

Abstract
The sphingosine‐1‐phosphate 1 receptor (S1P1R) is expressed by lymphocytes, dendritic cells, and vascular endothelial cells and plays a role in the regulation of chronic inflammation and lymphocyte egress from peripheral lymphoid organs. Ozanimod is an oral selective modulator of S1P1R and S1P5R receptors in clinical development for the treatment of chronic immune‐mediated, inflammatory diseases. This first‐in‐human study characterized the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of ozanimod in 88 healthy volunteers using a range of single and multiple doses (7 and 28 days) and a dose‐escalation regimen. Ozanimod was generally well tolerated up to a maximum single dose of 3 mg and multiple doses of 2 mg/d, with no severe adverse events (AEs) and no dose‐limiting toxicities. The most common ozanimod‐related AEs included headache, somnolence, dizziness, nausea, and fatigue. Ozanimod exhibited linear PK, high steady‐state volume of distribution (73–101 L/kg), moderate oral clearance (204–227 L/h), and an elimination half‐life of approximately 17 to 21 hours. Ozanimod produced a robust dose‐dependent reduction in total peripheral lymphocytes, with a median decrease of 65% to 68% observed after 28 days of dosing at 1 and 1.5 mg/d, respectively. Ozanimod selectivity affected lymphocyte subtypes, causing marked decreases in cells expressing CCR7 and variable decreases in subsets lacking CCR7. A dose‐dependent negative chronotropic effect was observed following the first dose, with the dose‐escalation regimen attenuating the first‐dose negative chronotropic effect. Ozanimod safety, PK, and PD properties support the once‐daily regimens under clinical investigation.
Keywords: sphingosine‐1‐phosphate receptor, ozanimod, pharmacodynamics, pharmacokinetics, safety, first‐in‐human study
Relapsing multiple sclerosis (RMS) and inflammatory bowel disease (IBD) are chronic immune‐mediated inflammatory disorders that are often treated with immunosuppressants and immune system modulators, with the intent to dampen aberrant immune responses.1, 2 RMS is characterized by inflammatory infiltration of the central nervous system, with resultant edema, demyelination, and oligodendrocyte and neuronal loss.3 IBD, including ulcerative colitis and Crohn's disease, are chronic gastrointestinal inflammatory disorders characterized by infiltration of lymphocytes, macrophages, and other immune cells.4 Both RMS and IBD have a lifelong chronic course of exacerbations and remissions, with impairment that accumulates over time.3, 5, 6
Currently approved medications for RMS and IBD have multiple limitations. Many drugs for RMS and IBD are administered by injection, presenting challenges associated with injection‐related adverse events, compliance, and convenience.7 Newer oral agents for RMS are more efficacious but carry significant tolerability and/or safety concerns that affect patient quality of life and treatment adherence.1 With respect to IBD, conventional oral immunomodulators (eg, thiopurines, methotrexate) have limited efficacy and often a delay in the onset of action, whereas biologics (eg, anti–tumor necrosis factor antibodies) are associated with serious adverse reactions such as opportunistic infections and lymphoma.8 Thus, a substantial unmet medical need exists for oral treatments that are highly effective, safe, and well tolerated in patients with these immune‐mediated diseases.
The sphingosine‐1‐phosphate receptor (S1P1R) is expressed by lymphocytes, dendritic cells, cardiomyocytes, and vascular endothelial cells and is involved in the regulation of chronic inflammation (via mediation of lymphocyte movement), heart rate, smooth muscle tone, and endothelial function.9, 10 Because RMS and IBD are characterized by the recirculation and accumulation of autoreactive lymphocytes,11 modulation of the S1P1R is a rational target to ameliorate immunopathologic processes associated with these diseases. Fingolimod, a nonselective S1P receptor modulator, is approved for the treatment of RMS, but has a number of safety concerns including cardiovascular events (eg, bradycardia, conduction abnormalities, hypertension), macular edema, and elevated liver transaminases.12, 13 Unlike the nonselective fingolimod, ozanimod is a selective and potent activator of only the S1P1R and S1P5R.14 The objectives of the current study were to characterize the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of a range of single and multiple oral doses of ozanimod in healthy volunteers.
Methods
Study Design and Subjects
This was a phase 1 single‐center, randomized, double‐blind, placebo‐controlled, ascending single‐ and multiple‐dose study in healthy volunteers. The study was conducted between January 2011 and February 2012 under a US investigational new drug application and was approved by the IntegReview Ethical Review Board (Austin, Texas). It was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practices guidelines. All subjects provided written informed consent prior to study participation.
Eligible subjects were nonsmoking adults aged 18 through 55 years, with a body mass index (BMI) of 18 to 30 kg/m2 and normal hematologic, hepatic, and renal function, with no clinically significant abnormalities according to laboratory test results. Subjects were not allowed to take any prescription or over‐the‐counter medication during the study that could alter metabolism by cytochrome P450 3A. Subjects were required to be surgically sterile, postmenopausal (for women), sexually abstinent, or using effective birth control methods throughout study participation.
The study included single ascending‐dose (SAD) cohorts, multiple ascending‐dose (MAD) cohorts for 7 days (MAD‐7) or 28 days (MAD‐28), and a dose‐escalation (DE) cohort (Figure 1). Each cohort included 8 subjects randomly assigned to receive either ozanimod (n = 6) or placebo (n = 2). In the SAD cohort, ozanimod was administered as a single oral dose of 0.3, 1, 2, or 3 mg. In the MAD‐7 cohort, ozanimod was administered orally as 0.3, 1, or 2 mg once daily. In the MAD‐28 cohort, ozanimod was administered orally as 0.3, 1, or 1.5 mg once daily. In the DE cohort, ozanimod was administered as follows: 0.3 mg once daily on days 1–3, 0.6 mg once daily on days 4–5, 1 mg once daily on days 6–7, and 2 mg once daily on days 8–10. The starting dose of ozanimod for evaluation in the SAD was selected following the Food and Drug Administration Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers (July 2005). In the 28‐day repeated‐dose toxicity studies, the no‐observed‐adverse‐effect levels (NOAELs) were 0.218 and 0.164 mg/kg/d ozanimod (hydrochloride salt) for rat and monkey, respectively. Based on the NOAEL of 0.164 mg/kg/d for monkey, the most sensitive and relevant toxicology species for ozanimod, the human equivalent dose was approximately 0.053 mg/kg/day. Using a safety factor of 10 and human body weight of 60 kg, the maximum recommended starting dose was determined to be 0.317 mg/d. The starting dose of 0.3 mg was chosen for investigation in the SAD (Part A).
