

Abstract
A biochemical system and method have been developed to enable the quantitative measurement of cytoplasmic versus nuclear localization within cells in whole blood. Compared with the analyses of nuclear localization by western blot or fluorescence microscopy, this system saves a lot of time and resources by eliminating the necessity of purification and culturing steps, and generates data that are free from the errors and artifacts associated with using tumor cell lines or calculating nuclear signals from 2D images. This user‐friendly system enables the analysis of cell signaling within peripheral blood cells in their endogenous environment, including measuring the kinetics of nuclear translocation for transcription factors without requiring protein modifications. We first demonstrated the efficiency and specificity of this system for targeting nuclear epitopes, and verified the results by fluorescence microscopy. Next, the power of the technique to analyze LPS‐induced signaling in peripheral blood monocytes was demonstrated. Finally, both FoxP3 localization and IL‐2‐induced STAT5 signaling in regulatory T cells were analyzed. We conclude that this system can be a useful tool for enabling multidimensional molecular‐biological analyses of cell signaling within endogenous peripheral blood cells by conventional flow cytometry. © 2017 The Authors. Cytometry Part A Published by Wiley Periodicals, Inc. on behalf of ISAC.
Keywords: flow cytometry, nuclear localization, cell signaling, peripheral blood monocytes, Tregs, IL‐2, LPS, NF‐kB, STAT5, FoxP3
Introduction
Cell signaling involves a complex cascade of intermolecular events that culminate in the induction of one or more functional cellular responses. Most exogenous stimuli induce the activation and nuclear translocation of transcription factors or transcriptional modifying proteins. Once in the nucleus, transcription factors modify transcriptional activity to alter the cellular proteome and adapt cells to their environment 1, 2, 3, 4.
The subcellular localization of a protein has an important role in regulating its activity or function. Most proteins can function in multiple cellular compartments, and their localization determines their function 5, 6, 7, 8. For example, under normal conditions, Protein Disulfide Isomerase (PDI), ERp57, functions in regulating protein folding within the lumen of the endoplasmic reticulum (ER) 9, 10; however, under stress conditions, ERp57 may translocate into the nucleus and function as a transcription factor 11, 12, 13. For typical transcription factors, localization within the cytoplasm is repressive to their function, while translocation to the nucleus enables them to modify transcriptional activity 14, 15. Moreover, pathological cells are typically characterized by imbalances of particular proteins in one or more subcellular compartments, which may be used for diagnosis or prognosis, such as p53 16, 17, 18 and NF‐κB 19, 20, 21.
A variety of techniques exist to analyze subcellular localization, including confocal microscopy and western blotting on sub‐fractionated protein extracts. However, most of these techniques rely on homogenous cell preparations. In highly complex cell mixtures, such as whole blood, analyzing cellular activation and subcellular localization becomes much more difficult. The cells of interest generally need to be sorted from the blood prior to analysis or the sample needs to be analyzed by flow cytometry. Assuming that the researcher has access to the expensive equipment necessary, cell sorting is time consuming, may be harsh on the samples, and the recovered cells require at least an overnight resting period prior to further experimentation. In the most optimal circumstances, the cells are no longer in their endogenous environment and the results obtained from such samples may have questionable physiological relevance 22, 23, 24, 25, 26, 27. In general, it is preferable to analyze complex samples directly in their endogenous environment by use of flow cytometry, but the availability of flow cytometry‐based molecular biological assays is limited.
Modern flow cytometry enables the multiparametric differentiation of greater than 10 parameters with standard cytometers, and even upward of 60 parameters with specialized instruments, such as mass cytometers 28. However, among these numerous parameters, all resulting data are binary and do not confer any information on protein localization. This limits the analysis of protein activation to protein modifications, typically phosphorylation, with the increased modification of particular transcription factors used as a surrogate for their nuclear localization, such as in the case of the Signal Transducer and Activator of Transcription (STAT) family 29, 30. The problem with reliance upon surrogate markers, rather than assessing the markers directly, is that dogma is not always true in different cell types, activation states, or pathological conditions. For example, persistently phosphorylated STAT5 has been found to localize to the cytoplasm in cancer cells from patients with acute myeloid leukemia 31, 32, while CMV infection has been shown to impair the transcriptional activity of STAT1 and STAT3, even though they are both present in their phosphorylated forms 30.
Imaging flow cytometry provides a workaround for determining subcellular localization by allowing researchers to analyze moderate‐resolution images of cells 33. In imaging flow cytometry, cells in suspension are multi‐parametrically analyzed by both flow cytometry and a CCD camera that operates by time‐delay integration. However, the images are 2D rather than 3D, and the assessment of nuclear localization is still mostly qualitative since the calculation of signals within a nuclear mask does not fully account for the 3D depth of the cell, including regions in front of, behind or immediately peripheral to the nucleus, such as the ER 34, 35. Still, imaging flow cytometry enables a variety of high‐throughput morphological and subcellular localization studies that cannot be done by traditional flow cytometry, and both the technology and masking algorithms are continually improving 36, 37. Similarly, another technology that enables the assessment of morphological characteristics and subcellular localization is laser‐scanning cytometry (LSC). LSC provides higher resolution images than imaging flow cytometry, and, since the sample is not discarded into a waste stream, it allows researchers to conduct time‐resolved studies on cells, including enzyme‐ and drug‐kinetic studies, analyses of changes in cell morphology or DNA condensation, and even the sequential analyses of individual cells with different probes 38.
In order to simplify the problem of quantifying subcellular localization in complex cell mixtures, without requiring specialized instruments or cumbersome techniques, we developed a proprietary kit to biochemically partition cytoplasmic versus nuclear epitopes in leukocytes and other cells within whole blood. In this study, we utilized our kit to analyze protein localization and cell signaling in peripheral blood leukocytes. We first confirmed the partitioning efficiency with typical organelle markers. We then demonstrated the usefulness of our kit by analyzing cell signaling following LPS stimulation of monocytes. Finally, we analyzed the localization of FoxP3 within Tregs and the induction of STAT5 nuclear translocation by IL‐2 stimulation. We conclude that our kit can be useful for the molecular‐biological analysis of protein localization and cell signaling within cells in whole blood by conventional flow cytometry.
