–26) | 4 (2–9) | 12*** (9–18) | 75.0*** (60.0–85.7) |
| Difference from placebo | – | −5 | 6 (5–8) | 28.0 (21.6–34.9) |
| Placebo | 18 (14–24) | 11 (6–19) | 7 (1–12) | 41.7 (6.3–66.7) |
***Median (interquartile range). Difference from placebo: Hodges–Lehmann estimator (95% confidence intervals). P < 0.001 vs placebo (Wilcoxon two‐sample test). BPO, benzoyl peroxide; FAS, full analysis set; IL, inflammatory lesions.
Secondary efficacy end‐point
The percentage reduction in the numbers of TL and non‐IL at the end of the study for FAS. Table 3 shows the reduction rate in the numbers of TL and non‐IL from the start to the end of the study. The median percentage reduction for TL was 62.2% for 2.5% BPO and 67.9% for 5% BPO, which were significantly different from placebo (28.6%, P < 0.001 Wilcoxon two‐sample test). The median percentage reduction in non‐IL was 56.5% for 2.5% BPO and 68.2% for 5% BPO, which were significantly larger than the 21.9% for placebo (Wilcoxon two‐sample test, P < 0.001).
Absolute reduction in number of TL, IL and non‐IL at the end of the study in FAS. The absolute reduction in the count of each type of lesion is shown in Tables 2 and 3. The median reduction in TL was 29 for 2.5% BPO, 31 for 5% BPO and 14 for placebo, suggesting that 2.5% and 5% BPO reduced TL significantly more than placebo (Wilcoxon two‐sample test, P < 0.001). The median reduction in number of IL was 12 for 2.5% BPO, 12 for 5% BPO and seven for placebo, suggesting that 2.5% and 5% BPO reduced IL significantly more than placebo (Wilcoxon two‐sample test, P < 0.001). Concerning the number of non‐IL, the median reduction was 16 for 2.5% BPO, 19 for 5% BPO and seven for placebo, similarly suggesting that the absolute reduction in number of non‐IL for 2.5% or 5% BPO groups was significantly larger than in the placebo group (Wilcoxon two‐sample test, P < 0.001).
- Percentage reduction in TL, IL and non‐IL over time in the FAS. The percentage reduction in each type of lesion shifted over time is shown in Figure 3. The median percentage reduction in number of TL for 2.5% BPO and 5% BPO was 22.6% versus 27.1% at week 2, 33.8% versus 45.5% at week 4, 43.8% versus 54.6% at week 6 and 62.5% versus 69.3% at week 12. Conversely, the median reduction in number of TL for placebo was 8.5% at week 2, 14.8% at week 4, 20.3 at week 6 and 28.8 at week 12, which were smaller than those for 2.5% BPO and 5% BPO at all evaluation time points.
Figure 3.