Figure 1.
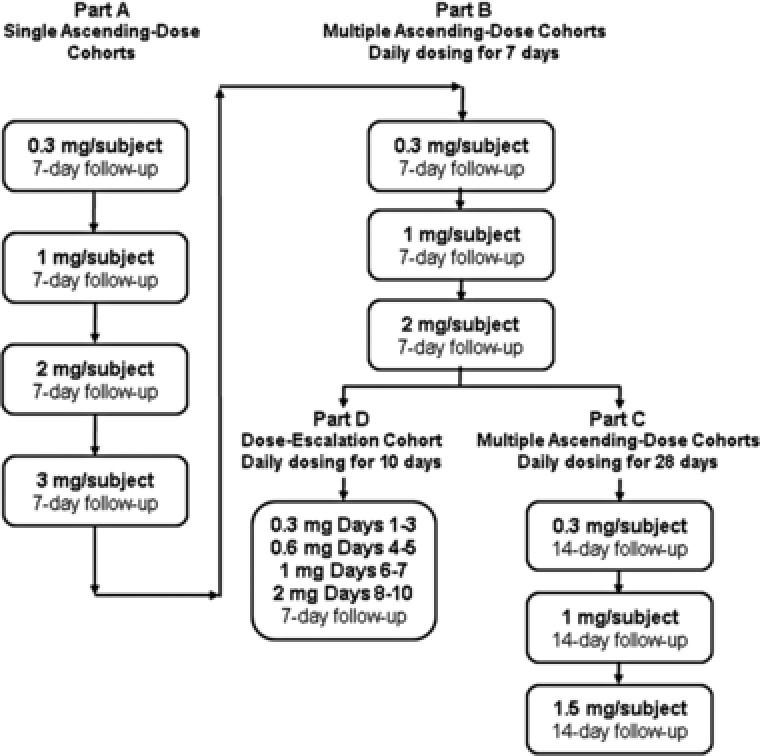
Study dosing schema.
Doses were escalated initially by approximately 3.3‐fold, followed by 2‐fold and/or 1.5‐fold. Dose escalation in Parts A, B, and C occurred only after a favorable review of safety data including follow‐up assessments from the previous dose by the sponsor and investigator. Based on the emerging clinical data (safety and PD) during the SAD (Part A), a dose higher than 3 mg was not evaluated. An acceptable safety and tolerability profile was to be established in Part A before enrollment commenced in Part B, and in Part B before enrollment commenced in Parts C and D.
Assessments
Safety
Safety was assessed by the reporting of treatment‐emergent adverse events (TEAEs), clinical laboratory abnormalities, electrocardiogram (ECG) findings, and vital signs. Continuous 24‐hour ECGs (via telemetry or Holter monitoring) were collected at baseline and after dosing. Based on the clinical safety experience of fingolimod, several categories of adverse events were considered of special interest, including cardiac (eg, bradycardia, conduction blocks), pulmonary (eg, decrease in pulmonary function), hepatic (eg, elevation of liver transaminases), and ophthalmologic (eg, macular edema) events.
Pharmacokinetics
In the SAD cohort, blood samples for the PK analysis were collected at baseline, on day 1 (0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 16 hours postdose) and on day 2 (24 and 36 hours postdose). In the MAD‐7 cohort, blood samples for PK analysis were collected at baseline (0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 16 hours postdose); immediately prior to dosing on days 2, 3, 4, and 7; on day 7 (0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 16 hours after final dose); and on day 8 (24 and 36 hours after final dose). In the MAD‐28 cohort, blood samples for PK analysis were collected at baseline (0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 16 hours postdose); immediately prior to dosing on days 2, 3, 4, 7, 14, 21, and 28; on day 28 (0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 16 hours after final dose); and on day 29 (24 and 36 hours after final dose). In the DE cohort, blood samples for PK analysis were collected immediately prior to dosing and 4, 6, 8, and 12 hours postdose on day 1; immediately before dosing on days 2 through 9; immediately prior to dosing and 4, 6, 8, and 12 hours postdose on day 10; and on day 11 (24 and 36 hours after final dose). Samples also were obtained each day during the follow‐up period for every cohort. Urine samples were collected for 24 hours on day 1 dosing for the SAD and MAD cohorts.
Plasma and urine concentrations of ozanimod were determined using a validated liquid chromatography–tandem mass spectrometry method by Ricerca Biosciences, LLC (Concord, Ohio). To measure plasma concentrations, ozanimod was extracted from a 250‐μL aliquot of human K2‐ethylenediaminetetraacetic acid plasma by protein precipitation with 0.1% formic acid in acetonitrile. To measure urine concentrations, ozanimod was extracted from a 1‐mL aliquot of urine by liquid–liquid extraction using methyl tert‐butyl ether. After evaporation of the solvent from the supernatant, the residual extract was reconstituted in a mixture of methanol, dimethylsulfoxide, and water and then subjected to high‐performance liquid chromatography (HPLC) on a reverse‐phase HPLC column combined with a mass spectrometer. Calibration was accomplished by weighted linear regression of the ratio of the peak area of the analyte to that of the added deuterated internal standard and plotting to the corresponding nominal concentration of ozanimod. The methods were validated over the concentration range of 0.0400 to 10.0 ng/mL for plasma concentrations and 0.0368 to 9.20 ng/mL for urine concentrations. The interassay precision values for plasma and urine ozanimod were ≤6.5% and ≤11.9%, respectively. The accuracy (%bias) values for plasma and urine ozanimod ranged from 1.6% to 7.3% and 0.8% to 3.6%, respectively.
Pharmacodynamics
Serial blood samples were collected from subjects for measurement of absolute lymphocyte count (ALC) and to assess the change from baseline. In the SAD cohort, blood samples were collected at baseline (‐24, ‐22, ‐20, ‐18, ‐16, ‐12, and ‐8 hours and immediately predose), on day 1 (2, 4, 6, 8, 12, and 16 hours postdose), and on day 2 (at 24 and 36 hours postdose). In the MAD‐7 cohort, blood samples for were collected at baseline (‐24, ‐22, ‐20, ‐18, ‐16, ‐12, and ‐8 hours, and immediately before the first dose); on day 1 (2, 4, 6, 8, 12, and 16 hours after the first dose); immediately prior to dosing on days 2, 3, 4, 5, and 7; on day 7 (2, 4, 6, 8, 12, and 16 hours after final dose); and on day 8 (24 and 36 hours after final dose). In the MAD‐28 cohort, blood samples were collected at baseline (‐24, ‐22, ‐20, ‐18, ‐16, ‐12, and ‐8 hours, and immediately before the first dose); on day 1 (2, 4, 6, 8, 12, and 16 hours after first dose); immediately prior to dosing on days 2, 3, 4, 5, 7, 14, 21, and 28; on day 28 (2, 4, 6, 8, 12, and 16 hours after final dose); and on day 29 (24 and 36 hours after final dose). In the DE cohort, blood samples were collected 24 hours prior to the first dose and immediately before dosing on days 1 through 10. Samples were also obtained daily for 7 days during the follow‐up period for every cohort and 14 days after the last dose for the MAD‐28 cohorts.