Materials and Methods
Blood
Fresh blood was collected onsite daily from normal donors by informed consent. All blood utilized in this study was collected using K3‐EDTA vacutainers (Becton Dickinson, Franklin Lakes, NJ). For Treg analyses, the donors were fasted for several hours prior to blood draw in order to minimize mitigating factors that may influence baseline activity levels.
Reagents
The Whole Blood Nuclear Localization Kit* was designed and prepared in our laboratory, and is currently distributed on demand through the Custom Design Service at Beckman Coulter Life Sciences. ERp57 (MaP.ERp57), GRP78 (A‐10), Smac (H‐177), Lamin B (C‐20), Lamin B (G‐1), Bcl‐xL (H‐5), α‐Tubulin (TU‐02), α‐Tubulin‐FITC (TU‐02‐FITC), β‐Tubulin‐AF647 (H‐235‐AF647), Mitofilin (B‐10), Cytochrome C (A‐8), and STAT5b‐FITC (G‐2) were from Santa Cruz Biotechnology (Dallas, TX). Goat Anti‐Mouse IgG1‐AF647 (1070‐31), Goat Anti‐Mouse IgG2a‐AF488 (1080‐30), Goat Anti‐Mouse IgG2b‐AF488 (1090‐30), Goat Anti‐Mouse IgG3‐PC7 (1100‐17), Goat Anti‐Mouse IgG3‐AF647 (1100‐31), Goat Anti‐Mouse IgM‐PE (1020‐09), and Goat Anti‐Mouse IgM‐AF488 (1020‐30) were from SouthernBiotech (Birmingham, AL). IκBα‐AF488 (L35A5), RelA‐PE (L8F6), phospho‐RelA S536‐AF647 (93H1), and phospho‐ERK T202/Y204‐AF647 (E10) were from Cell Signaling Technology (Danvers, MA). Phospho‐RelA S529‐PC7 (K10.895.12.50) and phospho‐CREB S133‐PE (J151‐21) were from BD Biosciences (San Jose, CA). FoxP3‐AF647 (259D), CD14‐Pacific Blue (RMO52), CD14‐Krome Orange (RMO52), CD3‐FITC (UCHT1), CD3‐PE (UCHT‐1), CD3‐PC7 (UCHT‐1), CD3‐APC (UCHT‐1), CD3‐PB (UCHT‐1), CD3‐Krome Orange (UCHT1), CD25‐PC7 (B1.49.9), CD45‐PC7 (J.33), CD45‐Krome Orange (J.33), IgG1‐FITC (IM0639U), IgG1‐PE (IM0670U), IgG1‐PC7 (6607099), IgG1‐APC (IM2475U), IgG2a‐FITC (A12690), IgG2a‐PC7 (A12692), IgG2b‐FITC (B36623), IgM‐FITC (IM1269U), and IgM‐RD1 (6604117) were from Beckman Coulter (Brea, CA). Chicken‐anti‐Mouse‐IgG‐AF647 (A21463), F(ab')2‐Donkey‐anti‐Goat IgG‐PE (31860), Chicken‐anti‐Rabbit‐IgG‐AF488 (A21441), DAPI, Goat IgG Isotype Control (02–6202), Rabbit IgG Isotype Control (02‐6102), Normal Mouse Serum (NMS), Normal Rabbit Serum (NRS), Normal Chicken Serum (NCS), Normal Goat Serum (NGS), Human AB Serum (HABS), LPS, rhIL‐2, and 10× PBS were from ThermoFisher Scientific (Waltham, MA). Mouse IgG1 (M5284), Mouse IgG2a (M5409), Mouse IgG2b (SAB4700729), Mouse IgG3 (SAB4700759), and Mouse IgM (M5909) isotype controls were from Sigma Aldrich (St. Louis, MO).
Differential Cell Permeabilization Using the Whole Blood Nuclear Localization Kit
The workflow used for sample preparation with the Whole Blood Nuclear Localization Kit is depicted in Figure 1A. Briefly, the blood was split into two 100 μL fractions for each sample condition, then 100 μL of fixative was added to each tube, and they were incubated for 10 min at room temperature (RT). Alternatively, samples that were stimulated were batched and then split after adding fixative. Next, 1 mL of lysis Buffer 1 or Buffer 2 were added to one tube of each pair, and the tubes were incubated for 30 min at RT. Finally, the tubes were each washed 2× by adding 2 mL of wash buffer, centrifuging at 500g in an Allegra 6R centrifuge with a GH‐3.8 Rotor (366816, Beckman Coulter, Brea, CA), and decanting the supernatant after each wash. Prior to the addition of antibodies, the tubes were incubated with normal serum for at least 10 min to block or reduce non‐specific and FC‐receptor binding, as described in the following section.
Figure 1.
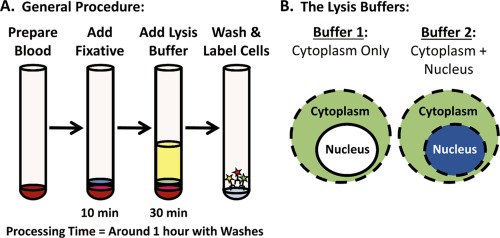
Workflow for the Whole Blood Nuclear Localization Kit. A) The workflow for the Nuclear Localization Kit is similar to most Fix/Perm kits for flow cytometry, comprised of a brief fixation step, followed by permeabilization and then labeling. B) What differentiates the Nuclear Localization Kit is that 2 aliquots of the sample are permeabilized side‐by‐side with 2 different lysis buffers, enabling the nuclear signal to be quantitatively calculated from the resulting data.