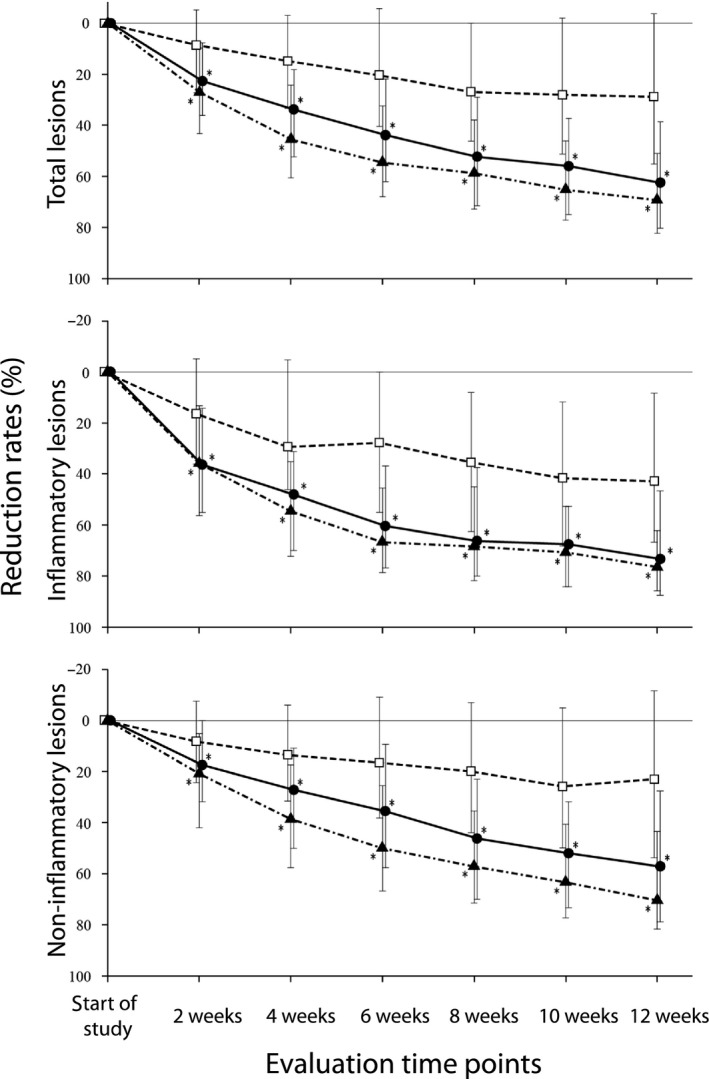 Percentage reduction in number of each type of lesion over time. (●) 2.5% benzoyl peroxide (BPO) gel, (▲) 5% BPO gel, (□) placebo, median (interquartile range). *P < 0.001 (two‐tailed test [α = 0.05], Bonferroni correction [testing total 36 times]). Wilcoxon two‐sample test (vs placebo).
Percentage reduction in number of each type of lesion over time. (●) 2.5% benzoyl peroxide (BPO) gel, (▲) 5% BPO gel, (□) placebo, median (interquartile range). *P < 0.001 (two‐tailed test [α = 0.05], Bonferroni correction [testing total 36 times]). Wilcoxon two‐sample test (vs placebo).
Table 3.
Absolute number and percentage reduction of total lesions and non‐inflammatory lesions (FAS)
| Cohorts | Baseline, no. of lesions | End of study, no. of lesions | Absolute reduction, no. lesions | Percentage reduction, % |
|---|---|---|---|---|
| No. of TL | ||||
| 2.5% BPO gel (203 cases) | 50 (40–63) | 21 (10–35) | 29*** (17 to 38) | 62.2*** (33.3 to 79.6) |
| Difference from placebo | – | −16 | 16 (12 to 20) | 29.4 (22.7 to 36.5) |
| 5% BPO gel (203 cases) | 51 (40–64) | 18 (8–30) | 31*** (23 to 43) | 67.9*** (48.6 to 81.8) |
| Difference from placebo | – | −18 | 20 (16 to 24) | 36.0 (29.3 to 42.9) |
| Placebo | 51 (41–67) | 36 (22–59) | 14 (−2 to 28) | 28.6 (−3.9 to 54.4) |
| No. of non‐IL | ||||
| 2.5% BPO gel (203 cases) | 29 (23–40) | 14 (7–25) | 16*** (7 to 24) | 56.5*** (26.3 to 78.3) |
| Difference from placebo | – | −10 | 10 (7 to 13) | 29.5 (21.4 to 37.6) |
| 5% BPO gel (203 cases) | 30 (23–41) | 11 (6–21) | 19*** (12 to 26) | 68.2*** (38.7 to 81.4) |
| Difference from placebo | – | −12 | 13 (10 to 16) | 37.5 (29.7 to 45.2) |
| Placebo | 30 (23–43) | 25 (14–41) | 7 (−4 to 17) | 21.9 (−13.0 to 53.3) |
***Median (interquartile range). Difference from placebo: Hodges–Lehmann estimator (95% confidence intervals). P < 0.001 vs placebo (Wilcoxon two sample test). Difference from placebo: Hodges‐Lehmann estimator (95% confidence intervals). P < 0.001 vs placebo (Wilcoxon two sample test). BPO, benzoyl peroxide; FAS, full analysis set; IL, inflammatory lesions; SD, standard deviation; TL, total lesions.