Evaluation of lymphocyte subsets via multicolor flow‐cytometric analysis was performed in the 1.5‐mg MAD‐28 cohort using a Becton Dickinson FACSCanto II cell analyzer. The lymphocyte subsets included CD4, CD8, CD20, CCR7, and CD45 to identify populations of naive, central, and effector memory T and B lymphocytes at baseline, and on days 28 and 42, following a 2‐week recovery period.
Analysis of safety, pharmacokinetic, and pharmacodynamic data
Adverse event data were summarized by system organ class and preferred terms within a system organ class for each treatment group. Clinical chemistry laboratory values and ECG parameters with changes from baseline (as applicable) were summarized by visit/time point and treatment group. Continuous ECG data were processed, and hourly average heart rates were calculated. Mean hourly heart rates were summarized by treatment groups for each day of dosing.
Plasma PK parameters were estimated using actual PK sample collection times, and noncompartmental analysis was performed with Phoenix WinNonlin, version 6.2 (Pharsight Corp, Palo Alto, California). Plasma PK parameters included the following: maximum drug concentration (Cmax), minimum drug concentration (Cmin), time to Cmax (Tmax), area under the plasma concentration–time curve from 0 extrapolated to infinity (AUC0–∞) or during the dosage interval (AUCτ), apparent oral clearance (CL/F), elimination half‐life (t1/2), and volume of distribution (Vd/F). The accumulation ratio (Rac) of ozanimod following multiple dosing was calculated as the ratio of AUCτ (day 7 or 28) to AUCτ (day 1).
Urine concentrations of ozanimod were used to determine the cumulative amount excreted in urine (Ae) over the 24‐hour collection period on day 1. The amount of ozanimod excreted in urine was expressed as a percentage of the administered dose (%Ae), and renal clearance (CLR) was calculated as the ratio of Ae to AUCτ on day 1.
ALCs of subjects receiving the study drug were compared with those of time‐matched placebo‐treated subjects to account for lymphocyte diurnal patterns, and data were expressed as mean changes and percent changes relative to baseline. Baseline for all trough samples is defined as the day 0, ‐24‐hour ALC values. Baseline for all nontrough samples collected after dosing is defined as the sample collected on day 0 at the same time of day.
Population PK analysis and PK/PD modeling of ALC will be described and presented in a separate publication.
Results
Subject Disposition
In this phase 1 SAD/MAD trial, 88 individuals were enrolled. The percentage of men and women were similar (51% male), and age ranged from 19 to 55 years. Most subjects were white, and the proportions of Hispanic Latinos and non‐Hispanic Latinos were comparable. Average BMI for placebo and ozanimod subjects was the same (25.2 kg/m2). No relevant differences in demographics or other baseline characteristics were observed within or across the study parts. All subjects completed the study except for 1 individual, who discontinued treatment from the 1.5‐mg MAD‐28 cohort because of a TEAE (type 1 second‐degree atrioventricular block) after a single dose (described in the Safety section).
Safety
TEAEs following administration of ozanimod were mild or moderate; no severe TEAEs or dose‐limiting toxicities were observed. The most common TEAEs (occurring in >1 subject in any cohort) are summarized in Table 1. The most common TEAEs for ozanimod (occurring in >5% of subjects) were contact dermatitis from the electrocardiogram pads (43%), headache (13%), somnolence (9%), nausea (9%), dizziness (7%), and fatigue (6%). Most events were deemed unrelated to study medication by the investigator and were mild or moderate in severity. One serious TEAE occurred in a subject who received ozanimod 0.3 mg in the MAD‐7 cohort (bronchoalveolar carcinoma of right lung), which was a previously undiagnosed, preexisting condition deemed unrelated to study medication. The investigator determined that the condition was present at screening but was not detected because of a poor‐quality chest x‐ray obtained at that visit.
Table 1.
Summary of Treatment‐Emergent Adverse Events Among All Cohorts (>1 Subject)
| Event | Placebo, n (%), n = 24 | Ozanimod, n (%), n = 68 |
|---|---|---|
| Any TEAE | 17 (70.8) | 51 (75.0) |
| Contact dermatitis (ECG pads) | 12 (50.0) | 29 (42.6) |
| Headache | 3 (12.5) | 9 (13.2) |
| Somnolence | 1 (4.2) | 6 (8.8) |
| Nausea | 0 | 6 (8.8) |
| Dizziness | 1 (4.2) | 5 (7.4) |
| Fatigue | 1 (4.2) | 4 (5.9) |
| Pruritus | 0 | 3 (4.4) |
| Abdominal pain | 0 | 3 (4.4) |
| Abnormal dreams | 1 (4.2) | 2 (2.9) |
| Dry mouth | 0 | 2 (2.9) |
| Vessel puncture‐site hematoma | 0 | 2 (2.9) |
| Sinus arrest | 0 | 2 (2.9) |
| Oral herpes | 0 | 2 (2.9) |
| Cough | 0 | 2 (2.9) |
| Nasal congestion | 0 | 2 (2.9) |
| Obstructive airways disorder | 0 | 2 (2.9) |
| Rhinorrhea | 0 | 2 (2.9) |
| Wheezing | 0 | 2 (2.9) |
| Decreased appetite | 0 | 2 (2.9) |
| Neck pain | 0 | 2 (2.9) |
TEAE, treatment‐emergent adverse event.
The effect on heart rate in the SAD cohort and after the first dose in the MAD‐7 and MAD‐28 cohorts is illustrated in Figure 2A. All subjects, including those who received placebo, initially experienced a reduction in heart rate over the first 24‐hour period following initial dose administration on day 1. The pattern of heart rate observed in all subjects, including those who received placebo, was consistent with the circadian rhythm, and heart rate reduction in the second 12 hours was also consistent with normal diurnal variation. Subjects who received ozanimod had a greater decrease in heart rate compared with placebo subjects, and ozanimod‐induced reductions in heart rate appeared to be dose dependent. Subjects who received ozanimod 3 mg had the largest reductions; their lowest mean hourly heart rate was 51.6 bpm in hour 21, consistent with the diurnal decrease in heart rate observed during sleep.