Antibody Labeling
For direct labeling with antibody conjugates, 50 μL of 10% NMS or a mixture of NMS and NRS in 1× PBS was added to each tube and they were incubated for 10 min at RT. Next, a premixed cocktail of the immunophenotyping, signaling and/or localization antibodies was added to each tube, and the tubes were incubated for 30 min in the dark at RT. For quantification of the background signal, an extra set of control tubes was stained with all of the immunophenotyping antibodies, but without the signaling or localization antibodies of interest. Instead, the background tubes were incubated with equivalently labeled isotype control antibodies. The tubes were then washed 2× with wash buffer as above, resuspended in 500 μL of resuspension buffer, and read on a flow cytometer.
For indirect labeling, 50 μL of 10% HABS and/or the appropriate serum for the secondary antibodies (i.e., NGS or NCS) in 1× PBS was added to each tube followed by incubation for 10 min at RT. Next, a premixed cocktail of the primary antibodies was added to each tube and they were incubated for 30 min to 1 hour at RT. After incubation, the tubes were washed 2× with wash buffer and then blocked for 10 min with 50 μL of 10% HABS or a mixture of HABS and NCS or NGS in 1× PBS. After 10 min, 200 μL of a premixed cocktail of the secondary antibodies in wash buffer was added to each tube and they were incubated for 30 min in the dark at RT. After secondary labeling, the tubes were washed 2× with wash buffer, and then incubated for 10 min with 50 μL of 10% NMS or a mixture of NMS and NRS in 1× PBS in order to block any residual binding activity of the secondary antibodies, as well as any residual non‐specific or FC‐receptor binding to the direct conjugates. Finally, a cocktail of the remaining direct antibody conjugates was added to the tubes and they were incubated for 30 min in the dark at RT. If DAPI was used in the particular experiment, it was added together with the last antibody cocktail at a final concentration of 0.25–2.5 ng/mL. After the final round of labeling, the tubes were washed 2× with wash buffer, resuspended in 500 μL of resuspension buffer, and then read on a flow cytometer. The background control for indirectly labeled samples was stained with the immunophenotyping antibodies and isotype controls for the primary antibodies, followed by incubation with the equivalent secondary antibodies; however, they were not stained with the signaling or localization antibodies of interest.
For the compensation panel, a set of samples was processed with Buffer 1 and then separately stained with only single‐color immunophenotyping or anti‐CD3 antibodies for each fluorophore utilized in the study.
Flow Cytometry
All experiments in this study were performed using a 13‐color, 4‐laser CytoFLEX S Flow Cytometer*, equipped with 375, 405, 488, and 638 nm lasers (B78557, Beckman Coulter, Brea, CA). The flow cytometer was operated as directed using CytExpert Software v1.2 (Beckman Coulter, Inc., Brea, CA). First, the compensation panel was acquired, and the compensation matrix was calculated and saved. Next, an experimental file was initiated, the gains and compensation values were imported from the compensation file, and all histograms and gating workflows were prepared. Finally, each experimental sample was acquired.
Data Analyses
The resulting flow cytometry data were further analyzed using CytExpert Software v1.2. First, the calculated compensation matrix was checked and fine‐tuned for the experimental samples, if necessary. Next, the population gates for each sample were adjusted, the data were collected into a statistics table, and then exported for the preparation of charts and graphs using Excel 2013 (Microsoft). For calculation of the Cytoplasmic MFI, the background signal for Buffer 1 was subtracted from the experimental data collected for the samples processed with Buffer 1. For calculation of the Nuclear MFI, the background signal for Buffer 2 was subtracted from the samples processed with Buffer 2, and the calculated Cytoplasmic MFI was then subtracted from these results, with the remaining signal consisting predominantly of the contribution from the nucleus.
Fluorescence Microscopy
For visualization of marker localization by microscopy, some samples were deposited onto microscope slides using a Cytospin 2 (Shandon, Runcorn, Cheshire, United Kingdom) after they were acquired on the flow cytometer. These samples were then allowed to air dry, mounted with Vectashield (H‐1500, Vector Laboratories, Burlingame, CA), and then coverslipped. Each slide was analyzed and images were acquired using an Axioskop 2 Plus fluorescence microscope, with an AttoArc 2 HBO 100 W halogen lamp and specific excitation and emission filters for FITC, PE, APC and DAPI (Carl Zeiss, Oberkochen, Germany). All images were captured using a 63× oil‐immersion lens together with AxioVision v4.7 software (Carl Zeiss MicroImaging, Jena, Germany).
Statistical Analyses
Student's t‐tests were performed using GraphPad Software (GraphPad Software, La Jolla, CA) in order to determine whether the data from test samples were significantly different from control samples. * = P‐values < 0.05. ** = P‐values < 0.01.
Results
Assessing the Subcellular Distribution of Organelle‐Specific Markers
The Whole Blood Nuclear Localization Kit was designed to differentially permeabilize WBCs in two fractions in order to enable the quantitative assessment of nuclear localization by conventional flow cytometry. Since different subcellular membranes are composed of differential lipid compositions 39, we hypothesized that we could find combinations of detergents that would specifically permeabilize the plasma membrane without affecting the nuclear envelope. We first set up a multimodal screen to test different combinations of detergents with MCF7 cells, varying the concentrations and buffer conditions. Next, we analyzed the staining efficiency of HSP60 and HDAC1 in the different samples, using fluorescence microscopy in order to assess the integrity of the plasma and mitochondrial membranes versus the nuclear membrane, respectively. We then tested our system on whole blood, beginning with the detergent combinations and concentration ranges that we identified in our proof‐of‐concept studies. In order to simplify the determination of plasma membrane permeabilization when working with whole blood, we preloaded the blood samples with calcein and utilized the loss of staining to precisely gauge both plasma and endosomal membrane permeabilization. Using these parameters, we identified optimal detergent conditions for whole blood. We also optimized the buffer composition to fully lyse RBCs in highly cross‐linked samples, while being compatible with and enabling the targeting of subcellular membranes with low concentrations of detergents that otherwise would not efficiently lyse whole blood samples. Data from the invention process have been included within a filed US Patent Application.