Concerning IL, the median percentage reduction for 2.5% BPO and 5% BPO were 36.4% versus 35.8% at week 2, 48.1% vs 54.6% at week 4, 60.4% versus 66.7% at week 6 and 73.3% versus 76.5% at week 12. Compared with these, the median percentage reduction for placebo was 16.4% at week 2, 29.4% at week 4, 27.8% at week 6 and 42.9% at week 12, and smaller than those for 2.5% BPO and 5% BPO at all evaluation time points.
The median percentage reduction in number of non‐IL for 2.5% BPO and 5% BPO was 17.4% versus 20.8% at week 2, 27.2% versus 38.6% at week 4, 35.5% versus 50.0% at week 6 and 57.1% versus 70.4% at week 12. The median percentage reduction in the number of non‐IL for placebo was 8.3% at week 2, 13.6% at week 4, 16.7% at week 6 and 23.1% at week 12, which were smaller than those for 2.5% BPO and 5% BPO at all evaluation time points.
Evaluation of subpopulations
To evaluate the primary efficacy end‐point further, ancova was performed with values converted to the median percentage reduction in the number of IL by the “joint‐ranking” procedure as a response variable and “treatment group” and “age” as explanatory variables. The analysis suggests that the reduction rate in the number of IL at the end of the study may increase with increasing patient age. This tendency was observed in the placebo group, but not in the 2.5% or 5% BPO groups (Table 4).
Table 4.
Percentage reduction in IL at the end of the study by subpopulations (FAS)
| Age | 12–15 years | 16–18 years | 19–22 years | 23–49 years |
|---|---|---|---|---|
| 2.5% BPO gel | ||||
| No. of cases | 52 | 57 | 46 | 48 |
| Median | 73.9 | 66.7 | 69.3 | 80.4 |
| Interquartile | 40.3 to 89.7 | 42.1 to 84.2 | 47.8 to 80.0 | 59.4 to 93.3 |
| 5% BPO gel | ||||
| No. of cases | 46 | 51 | 52 | 54 |
| Median | 72.8 | 72.0 | 71.9 | 78.8 |
| Interquartile | 57.7 to 84.6 | 46.2 to 83.3 | 54.7 to 85.2 | 66.7 to 86.4 |
| Placebo | ||||
| No. of cases | 57 | 54 | 58 | 32 |
| Median | 30.4 | 36.7 | 45.8 | 60.6 |
| Interquartile | −2.7 to 63.6 | 0.0 to 64.7 | 15.4 to 66.7 | 36.9 to 77.1 |
BPO, benzoyl peroxide; FAS, full analysis set; IL, inflammatory lesions.
Safety
Safety assessment included 609 patients who applied the study drug at least once (204 for 2.5% BPO, 204 for 5% BPO and 201 for placebo).
Adverse events
Table 5 summarizes the incidence of AE in each group with or without a causal relation with the study drug. The percentage of patients who experienced AE was 56.4% (115/204) for 2.5% BPO, 58.8% (120/204) for 5% BPO and 47.3% (95/201) for placebo. The incidences of mild AE in the 2.5% BPO, 5% BPO and placebo groups were 112, 115 and 92, respectively, and those of moderate AE were five, 11 and five, respectively. No severe AE were observed in any groups. The incidence of AE with a possible causal relation with the study drug was 37.3% (76/204) for 2.5% BPO, 38.7% (79/204) for 5% BPO and 12.9% (26/201) for placebo. Among them, mild AE were 73, 75 and 25, and moderate events were four, seven and one, respectively, in each group.
Table 5.