Figure 2.
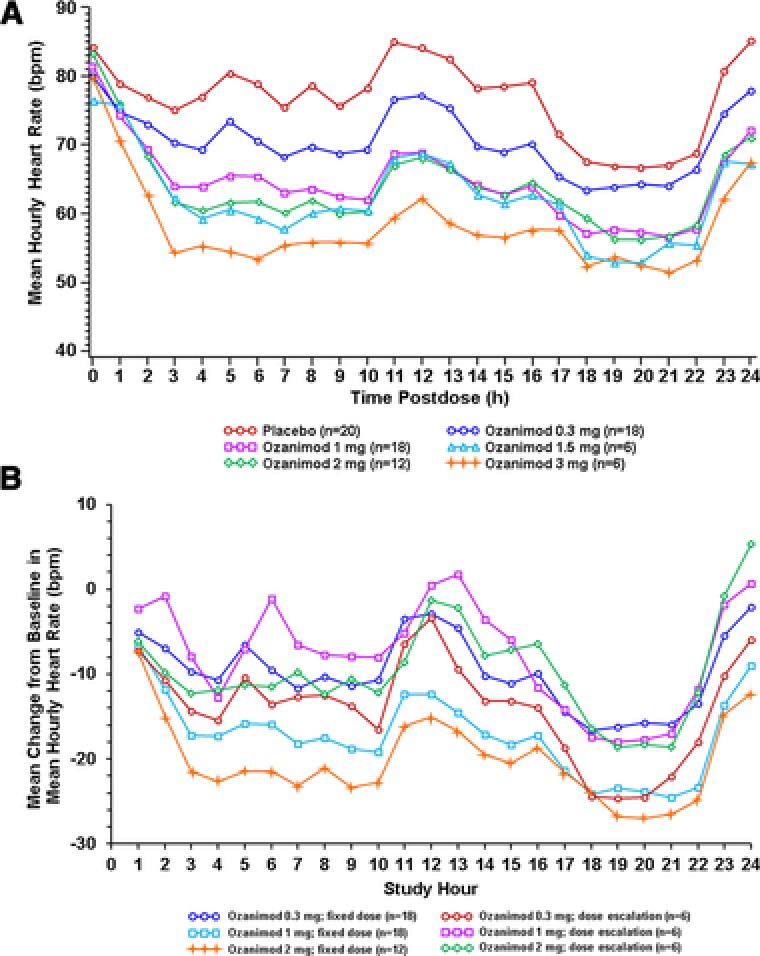
Mean hourly heart rate observed on day 1 in all fixed‐dose cohorts (A) or mean changes from daily baseline in mean hourly heart rate for fixed‐dose cohorts versus dose‐escalation cohort (B).
The use of a gradual dose‐escalating regimen to mitigate larger reductions in heart rate observed with fixed‐dose administration was tested. Although heart rate reductions in the DE cohort at the 0.3‐mg dose on day 1 were similar to those who received 0.3 mg in the fixed‐dose cohorts on day 1, as the dose was escalated to 1 mg (day 6) and 2 mg (day 8), the decreases in heart rate observed for the DE cohort were smaller than those on day 1 for the 1‐ and 2‐mg fixed‐dose groups (Figure 2B).
Four subjects experienced grade 2 cardiac TEAEs of bradycardia and/or cardiac conduction abnormalities detected within the first 6 hours of dosing. Of these 4 subjects, 3 were in the 0.3‐mg SAD cohort and included 2 subjects who experienced a 2‐second sinus pause during periods of bradycardia (heart rates in low to mid 40s in 1 case and low 30s in the other) and 1 subject who experienced intermittent bradycardia, with heart rates in the upper 30s. All 3 of these subjects with TEAEs were asymptomatic, and they resolved without medical intervention. The fourth subject experienced a second‐degree Mobitz type 1 atrioventricular block after the first dose of ozanimod 1.5 mg, which resolved without intervention within 24 hours; however, the subject discontinued participation in the study because of this event. There were no cardiac‐related TEAEs detected in the dose‐escalation groups.
Two subjects in the 2‐mg MAD‐7 cohort had mild, asymptomatic, transient decreases in pulmonary function (forced expiratory volume in 1 second [FEV1] and forced vital capacity [FVC]) that resolved 7 days after the last dose without medical intervention. No clinically significant changes in blood pressure were observed. However, a dose‐related decrease in systolic blood pressure was observed on day 1, which coincided temporally with the lowest heart rate but was not deemed clinically important. No ozanimod‐related effects were observed on liver function tests (ie, levels of alanine aminotransferase, aspartate aminotransferase, and total bilirubin were normal) or ophthalmologic examinations.
Pharmacokinetics
Mean ozanimod plasma concentration–time profiles following administration of single oral doses and daily oral doses for 7 or 28 days, under fasting conditions are presented in Figure 3. Ozanimod PK parameters following single and multiple doses are summarized in Tables 2 and 3, respectively. Following oral administration under fasting condition, median Tmax values were approximately 8.0 to 12.0 hours (single doses) and 8.0 hours (multiple doses). Dose‐proportional increases in Cmax and AUC0–∞ or AUCτ were observed. The percent coefficients of variation for Cmax and AUC0–∞ or AUCτ were approximately 17% to 32% (single doses) and 10% to 48% (multiple doses). Ozanimod exposure (AUCτ, Cmax, Cmin) was similar after 7 and 28 days of dosing of 0.3 or 1 mg (Table 3), indicating that steady state was attained by day 7, which also was supported by mean Cmin for 1 mg throughout the dosing days (0.0749 ng/mL [day 2], 0.104 ng/mL [day 3], 0.110 ng/mL [day 4], 0.160 ng/mL [day 7], 0.176 ng/mL [day 14], 0.137 ng/mL [day 21], and 0.145 ng/mL [day 28]). Mean t1/2 values also were similar across doses, ranging from 15.8 to 17.8 hours. High mean Vd/F values (73–102 L/kg) were observed, likely reflecting extensive distribution of ozanimod into tissues. Multiple dosing of ozanimod for 7 or 28 days resulted in approximately 2‐fold drug accumulation (Rac) and 2‐fold peak‐to‐trough concentration ratio at steady state.
Figure 3.