The Whole Blood Nuclear Localization Kit consists of 2 lysis buffers, together with a wash buffer, a resuspension buffer, and a fixative. The first buffer (Buffer 1) lyses RBCs and permeabilizes the plasma membrane together with the endosomes, lysosomes, the outer mitochondrial membrane, and the endoplasmic reticulum. The second buffer (Buffer 2) permeabilizes everything that the first buffer does, but also permeabilizes the nuclear membrane (Fig. 1B). This kit is easy to use, works with small volumes of blood, takes no longer to perform than a standard immunophenotyping experiment, and can be used with conventional flow cytometry, imaging flow cytometry, or even fluorescence microscopy.
We first tested the efficiency of the kit by analyzing the subcellular distribution of organelle‐specific markers. For immunophenotyping, we stained the blood with DAPI in order to detect the DNA‐containing WBCs, and then specifically labeled T cells and monocytes with CD3 and CD14, respectively. The CD14 staining also enabled separation of the lymphocyte, monocyte and neutrophil populations for further analyses (Fig. 2A). Once we were able to identify our populations of interest, we analyzed the partitioning efficiency of a variety of organelle markers (Fig. 2B). The markers we tested were: Lamin B in the nucleus 40, 41, α‐Tubulin in the cytoplasm, GRP78 in the ER 42, and Smac/Diablo in the mitochondrial intermembrane space 43. These results clearly demonstrate the permeabilization of the cytoplasm, ER and mitochondria by Buffer 1, with the nucleus only permeabilized by Buffer 2.
Figure 2.
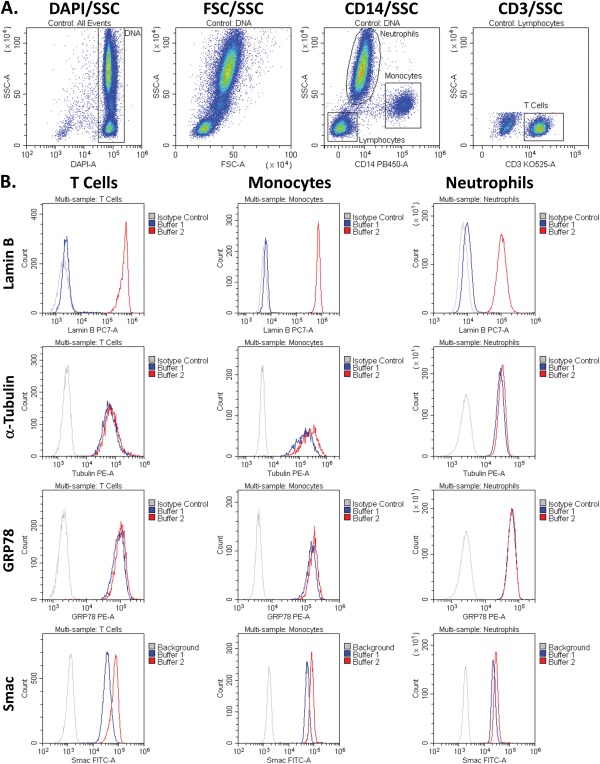
The staining of organelle‐specific markers following permeabilization with Buffer 1 or 2. A) Gating workflow. B) Histogram overlays of the MFIs of the different markers in T cells, monocytes, and neutrophils following permeabilization with either Buffer 1 or Buffer 2. The cytoplasmic and ER markers, Tubulin and GRP78, respectively, were near maximal following permeabilization with Buffer 1. The mitochondrial inner membrane and intermembrane space marker, Smac, was also near maximal following treatment with Buffer 1. The nuclear marker, Lamin B, was only accessible following permeabilization with Buffer 2.
The relative percentages of MFIs for a variety of organelle markers within the Buffer 1 fraction versus the MFIs specific to the Buffer 2 fraction (i.e., the difference between the 2 fractions) are shown in Figure 3, including Lamin B, α‐Tubulin, and GRP78, together with: Bcl‐xL, a Bcl‐2 family member that is present in both the cytosol and the mitochondrial membranes 44; Mitofilin, a mitochondrial cytoskeletal protein within the intermembrane space 45; and Cytochrome C, a component of the electron transport chain that anchors to the inner mitochondrial membrane 46. These results were quantified by calculating the cytoplasmic (Buffer 1) versus nuclear (Buffer 2 ‐ Buffer 1) MFIs and then determining their relative percentage of the whole‐cell MFI (Buffer 2).
Figure 3.