Frequently observed adverse events with or without a causal relation with the study drugs (more than 2% incidence in FAS)
| System organ class (preferred term) | No causal relation with the study drug | Possible causal relation with the study drug | ||||
|---|---|---|---|---|---|---|
| 2.5% BPO, no. cases (%) | 5% BPO, no. cases (%) | Placebo, no. cases (%) | 2.5% BPO, no. cases (%) | 5% BPO, no. cases (%) | Placebo, no. cases (%) | |
| No. of subjects analyzed | 204 | 204 | 201 | 204 | 204 | 201 |
| Adverse events | 115 (56.4) | 120 (58.8) | 95 (47.3) | 76 (37.3) | 79 (38.7) | 26 (12.9) |
| General disorders and administration site conditions | – | – | – | – | – | – |
| Application site irritation | 17 (8.3) | 25 (12.3) | 2 (1.0) | 17 (8.3) | 25 (12.3) | 2 (1.0) |
| Application site erythema | 28 (13.7) | 22 (10.8) | 5 (2.5) | 28 (13.7) | 22 (10.8) | 4 (2.0) |
| Application site pruritus | 7 (3.4) | 5 (2.5) | 0 | 7 (3.4) | 5 (2.5) | 0 |
| Infections and infestations | – | – | – | – | – | – |
| Nasopharyngitis | 17 (8.3) | 31 (15.2) | 20 (10.0) | 0 | 0 | 0 |
| Laboratory test | – | – | – | – | – | – |
| White blood cell count increase | 11 (5.4) | 6 (2.9) | 9 (4.5) | 1 (0.5) | 1 (0.5) | 0 |
| Blood cholesterol decrease | 4 (2.0) | 5 (2.5) | 5 (2.5) | 0 | 1 (0.5) | 0 |
| Blood bilirubin increase | 3 (1.5) | 2 (1.0) | 7 (3.5) | 2 (1.0) | 1 (0.5) | 2 (1.0) |
| Aspartate aminotransferase increase | 2 (1.0) | 0 | 6 (3.0) | 0 | 0 | 4 (2.0) |
| Nervous system disorders | – | – | – | – | – | – |
| Headache | 5 (2.5) | 1 (0.5) | 1 (0.5) | 0 | 0 | 0 |
| Skin and subcutaneous tissue disorders | – | – | – | – | – | – |
| Skin exfoliation | 42 (20.6) | 49 (24.0) | 19 (9.5) | 39 (19.1) | 48 (23.5) | 16 (8.0) |
| Contact dermatitis | 6 (2.9) | 7 (3.4) | 0 | 5 (2.5) | 3 (1.5) | 0 |
| Eczema | 4 (2.0) | 5 (2.5) | 2 (1.0) | 0 | 0 | 0 |
BPO, benzoyl peroxide; FAS, full analysis set.
Frequently observed adverse events
Adverse events found in 2% or more of patients using 2.5% BPO were skin exfoliation in 20.6% (42/204), application site erythema in 13.7% (28/204), application site irritation in 8.3% (17/204), nasopharyngitis in 8.3% (17/204), white blood cell count increase in 5.4% (11/204), application site pruritus in 3.4% (7/204), contact dermatitis in 2.9% (6/204) and headache in 2.5% (5/204). Among these, AE with a possible causal relation with the study drug were skin exfoliation in 19.1% (39/204), application site erythema in 13.7% (28/204), application site irritation in 8.3% (17/204), application site pruritus in 3.4% (7/204) and contact dermatitis in 2.5% (5/204).
Adverse events found in 2% or more of patients using 5% BPO, were skin exfoliation in 24.0% (49/204), nasopharyngitis in 15.2% (31/204), application site irritation in 12.3% (25/204), application site erythema in 10.8% (22/204), contact dermatitis in 3.4% (7/204), white blood cell count increase in 2.9% (6/204), blood cholesterol decrease in 2.5% (5/204), application site pruritus in 2.5% (5/204) and eczema in 2.5% (5/204). Among these, AE with a possible causal relation with the study drug were skin exfoliation in 23.5% (48/204), application site irritation in 12.3% (25/204), application site erythema in 10.8% (22/204) and application site pruritus in 2.5% (5/204).