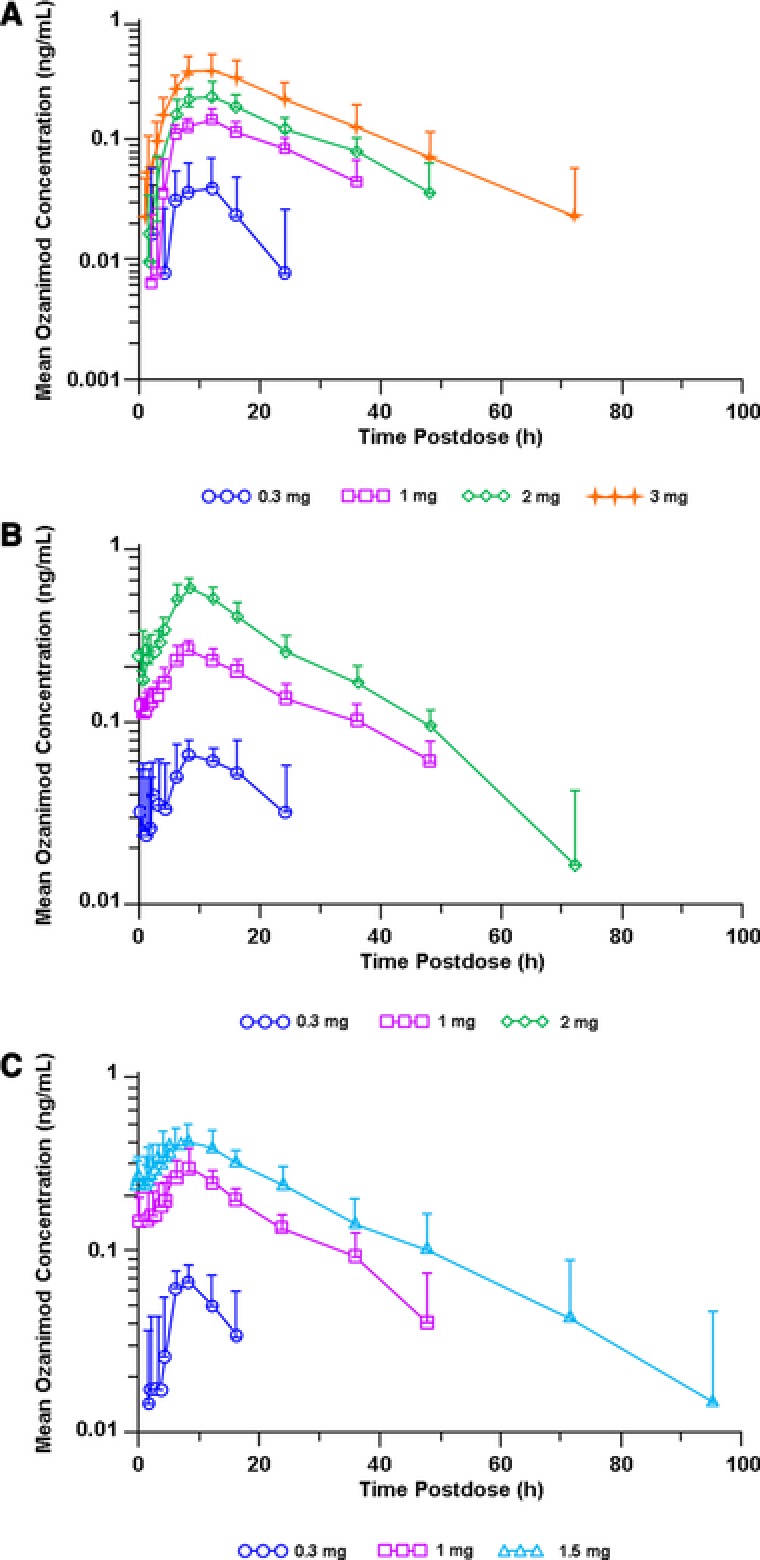
Mean ± SD ozanimod plasma concentration–time profiles for single oral doses (A) and at steady state following multiple daily oral doses for 7 days (B) or 28 days (C) under fasting conditions.
Table 2.
Single‐Dose Pharmacokinetic Parameters of Ozanimod Under Fasting Conditions
| Ozanimod Dose | ||||
|---|---|---|---|---|
| Parameter | 0.3 mg (n = 4) | 1.0 mg (n = 6) | 2.0 mg (n = 6) | 3.0 mg (n = 6) |
| AUC0–∞, ng·h/mL | NC | 4.40 (0.764) | 6.56 (1.95) | 11.6 (4.92) |
| Cmax, ng/mL | 0.0707 (0.0181) | 0.148 (0.0313) | 0.231 (0.0732) | 0.392 (0.140) |
| Tmax, h | 10.0 (2.00–12.0) | 12.0 (6.00–12.0) | 8.00 (8.00–12.0) | 10.0 (8.00–12.0) |
| Vd/F, L/kg | NC | 81.9 (21.8) | 102 (25.2) | 92.0 (20.0) |
| CL/F, L/h | NC | 233 (36.9) | 333 (116) | 303 (139) |
| t1/2, h | NC | 17.8 (3.32) | 15.8 (1.95) | 16.6 (5.11) |
| %Ae | 0.0432 (0.0265) | 0.0262 (0.0164) | 0.0250 (0.00433) | 0.0625 (0.0325) |
| CLR, L/h | 0.126 (0.0884) | 0.116 (0.0684) | 0.147 (0.0476) | 0.287 (0.132) |
Data are presented as mean (standard deviation), except for Tmax, which is presented as median (minimum–maximum). %Ae, cumulative amount of drug excreted unchanged in urine (expressed as a percentage of the administered dose); AUC0–∞, area under the plasma study drug concentration–time curve from time 0 to infinity; Cmax, maximum drug concentration; CL/F, apparent oral clearance; CLR, renal clearance; NC, not calculated because of insufficient data; Tmax, time to Cmax; t1/2, elimination half‐life; Vd/F, volume of distribution.
Table 3.