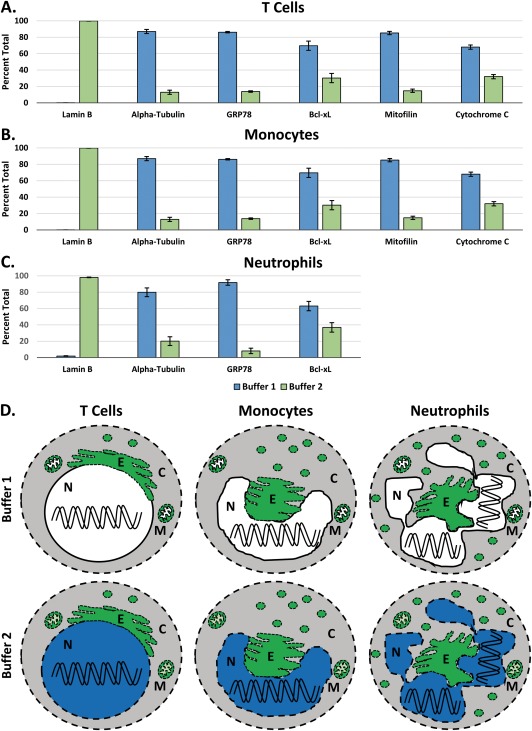
Quantifying the subcellular distribution of organelle‐specific markers. A) The distribution of organelle‐specific makers in CD3+ T cells, calculated using the Whole Blood Nuclear Localization Kit. The percentages were calculated by determining the fraction of the Buffer 1 (Cytoplasm) signal that is within Buffer 2 (Whole Cell) signal. These data represent the average percentage of marker localization from 3 different blood donors ± the SEM. B) The distribution of organelle‐specific markers in CD14+ monocytes. C) The distribution of organelle‐specific markers in neutrophils. D) A summary of the efficiency of permeabilization in the 3 cell populations seen in A, B and C. The cytoplasm was permeabilized by both buffers and is colored gray for contrast. The nucleus is colored either blue when permeabilized or white if not, and the organelles are green when permeabilized. C = Cytoplasm, N = Nucleus, E = Endoplasmic Reticulum, and M = Mitochondria.
In T cells, greater than 99% of Lamin B was calculated to be within the Buffer 2 fraction, showing a clear delineation for the nucleus between the 2 fractions (Fig. 3A). In contrast, α‐Tubulin, GRP78, Bcl‐xL, Mitofilin, and Cytochrome C predominantly localized within the Buffer 1 fraction. This demonstrates the clear partitioning of the cytoplasmic components to the Buffer 1 fraction. Approximately 30% of residual Bcl‐xL and Cytochrome C were detected in the Buffer 2 fraction. This is likely due to further solubilization of the mitochondrial membrane proteins exposing additional epitopes. The very presence of Mitofilin and Cytochrome C staining in the cytoplasmic fraction suggests that the outer mitochondrial membrane was indeed permeabilized by Buffer 1.
In monocytes, greater than 99% of Lamin B was present within the Buffer 2 fraction (Fig. 3B). Similar to T cells, we found α‐Tubulin, GRP78, Bcl‐xL Mitofilin and Cytochrome C predominantly within the Buffer 1 fraction.
In neutrophils, greater than 98% of Lamin B was present within the Buffer 2 fraction, while α‐Tubulin, GRP78, and Bcl‐xL were mostly present within the Buffer 1 fraction (Fig. 3C). The neutrophils largely partitioned similar to both T cells and monocytes, although approximately 37% of Bcl‐xL partitioned to the Buffer 2 fraction. Mitofilin and Cytochrome C were not able to be measured in neutrophils because the neutrophils had a very high level of non‐specific binding to these antibodies: IgG2a and IgG2b isoforms. A variety of methods were employed to block and or reduce such binding, but to no avail. In any case, the remaining antibodies in the panel suggests that this system performs similar with granulocytes as it does with lymphocytes and monocytes.
Collectively, the data from Figures 3A–3C are summarized pictorially in Figure 3D. The primary difference is that Buffer 2 permeabilizes the nucleus in addition to everything else permeabilized by Buffer 1. It also begins to permeabilize the inner mitochondrial membrane.
Comparing the Flow Cytometry Results to Fluorescence Microscopy
In order to visually confirm the staining found by flow cytometry, we prepared samples that were analyzed by both flow cytometry and fluorescence microscopy. In these tests, the blood was first split into two fractions and permeabilized with either Buffer 1 or Buffer 2. These cells were then labeled with organelle‐specific antibodies, ERp57 and Lamin B, as well as DAPI and CD3 in order to identify WBCs and T cells, respectively. In addition, the samples were stained with CD14 Krome Orange to enhance the separation of lymphocytes, monocytes, and neutrophils by flow cytometry. The Krome Orange was not detectable with the filter setup on the fluorescence microscope. After staining, each sample was read on the flow cytometer until 50K events were collected, and then the remaining sample fraction was deposited onto a glass slide using a Cytospin 2, followed by fluorescence microscopy. For the flow cytometry experiment, the gating workflow was similar to Figure 2, except that CD3 was moved to the FITC channel in order to enable it to be visualized by the fluorescence microscope (Fig. 4A). The histogram overlays for ERp57 and Lamin B in each of the different populations demonstrate that the kit performed as expected (Fig. 4B). In particular, Buffer 1 permeabilized the plasma membrane, including the ER, while Buffer 2 further permeabilized the nucleus. When analyzing the sample by fluorescence microscopy, ERp57 can be seen to be equally present within cells that were processed using either Buffer 1 or Buffer 2, while Lamin B is specifically seen only in cells that were processed with Buffer 2 (Fig. 4C). Note that both of the buffer fractions were stained with the same primary antibody cocktail, and all of the cells, including the background samples, were stained with the same secondary antibody cocktail. ERp57 sometimes has a similar pattern of staining as Lamin B, but this is simply because Lamin B is the nuclear cytoskeleton on the inside of the nuclear membrane, while ERp57 labels the ER, which wraps around the outside of the nuclear membrane. In each case, the Lamin B staining is identical in morphology to the DAPI staining, while ERp57 is more similar to the shape of the cell. In these images, the CD3 T cells are brightly labeled green by the CD3 FITC antibody, while the majority of the remaining cells that can be seen in the background are neutrophils, dimly labeled with the CD3 antibody. The neutrophils demonstrate a lower expression of Lamin B than the lymphocytes, and this is also seen by flow cytometry (Fig. 4B).
Figure 4.