Adverse events found in 2% or more of patients using placebo were nasopharyngitis in 10.0% (20/201), skin exfoliation in 9.5% (19/201), white blood cell count increase in 4.5% (9/201), blood bilirubin increase in 3.5% (7/201), aspartate aminotransferase increase in 3.0% (6/201), application site erythema in 2.5% (5/201) and blood cholesterol decrease in 2.5% (5/201). Among these, the only AE with a possible causal relation with the study drug was skin exfoliation in 8.0% (16/201).
Deaths and other severe or noteworthy adverse events
There were no cases of death or severe AE in this study. The number of patients who discontinued due to AE was 13 (six, five and two for 2.5% BPO, 5% BPO and placebo, respectively). All events with 2.5% and 5% BPO had a possible causal relation with the study drug, namely, application site erythema (three cases), contact dermatitis (three cases), application site pain and application site swelling (one case each) and skin exfoliation (one case) with 2.5% BPO gel, and application site erythema (two cases), contact dermatitis (two cases), application site irritation and skin exfoliation (one case each) with 5% BPO. All AE were mild or moderate, and resolved spontaneously or with medication.
Evaluation of local skin tolerability scores for scaling and erythema and laboratory test values
Local skin tolerability scores over time from the start to the end of the study are shown in Tables S5 and S6. Most participants scored 0 in all groups (>80% for scaling and ~90% for erythema). Participants with scores of 1 or 2 were few. The number of patients with an increase in the local skin tolerability score from the start of the study was more for 2.5% and 5% BPO than placebo.
Clinical laboratory tests showed no significant changes during the study. Regarding AE with a possible causal relation with the study drugs, most were mild or moderate and occurred at the application sites. Most patients with such reactions completed the study without discontinuation, and AE observed in discontinued cases resolved quickly. Therefore, we concluded that the tolerability of the study drug is high in actual use settings.
Discussion
This study was a placebo‐controlled, randomized, double‐blinded, parallel‐group comparative, multicenter study, to confirm safety and efficacy, and determine the clinically recommended dose by administration of either 2.5% or 5% BPO once daily for 12 weeks to participants with acne vulgaris.
In all treatment groups in this study, the number of female patients was greater than that of male patients, and more than 80% were between 12 and 25 years old. This distribution is similar to the results of a multicenter seasonal national survey conducted by the Japanese Dermatological Association,7 and a substantial investigation of acne patients we performed at medical institutions that specialized in dermatology.8 Therefore, the population would reflect the target population of patients for the study drugs when marketed.
At the end of the study, the median percentage reduction in the number of IL was 72.7% for 2.5% BPO and 75.0% for 5% BPO versus 41.7% for placebo. The reduction rate for 5% and 2.5% BPO compared with placebo at the end of the study was significantly larger. The number of IL in the 2.5% and 5% BPO groups was both 18 at the start of the treatment period, and five and four at the end, respectively, suggesting that the severity of acne vulgaris shifted from moderate to mild. Therefore, the reduction in the number of IL was clinically meaningful.
Concerning non‐IL, the median percentage reduction was 56.5% for 2.5% BPO, 68.2% for 5% BPO versus 21.9% for placebo. The percentage reduction with both 5% and 2.5% BPO was significantly larger than with placebo. The differences in the percentage reduction for 2.5% and 5% BPO from that for placebo were in a range (95% confidence interval) of 21.4–37.6% (2.5% BPO vs placebo) and 29.7–45.2% (5% BPO vs placebo). The European medical guideline6 recognizes a more than 10% difference between groups as a beneficial effect. Thus, the reduction in number of non‐IL with 2.5% and 5% BPO were clinically meaningful.