Steady‐State Pharmacokinetic Parameters of Ozanimod After Multiple Oral Dosing for 7 or 28 Days Under Fasting Conditions
| Ozanimod Once‐Daily Dosing for 7 Days | Ozanimod Once‐Daily Dosing for 28 Days | |||||
|---|---|---|---|---|---|---|
| Parameter | 0.3 mg (n = 6) | 1.0 mg (n = 6) | 2.0 mg (n = 6) | 0.3 mg (n = 6) | 1.0 mg (n = 6) | 1.5 mg (n = 5) |
| Cmax, ng/mL | 0.0708 (0.0105) | 0.255 (0.0247) | 0.551 (0.0713) | 0.0666 (0.0174) | 0.288 (0.0919) | 0.409 (0.0947) |
| Cmin, ng/mL | 0.0320 (0.0249) | 0.136 (0.0269) | 0.242 (0.0548) | 0.00 (0.00) | 0.132 (0.0230) | 0.229 (0.0627) |
| AUCτ, ng·h/mL | 1.22 (0.301) | 4.45 (0.461) | 8.95 (1.43) | 0.793 (0.383) | 4.82 (0.979) | 7.66 (1.86) |
| Tmax, h | 8.00 (8.00–12.0) | 8.00 (6.00–8.28) | 8.00 (6.00–12.0) | 8.00 (6.05–12.0) | 8.00 (6.00–12.0) | 8.00 (6.00–8.03) |
| Vd/F, L/kg | NC | 101 (22.5) | 81.4 (11.5) | NC | 73.3 (14.2) | 88.8 (14.8) |
| CL/F, L/h | NC | 227 (23.6) | 228 (37.6) | NC | 214 (36.8) | 204 (44.6) |
| t1/2, h | NC | 20.2 (1.93) | 17.0 (2.26) | NC | 17.7 (3.14) | 21.4 (9.48) |
| Rac | NC | 1.88 (0.283) | 2.14 (0.597) | NC | 2.34 (0.669) | 3.16 (0.676) |
| %Aea | 0.0433 (0.0147) | 0.0926 (0.0885) | 0.0910 (0.0507) | 0.0282 (0.0252) | 0.0594 (0.0272) | 0.0366 (0.0272) |
| CLR,a L/h | NC | 0.419 (0.449) | 0.435 (0.311) | NC | 0.271 (0.128) | 0.229 (0.204) |
Data are presented as mean (standard deviation), except for Tmax, which is presented as median (minimum–maximum).
%Ae, cumulative amount of drug excreted unchanged in urine (expressed as a percentage of the administered dose); AUCτ, area under the plasma concentration–time curve during the dosage interval; Cmax, maximum drug concentration; Cmin, minimum drug concentration; CL/F, apparent oral clearance; CLR, renal clearance; NC, not calculated because of insufficient data; Rac, accumulation ratio; Tmax, time to Cmax; t1/2, elimination half‐life; Vd/F, volume of distribution.
aParameters were calculated based on day 1 data (multiple dosing data were not collected).
Mean ozanimod urinary excretion (as a percentage of administered dose) ranged from 0.03% to 0.06% for single oral doses, 0.04% to 0.09% for 7‐day dosing regimens, and 0.03% to 0.06% for 28‐day dosing regimens. Ozanimod renal clearance ranged from 0.116 to 0.287 L/h for single oral doses, 0.189 to 0.435 L/h for 7‐day dosing regimens, and 0.229 to 0.291 L/h for 28‐day dosing regimens.
Pharmacodynamics
In the MAD‐7 and MAD‐28 cohorts, ozanimod induced a rapid, dose‐dependent, and reversible selective reduction in ALC. In the MAD‐7 cohort, median ALC decreased in a dose‐dependent manner after the first dose (0.3, 1, or 2 mg), with further reductions on repeated administration. Median reductions in ALC were 14%, 49%, and 68% in the ozanimod 0.3‐, 1‐, and 2‐mg dose groups, respectively, on day 7, compared with a reduction of 3% in the placebo group. The ALC for all subjects was within the normal range by day 13 (6 days after final dose).
In the MAD‐28 cohort, median ALC reductions after 28 days were 34%, 65%, and 68% at ozanimod doses of 0.3, 1, and 1.5 mg, respectively (Figure 4A). Recovery of lymphocyte counts into the normal range occurred within 48 to 72 hours after the last dose for all subjects in the MAD‐28 cohort who received the 1‐ or 1.5‐mg dose. In the DE cohort, decreases in ALC were observed throughout dosing, with the largest median decreases (67.4%) noted 24 hours after the final dose (Figure 4B). In this cohort, ALC did not fully recover to baseline values during the 7‐day postdosing observation period.
Figure 4.
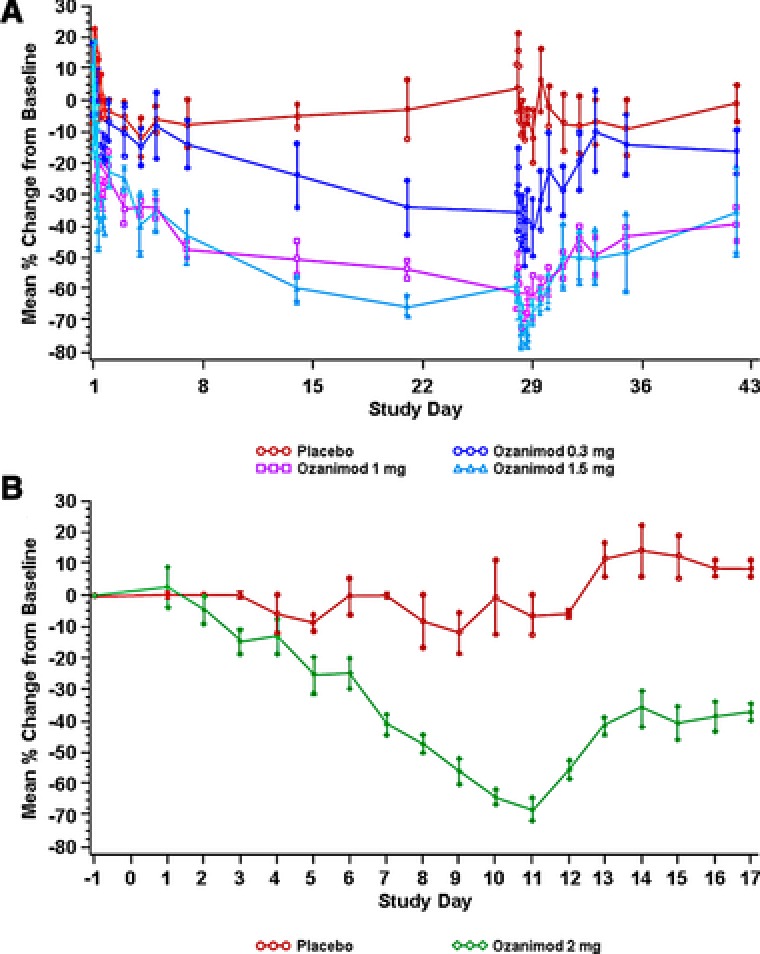
Mean percent change in absolute lymphocyte count ± SEM in subjects receiving fixed doses of ozanimod 0.3, 1, or 1.5 mg/d for 28 days (A) or after dose escalation of ozanimod (0.3 mg/d on days 1–3; 0.6 mg/d on days 4 and 5; 1 mg/d on days 6 and 7; and 3 mg/d on days 8–10) or placebo (B). Baseline for all trough samples is defined as the day 0, ‐24‐hour ALC values. Baseline for all nontrough samples collected after dosing is defined as the sample collected on day 0 at the same time. ALC, absolute lymphocyte count; SEM, standard error of the mean.