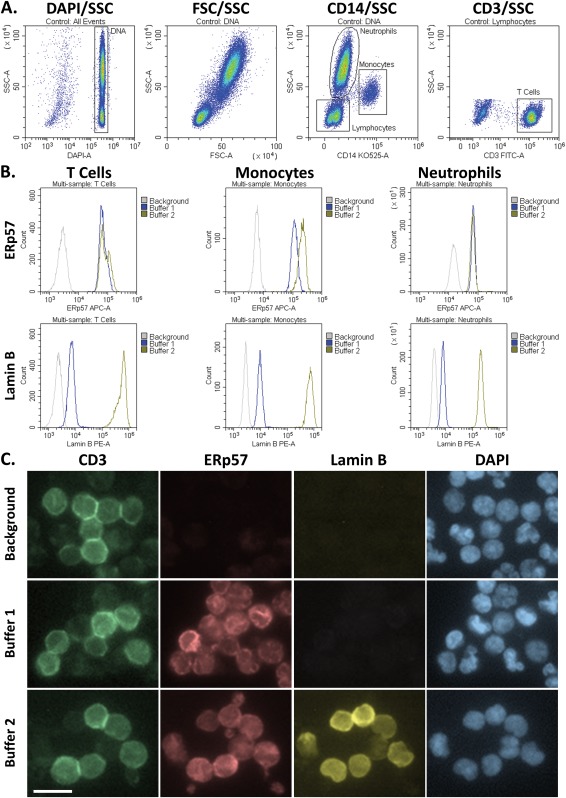
Comparison of the flow cytometry results to fluorescence microscopy. A) Gating workflow. B) MFIs of the ER or Nuclear markers, ERp57 or Lamin B, within T cells, monocytes and neutrophils, as seen by flow cytometry. C) Fluorescence micrographs of the same cells seen in B. Following the analyses by flow cytometry, the samples were deposited onto glass slides with a CytoSpin 2, and images were taken using a fluorescence microscope equipped with a 63× objective. The background control was stained with secondary and immunophenotyping antibodies, but was not stained with the primary antibodies for the organelle markers. The white bar in the lower‐left image represents 10 μm. All of these images are in the same scale.
LPS‐Induced Signaling in Monocytes
Next, we utilized our system to analyze the cytoplasmic versus nuclear localization of a variety of signaling proteins in monocytes, as well as the changes in their distributions following LPS stimulation. Specifically, we analyzed NF‐κB RelA, phospho‐RelA S529, phospho‐RelA S536, IκBα, phospho‐ERK T202/Y204, and phospho‐CREB S133. We used β‐tubulin and Lamin B as controls to verify the performance of the 2 buffers. The gating workflow for this experiment is shown in Figure 5A. In these experiments, CD45 was used to discriminate WBCs, while CD14 was used to discriminate monocytes. All of the signaling data are presented as the average MFIs ± the SEM. As seen in Figure 5B, unstimulated RelA was localized mostly in the cytoplasm, while LPS stimulation induced a 2.16‐fold increase in the nuclear localization of RelA by 10 min. Phospho‐RelA S529 also increased maximally by 10 min, and this increase closely correlated with RelA translocation into the nucleus. In contrast, phospho‐RelA S536 increased maximally by 5 min and was exclusively localized within the cytoplasm. The timing of these events indicates that the phosphorylation of S536 precedes S529, and, together with the spatial localization, suggests that S536 may regulate nuclear translocation, while S529 likely regulates transcriptional activation. In addition, the nucleocytoplasmic shuttle protein, IκBα, was localized in both the cytoplasm and nucleus, and LPS stimulation induced its maximal degradation by 10 min. This occurred within both the cytoplasm and nucleus, but, while greater than 80% of IκBα was degraded in the cytoplasm, this had a more profound impact on nuclear IκBα, reducing it by greater than 95%.
Figure 5.
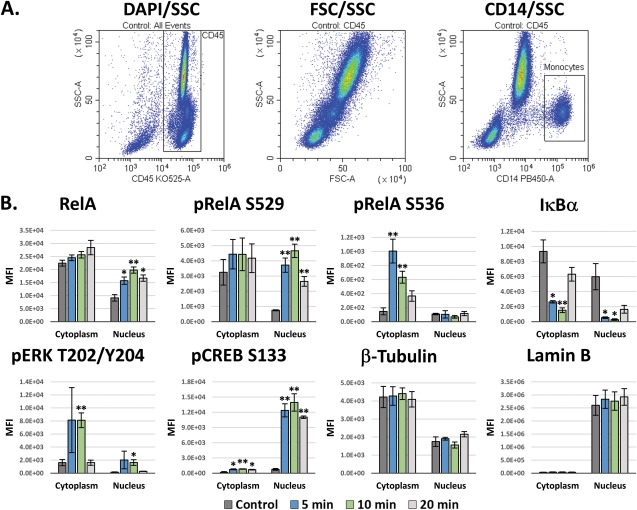
Subcellular signaling in LPS‐stimulated monocytes. A) Gating workflow. B) Intracellular signaling in monocytes following LPS stimulation. The RelA, phospho‐RelA S529 and S536, and IκBα signaling responses are classical NF‐κB signaling downstream of the TLR4 receptor. The ERK and CREB responses are due to activation of MAPK and Ca2+ signaling, respectively. Tubulin and Lamin B are a cytoskeletal proteins that were utilized as a controls for the Cytoplasm and Nucleus, respectively. These data are the averages of 3 different blood donors ± the SEM. Unpaired t‐tests were performed versus the control for each respective fraction in order to determine significance in B. * = P values < 0.05. ** = P values < 0.01.
Meanwhile, ERK1/2 T202/Y204 was maximally phosphorylated by 5 min, primarily within the cytoplasm. Phospho‐ERK also increased in roughly equivalent proportions within the nucleus, peaking around 5 min, though to a smaller degree. The transcription factor, CREB, was phosphorylated at S133 near maximally by 5 min, and this was localized exclusively within the nucleus. Finally, β‐Tubulin was present predominantly in the cytoplasm, though a small amount was partitioned with the nuclear fraction. As a marker for verification of the proper performance the buffer system, Lamin B correctly partitioned exclusively within the Buffer 2 fraction.