The incidences of AE in the 2.5% and 5% BPO groups were 56.4% and 58.8%, respectively. There was no difference in the incidence between the two groups, although both were higher than that of placebo. The AE observed in the 2.5% and 5% groups were mild to moderate in severity and most subjects continued the treatment without discontinuation. AE that led to study discontinuation resolved soon without treatment or with medication, suggesting high tolerability for the study drugs under actual use settings.
Concerns among acne patients are scarring, inflammatory and non‐IL; most patients are anxious to resolve the acne without leaving scars,9 and patients choose treatment for cosmetic reasons. It has also been reported that patients with acne suffer from low quality of life concerning emotional condition.10 Therefore, it is important to start appropriate treatment at an early stage to avoid the formation of IL and non‐IL and to prevent transition to nodules/cysts and scarring.1 It was observed that treatment with 2.5% and 5% BPO gel reduced not only the number of IL but also non‐IL after 2 weeks of treatment. There were no differences between the two BPO groups in the percentage reductions and absolute reductions in the numbers of IL and non‐IL at the end of the study. The results were similar to those of a comparable clinical study of topical BPO performed in the USA.11 Both 2.5% and 5% BPO gel can prevent the formation of IL and non‐IL at the start of treatment and improve the quality of life of patients.
These results confirmed the safety of 2.5% and 5% BPO gels, and suggest that they effectively reduce not only the number of IL but also non‐IL compared with placebo. In conclusion, both 2.5% and 5% BPO gels can be a beneficial treatment option for Japanese patients with acne vulgaris.
Conflict of Interest
Maruho Co. Ltd covered all expenses for this study. All drugs used for this study were provided by the company. M. K. was the medical advisor of the study and S. S., F. F., K. M., H. A., A. I., Y. T., N. H. and Y. Y. were coordinating investigators of the study. M. K. and Y. T. received fees for consultation, lectures and writing from the company. S. S .received fees for lectures and research grants from the company. A. I. received fees for consultation, lectures and writing, and research grants from the company. N. H. received fees for lectures and writing from the company. T. N. and T. K. are employees of the company.
Supporting information
Table S1. Schedule for investigations and observations.
Table S2. Definitions of inflammatory and non‐inflammatory lesions.
Table S3. Grading criteria for local skin tolerability scores.
Table S4. Profiles of withdrawals after randomization.
Table S5. Shift in local skin tolerability scores for scaling over time.
Table S6. Shift in local skin tolerability scores for erythema over time.
Acknowledgments
We would like to acknowledge the contributions of 58 investigators including the following principal investigators (alphabetical order of centers): Jun Adachi, Adachi Dermatological Clinic; Yoko Todoroki, Clinique Dermatologique Todoroki; Yoshihide Fukaya, Fukaya Dermatologic Clinic; Sakae Harada, Harada Hifuka Clinic; Yuki Suzuki and Mami Chiba, Iderea Skin Clinic Daikanyama; Masaaki Isonokami, Isonokami Dermatological Clinic; Akiko Ishikou, Kaminoge Hifuka Clinic; Takeo Kato, Kato Dermatology Plastic surgery; Yasuhiro Kawabata, Kawabata Dermatology Clinic; Miwako Kinoshita, Kinoshita Dermatology Clinic; Hiroyuki Yamao, Kitaayase Dermatology Clinic; Takako Kitano, Kitano Skin Clinic; Tetsufumi Koyama, Koyama Dermatology Clinic; Mari Suzuki, Mari Skin Care Clinic; Ryuji Maruyama, Maruyama Dermatology Clinic; Tomohiko Matsuyama, Matsuyama Dermatology Clinic; Ichiro Kurokawa, Meiwa Hospital; Yoshiyuki Murakami, Mildix Skin Clinic; Naoko Hattori, Naoko Dermatology Clinic; Tokio Numata, Numata Clinic; Tokuya Omi, Queen's Square Medical Facilities; Tamotsu Ebihara, Samoncho Clinic; Kunihiko Yoshikawa, Senrichuo Hospital; Toshitatsu Nogita, Shinjuku Minamiguchi Hifuka; Tomoko Murata, and Ikuko Kimura, Tsubasa Clinic; Masahiko Tsutsumi, Tsutsumi Clinic; Kaoru Watanabe, Watanabe Dermatology Clinic; Takeshi Yoshikawa, Yoshikawa Derma Clinic; Mina Yamada, Yotsuya Sanchome Hifuka; and Yuki Kuno, Yuki Skin Clinic.