The effects of ozanimod 1.5 mg on lymphocyte subsets on days 28 and 42 are summarized in Table 4. Reductions in peripheral circulating lymphocytes by ozanimod were subset specific, with marked decreases in the subsets expressing chemokine receptor 7 (CCR7+), including naive and central memory T cells (CD4+, CD8+). CCR7‐ lymphocytes were variably affected, and there was no effect on circulating numbers of CCR7‐ CD4+ TEMRA, CCR7‐ CD8+ TEMRA, or natural killer cells. Together, these results demonstrate selectivity of ozanimod for reducing particular lymphocyte subpopulations.
Table 4.
Effect of Ozanimod 1.5 mg/d on Lymphocyte Subsets on Days 28 and 42
| Ozanimod (n = 4) | Placebo (n = 2) | ||||
|---|---|---|---|---|---|
| Lymphocyte Subtype | Phenotypea | % Change on Day 28b | % Change on Day 42c | % Change on Day 28b | % Change on Day 42c |
| Naive T cells | CD4+ CCR7+ 45RA+ | −86 | −38 | +8 | +7 |
| Central memory | CD4+ CCR7+ 45RO+ | −88 | −52 | +8 | −6 |
| TEMRA | CD4+ CCR7‐ 45RA+ | −3 | +54 | +35 | +66 |
| Effector memory | CD4+ CCR7‐ 45RO+ | −69 | −41 | −19 | −5 |
| Naive T cells | CD8+ CCR7+ 45RA+ | −85 | −40 | +51 | +51 |
| Central memory | CD8+ CCR7+ 45RO+ | −83 | −55 | +58 | +34 |
| TEMRA | CD8+ CCR7− 45RA+ | +7 | −6 | +58 | +53 |
| Effector memory | CD8+ CCR7‐ 45RO+ | −39 | −33 | +22 | +29 |
| Total T cells | CD3+ CD20‐ | −68 | −39 | −5 | −7 |
| B cells | CD3− CD20+ | −77 | −49 | −3 | 0 |
| Natural killer cells | CD3− CD16/56+ | +18 | +2 | +33 | −9 |
TEMRA, subset of effector memory T cells that are 45RA positive.
aEvaluated by flow cytometry.
bMean percent change in cell count on day 28 relative to pretreatment.
cMean percent change in cell count following 14‐day recovery.
Discussion
Ozanimod is a selective S1P1R and S1P5R modulator being developed for the treatment of RMS and IBD. This was a first‐in‐human study evaluating the safety, PK, and PD of a range of single and multiple oral doses of ozanimod. Ozanimod was well tolerated up to the maximum dose of 3 mg as a single dose used in this study. No severe or dose‐limiting toxicities were observed, and most TEAEs were not considered related to the drug. Target TEAEs of special interest were based on the safety profile of fingolimod. Potential dose‐dependent safety findings in this study included cardiovascular (eg, asymptomatic bradycardia, cardiac conduction abnormalities) and pulmonary events (eg, transient decline in FEV1 and FVC) that occurred at doses of 1.5 mg and higher. However, none of the observed events was considered of clinical concern. No significant changes or abnormalities in liver function tests were observed. A dose‐dependent negative chronotropic effect was observed after the first dose of ozanimod, which was also observed with other S1P receptor modulators.15, 16, 17 The negative chronotropic effect of S1P receptor modulators may attenuate over time, secondary to S1P receptor desensitization on atrial myocytes.18 Because the negative chronotropic effect appears to occur with increasing exposure to ozanimod on day 1, the DE cohort demonstrated that gradual dose escalation of ozanimod (over several days) rather than starting with a high dose on day 1 appears to mitigate larger reductions in heart rate. Because gradual escalation may represent a safer approach to dosing agents in this class, it has been carried forward into completed phase 2 and ongoing phase 3 clinical trials of ozanimod.19, 20
Ozanimod exhibited linear PK, with dose‐proportional increases in exposure and low to moderate intersubject variability. The desirable PK properties of ozanimod include high steady‐state volume of distribution, moderate apparent oral clearance, and an elimination half‐life of approximately 20 hours, supporting the use of once‐daily oral therapy. Ozanimod high steady‐state volume of distribution likely reflects extensive distribution of ozanimod into tissues, which is also consistent with high tissue‐to‐blood ratios in rat (data on file). Once‐daily dosing regimens resulted in a steady‐state peak‐to‐trough concentration ratio and a drug accumulation ratio of approximately 2. Renal clearance is not an important excretion pathway for ozanimod.
S1P1R modulators limit lymphocyte egress from peripheral lymphoid organs, resulting in reductions in ALCs.21 Because this effect is likely an important driver of the efficacy observed in clinical trials with other S1P modulators,13 lymphocyte counts were included in this study as a PD marker. Robust dose‐dependent reductions in ALC were observed at all ozanimod doses studied, and lymphocyte counts normalized within 3 days after cessation of dosing. After 28 days of dosing, lymphocyte count reductions were 65% and 68% for the 1‐ and 1.5‐mg doses, respectively, suggesting a plateau effect at 1 mg. When a dose‐escalation regimen was used, lymphocyte counts continued to decrease throughout the 10‐day dosing period. The decrease in lymphocyte counts observed with ozanimod was similar to those observed with other S1P receptor agonists; decreases ranged from 34% to 68% in the current study and from 35% to 76% in studies with other drugs.18, 22, 23, 24 However, recovery of lymphocyte counts to the normal range occurred more rapidly in the present study (within 2–3 days of drug cessation) than in a study of the 0.5‐mg dose of fingolimod (6 weeks),25 which is a likely consequence of the shorter t1/2 of ozanimod.
The PD effects of ozanimod showed selectivity for lymphocyte subtypes, with greater effects on CD4+ CCR7+ and CD8+ CCR7+ T cells, and a lesser effect on effector memory cells than central memory cells. This might indicate that ozanimod can enable maintenance of protective immunity while targeting the T cells believed to be part of the immunopathologic process in RMS and IBD. Lymphocyte subset selectivity also has been reported for other drugs that target S1P receptors,18, 22 but further investigation of this apparent selectivity will be required to assess whether differences exist between agents or diseases and whether the differences are of clinical significance.