FoxP3 and STAT5 Signaling in Tregs
Finally, we tested the effectiveness of our kit with Tregs, a low‐abundance T‐cell population within peripheral blood. Traditional methods for working with Tregs typically necessitate sorting and expansion of this population prior to experimentation. In this experiment, we analyzed Tregs directly in whole blood, using CD45 to gate WBCs, CD4 to identify CD4 T cells, and then CD25hi/CD4l°w to discriminate Tregs from the CD25l°w/+/CD4+ conventional T cell (Tcons) population (Fig. 6A). FoxP3 was found to be expressed predominantly within the Treg population (Fig. 6B). While experimenting with Tregs, we consistently noticed that the levels and localization of FoxP3 within Tregs tends to change depending on how long after blood collection the experiment is initiated (i.e., the “age of the blood”). When the blood was fresh, 80+% of FoxP3 was localized within the nucleus of Tregs; however, the FoxP3 within Tregs progressively translocated into the cytoplasm following blood collection, leveling off after 4 hours (Fig. 6B). At that point, the FoxP3 within the cytoplasm appears to degrade back to control levels, resulting in an overall reduction in FoxP3 that reaches significance around 4–6 hours after blood collection (Fig. 6C). This was not due to any changes in nuclear permeabilization, as the analyses of Lamin B localization indicated that the kit performed as expected (Fig. 6D), and this effect did not appear to affect the response of Tregs to IL‐2 stimulation.
Figure 6.
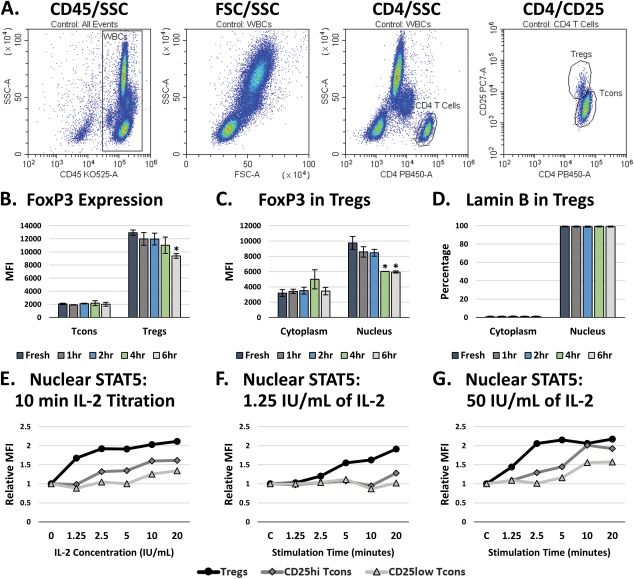
FoxP3 localization and IL‐2 stimulation of STAT5 signaling in Tregs. A) Gating workflow. B) Total FoxP3 in Tregs over time following blood collection. These data are the average of 2 independent experiments ± the SEM. C) Subcellular distribution of FoxP3 in Tregs over time following blood collection. These data the average of 2 independent experiments ± the SEM. D) Analysis of Lamin B within the Tregs in B and C, indicating that the permeabilization of the nucleus was not affected over time following blood collection. E) Analysis of the nuclear translocation of the whole STAT5b protein in Tregs and CD25hi or CD25l°w Tcons after a 10 min stimulation with a titration of IL‐2. F) Analysis of the nuclear translocation of STAT5b over time following a 1.25 IU/mL IL‐2 stimulation. G) Analysis of the nuclear translocation of STAT5b over time following a 50 IU/mL IL‐2 stimulation. Unpaired t‐tests were performed versus the control of each respective fraction in order to determine significance in B–D. * = P values < 0.05.
Stimulating Tregs with IL‐2 induces the phosphorylation of STAT5, which activates it to translocate into the nucleus and initiate transcriptional activity. Without the ability to properly assess nuclear STAT5 levels, traditional flow cytometry assays utilize the phosphorylation of STAT5 as a surrogate marker for the nuclear localization of STAT5. However, phospho‐STAT5 is difficult to analyze and requires rather harsh permeabilization conditions to expose the phospho‐epitope. Using this kit, we were able to analyze the nuclear translocation of the actual STAT5 protein. In Figure 6E, we stimulated whole blood with a titration of IL‐2 and analyzed the nuclear localization of STAT5 following 10 min of stimulation. We found that even the lowest concentration tested, 1.25 IU/mL (625 pg/mL), produced near maximal nuclear STAT5 levels in Tregs after 10 min, while the CD25hi and CD25l°w Tcons populations required higher doses of IL‐2 to induce a comparable response. These results are consistent with previous research regarding the optimal induction of STAT5 signaling in Tregs by IL‐2 47. A stimulation time course for 1.25 IU/mL of IL‐2 indicated that this concentration would be effective to more selectively stimulate Tregs rather than Tcons in assays such as Treg suppression assays, where the stimulation of cytotoxic and effector Tcons is not desirable (Fig. 6F). A more traditional STAT5 response can be seen by stimulating blood with 50 IU/mL of IL‐2, resulting in maximal nuclear translocation of STAT5 in Tregs within 2.5 min, followed by Tcons around 10 min (Fig. 6G).
Discussion
Collectively, these results demonstrate that the Whole Blood Nuclear Localization Kit is a useful tool for studying cell signaling and nuclear localization within cells in whole blood. This kit provides multiple advantages over existing approaches. One of the primary advantages is that the cells can be analyzed directly in their endogenous environment, meaning that the cells are not adulterated by purification and incubation steps, which results in more physiologically relevant data. Indeed, this effect can be seen in Figure 6C, where the length of time that blood is incubated following collection, without any other mitigating factors, can affect signaling events and the subcellular distributions of proteins. In fact, since Tregs are defined by the nuclear localization of FoxP3, such changes can have a profound impact on experimental results and conclusions due to misidentification or incomplete classification of the Treg population. Moreover, by eliminating the necessity of extensive purification and incubation steps for WBCs, a lot of time and resources can be saved while also resulting in faster data collection and increased efficiency. From start to finish, this system takes approximately 1 hour to prepare whole blood samples for staining, which is more rapid than some of the most optimized immunocytochemistry protocols. Conversely, some purification and expansion protocols take weeks, often resulting in failed or questionable data once the experiment is complete. Since rare and low‐abundance cells, such as Tregs, can be studied directly in situ, smaller sample quantities are required, which also enables the study of cell signaling in rare and precious samples. And, perhaps most importantly, this system does not rely on protein modifications to determine the activation states of cells and pathways, enabling the study of alterations in subcellular distributions of either whole proteins or protein modifications.