This is a secondary publication of an article originally published in Japanese in the Journal of Clinical Therapeutics & Medicines (“A 12‐week, multi‐center, placebo‐controlled, randomized, double‐blind, parallel‐group, comparative phase II/III study of benzoyl peroxide gel in patients with acne vulgaris,” 2014; 30(8): 651–668). Our submission for secondary publication in English was approved by the Editor‐in‐Chief of the Journal of Clinical Therapeutics & Medicines.
References
- 1. Kurokawa I, Nishijima S. Acne vulgaris. Comprehensive Handbook of Clinical Dermatology 2002; 17: 117–130 (In Japanese). [Google Scholar]
- 2. Decker LC, Deuel DM, Sedlock DM. Role of lipids in augmenting the antibacterial activity of benzoyl peroxide against Propionibacterium acnes . Antimicrob Agents Chemother 1989; 33: 326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eady EA, Farmery MR, Ross JI, Cove JH, Cunliffe WJ. Effects of benzoyl peroxide and erythromycin alone and in combination against antibiotic‐sensitive and ‐resistant skin bacteria from acne patients. Br J Dermatol 1994; 131: 331–336. [DOI] [PubMed] [Google Scholar]
- 4. Burkhart CG, Butcher C, Burkhart CN, Lehmann P. Effects of benzoyl peroxide on lipogenesis in sebaceous glands using an animal model. J Cutan Med Surg 2000; 4: 138–141. [DOI] [PubMed] [Google Scholar]
- 5. Strauss JS, Krowchuk DP, Leyden JJ et al Guidelines of care for acne vulgaris management. J Am Acad Dermatol 2007; 56: 651–663. [DOI] [PubMed] [Google Scholar]
- 6. Nast A, Dréno B, Bettoli V et al European evidence‐based (S3) guidelines for the treatment of acne. J Eur Acad Dermatol Venereol 2012; 26(Suppl 1): 1–29. [DOI] [PubMed] [Google Scholar]
- 7. Furue M, Yamazaki S, Jimboh K et al Nationwide cross‐sectional and seasonal multicenter study of dermatological patients in Japan. J Dermatol 2009; 119: 1795–1809 (In Japanese). [DOI] [PubMed] [Google Scholar]
- 8. Kawashima M, Akamatsu H, Hayashi N et al Survey of the patients with acne at dermatological clinics. Jpn J Clin Dermatol 2008; 62: 673–682 (In Japanese). [Google Scholar]
- 9. Hayashi N, Kawashima M, Watanabe S et al An epidemiological study of acne vulgaris in Japan by questionnaire. J Dermatol 2001; 111: 1347–1355 (In Japanese). [Google Scholar]
- 10. Hayashi N, Higaki Y, Kawamoto K, Kamo T, Shimizu S, Kawashima M. A cross‐sectional analysis of quality of life in Japanese acne patients using the Japanese version of Skindex‐16. J Dermatol 2004; 31: 971–976. [DOI] [PubMed] [Google Scholar]
- 11. Mills OH Jr, Kligman AM, Pochi P, Comite H. Comparing 2.5%, 5%, and 10% benzoyl peroxide on inflammatory acne vulgaris. Int J Dermatol 1986; 25: 664–667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Schedule for investigations and observations.
Table S2. Definitions of inflammatory and non‐inflammatory lesions.
Table S3. Grading criteria for local skin tolerability scores.
Table S4. Profiles of withdrawals after randomization.
Table S5. Shift in local skin tolerability scores for scaling over time.
Table S6. Shift in local skin tolerability scores for erythema over time.