Conclusions
This study demonstrates that ozanimod is generally well tolerated after single and multiple doses and with dose escalation and has PK and PD properties that make it a promising immunomodulatory agent for the treatment of chronic inflammatory diseases.
Declaration of Conflicting Interests
J.Q. Tran, J.L. Brooks, and A. Olson are current employees of Receptos Services, a wholly owned subsidiary of Celgene Corporation, and are stockholders in Receptos and Celgene. At the time of the study and analysis, J.P. Hartung, R.J. Peach, M. Boehm, H. Smith, G.A. Timony, S. Gujrathi, and P.A. Frohna were employees of Receptos. J.P. Hartung is currently employed by JPH Clinical Development, Inc (San Diego, California). R.J. Peach is currently self‐employed. M.F. Boehm is currently employed by Boehm Projects, LLC. H. Smith is currently employed by Nuvelution Pharma, Inc (South San Francisco, California). G.A. Timony is currently self‐employed as a pharmaceutical consultant. S. Gujrathi is currently self‐employed. P.A. Frohna is currently employed by Frohna Biotech Consulting (Solana Beach, California). H. Rosen is an employee of the Scripps Research Institute and is a scientific founder of Receptos. The authors received writing and editorial support for manuscript preparation from Susan Martin, PhD, and Philip Sjostedt, BPharm, MPH, from the Medicine Group, which was paid for by Receptos. The authors, however, directed and are fully responsible for all content and editorial decisions for this article.
Funding
This study was supported by Receptos, a wholly owned subsidiary of Celgene Corporation.
References
- 1. Tullman MJ. A review of current and emerging therapeutic strategies in multiple sclerosis. Am J Manag Care. 2013;19(2 Suppl):S21–S27. [PubMed] [Google Scholar]
- 2. Ng SC, Kamm MA. Therapeutic strategies for the management of ulcerative colitis. Inflamm Bowel Dis. 2009;15(6):935–950. [DOI] [PubMed] [Google Scholar]
- 3. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952. [DOI] [PubMed] [Google Scholar]
- 4. Stenson W. Inflammatory bowel disease In: Bennett JC, Goldman L, eds. Cecil Textbook of Medicine. Vol 1. 21st ed Philadelphia, PA: W. B. Saunders; 2001:722–729. [Google Scholar]
- 5. Glickman R. Inflammatory bowel disease: ulcerative colitis and Crohn's disease In: Fauci AS, Braunwald E, eds. Harrison's Principles of Internal Medicine. 14th ed. New York, NY: McGraw‐Hill, Health Professions Division; 1998:1633–1645. [Google Scholar]
- 6. Van Limbergen J, Russell RK, Drummond HE, et al. Definition of phenotypic characteristics of childhood‐onset inflammatory bowel disease. Gastroenterology. 2008;135(4):1114–1122. [DOI] [PubMed] [Google Scholar]
- 7. Lugaresi A. Addressing the need for increased adherence to multiple sclerosis therapy: can delivery technology enhance patient motivation? Expert Opin Drug Deliv. 2009;6(9):995–1002. [DOI] [PubMed] [Google Scholar]
- 8. Bousvaros A. Use of immunomodulators and biologic therapies in children with inflammatory bowel disease. Expert Rev Clin Immunol. 2010;6(4):659–666. [DOI] [PubMed] [Google Scholar]
- 9. Karuppuchamy T, Behrens EH, Gonzalez‐Cabrera P, et al. Sphingosine‐1‐phosphate receptor‐1 (S1P) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017;10(1):162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Subei A, Cohen J. Sphingosine 1‐phosphate receptor modulators in multiple sclerosis. CNS Drugs. 2015;29(7):565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scott FL, Clemons B, Brooks J, et al. Ozanimod (RPC1063) is a potent sphingosine‐1‐phosphate receptor‐1 (S1P1) and receptor‐5 (S1P5) agonist with autoimmune disease‐modifying activity. Br J Pharmacol. 2016;173(11):1778–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cohen J, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. [DOI] [PubMed] [Google Scholar]
- 13. Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124–1140. [DOI] [PubMed] [Google Scholar]
- 14. Sorensen PS. Ozanimod: a better or just another S1P receptor modulator? Lancet Neurol. 2016;15(4):345–347. [DOI] [PubMed] [Google Scholar]
- 15. Schmouder R, Serra D, Wang Y, et al. FTY720: placebo‐controlled study of the effect on cardiac rate and rhythm in healthy subjects. J Clin Pharmacol. 2006;46(8):895–904. [DOI] [PubMed] [Google Scholar]
- 16. Hoch M, Darpo B, Brossard P, et al. Effect of ponesimod, a selective S1P1 receptor modulator, on the QT interval in healthy individuals. Basic Clin Pharmacol Toxicol. 2015;116(5):429–437. [DOI] [PubMed] [Google Scholar]
- 17. Legangneux E, Shakeri‐Nejad K, Aslanis V, et al. Cardiac effects of siponimod (BAF312) re‐initiation after variable periods of drug discontinuation in healthy subjects. Clin Ther. 2016;38(3):631–645.e631. [DOI] [PubMed] [Google Scholar]
- 18. Kovarik JM, Schmouder R, Barilla D, Wang Y, Kraus G. Single‐dose FTY720 pharmacokinetics, food effect, and pharmacological responses in healthy subjects. Br J Clin Pharmacol. 2004;57(5):586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine1‐phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo‐controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373–381. [DOI] [PubMed] [Google Scholar]
- 20. Sandborn WJ, Feagan BG, Wolf DC, et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N Engl J Med. 2016;374(18):1754–1762. [DOI] [PubMed] [Google Scholar]
- 21. Sanna MG, Wang SK, Gonzalez‐Cabrera PJ, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2(8):434–441. [DOI] [PubMed] [Google Scholar]
- 22. Brossard P, Derendorf H, Xu J, et al. Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first‐in‐human study. Br J Clin Pharmacol. 2013;76(6):888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brossard P, Scherz M, Halabi A, et al. Multiple‐dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: favorable impact of dose up‐titration. J Clin Pharmacol. 2014;54(2):179–188. [DOI] [PubMed] [Google Scholar]
- 24. Kovarik JM, Schmouder R, Barilla D, et al. Multiple‐dose FTY720: tolerability, pharmacokinetics, and lymphocyte responses in healthy subjects. J Clin Pharmacol. 2004;44(5):532–537. [DOI] [PubMed] [Google Scholar]
- 25. Francis G, Kappos L, O'Connor P, et al. Temporal profile of lymphocyte counts and relationship with infections with fingolimod therapy. Mult Scler. 2014;20(4):471–480. [DOI] [PubMed] [Google Scholar]