In our results from analyzing the distributions of organelle markers, each of the cells were found to have a little Bcl‐xL and Cytochrome C within the Buffer 2 fraction. These proteins were utilized as markers for permeabilization of the outer mitochondrial membrane and exposure of the mitochondrial intermembrane space. This occurs because the boundary for the total permeabilization of both the ER and outer mitochondrial membrane is right on the cusp of the initiation of permeabilization for the nuclear membrane. The efficiency of partitioning can be improved further, but would come at the cost of occasionally initiating nuclear permeabilization in a small subset of cells. In any case, the near‐complete partitioning of the ER and mitochondria with the Buffer 1 fraction is an added benefit for the specificity of the calculated nuclear signal, but likely would not be an issue for most cell‐signaling applications: the actual cytoplasmic proteins are completely exposed by Buffer 1, while the nuclear proteins are completely exposed by Buffer 2. Furthermore, most cell‐signaling pathways induce relatively rapid nuclear translocation, but do not induce equivalent protein translocation into either the ER or the mitochondria. Indeed, the system for translocation into the mitochondria requires proteins to be denatured and then fold or refold once inside the mitochondrial matrix 48, or to pass out of the mitochondria through pores during apoptotic signaling 49, which are different systems altogether. So, while the basal levels of a particular organelle‐specific protein may be slightly altered due to a low level of spillover from the ER or mitochondria in some cells, the alterations in signaling responses would still be expected to be predominantly due to nuclear translocation, nucleocytoplasmic transport, compartmentalization of protein modifications, or de novo protein synthesis, particularly during earlier signaling timeframes. However, if such results are found when analyzing proteins expected to be primarily localized within the spillover organelles, the results should definitely be verified. In fact, if there are any concerns over partitioning artifacts with this system, the traditional techniques could still be utilized for verification and support, but in a more focused and predictable manner, saving both time and resources. Ultimately, even with a little partitioning inefficiency in some cases, this system is still a significant quantitative improvement over attempts to quantify subcellular localization from fluorescence micrographs using masks and algorithms. In fact, this system would work well together with imaging flow cytometry in order to clearly define nuclear localization, functioning as a biochemical mask.
Whereas imaging flow cytometry is akin to fluorescence microscopy by flow cytometry, we envision the Whole Blood Nuclear Localization Kit as similar to performing a quantitative western blot on subcellular protein fractions by flow cytometry, without requiring the cells to be purified from complex, heterogeneous cell mixtures. This was illustrated by evaluating cell signaling in LPS‐stimulated monocytes and IL‐2‐stimulated Tregs, both of which responded as would be expected with western blots performed on cytoplasmic versus nuclear protein fractions. Interestingly, in the case of LPS‐induced RelA phosphorylation, there was a clear spatial and temporal separation of phosphorylation at either S529 or S536. While researchers have analyzed these sites for decades to attribute a functional role to each site, a variety of conflicting data has been presented suggesting that one controls nuclear translocation while the other regulates transactivation and vice versa 50, 51. A lot of the conflict in such situations comes down to the artificial nature of the systems being tested, which typically consist of tumor cells lines that have multiple proteins overexpressed within the cells. By analyzing unadulterated monocytes in their endogenous peripheral blood environment, the order and localization of these events clearly suggests that phosphorylation of RelA at S536 plays a larger role in initiating the nuclear translocation of RelA, while phosphorylation of RelA at S529 likely plays a larger role in regulating transcriptional activity. These findings support previous publications indicating that S536 is important for the kinetics of nuclear import 52, while S529 is important for transactivation potential 53, 54, but does not affect nuclear translocation 33. Furthermore, this underscores that caution should be used when strictly using protein‐modification events as surrogate markers for cellular activation instead of actually analyzing the underlying events.
In conclusion, we have presented a new system and method for analyzing cellular activation in complex cell mixtures that will save both time and money when compared to existing technologies. Use of this system can help to improve our collective understanding of the normal and aberrant cellular mechanisms within endogenous cells in whole blood. Our future work will focus on tuning this system to function optimally with cell cultures and other purified cells, and we are interested in collaborating with other researchers to enhance the applications and potential for utilizing high‐throughput, multiparametric flow cytometry for basic science research.
Disclaimer
Beckman Coulter and the Beckman Coulter product and service marks mentioned herein are trademarks or registered trademarks of Beckman Coulter, Inc. in the United States and other countries. All other trademarks are the property of their respective owners. *The Whole Blood Nuclear Localization Kit and the CytoFLEX S are for Research Use Only.
Competing Interests
The authors are employees of Beckman Coulter, Inc.
Supporting information
Supporting Information
Acknowledgments
The authors would like to thank Jack Figg for his technical help with analyzing antibody efficiencies and for help with validating the kit with different donors, sample types and signaling pathways. GB and SG conceptualized the system; GB designed and conducted the experiments; GB and SG analyzed the data and wrote the manuscript.
The copyright line was altered in July 2017
Literature Cited
- 1. O'Shea JJ, Gadina M, Kanno Y. Cytokine signaling: Birth of a pathway. J Immunol 2011;187:5475–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barolo S, Posakony JW. Three habits of highly effective signaling pathways: Principles of transcriptional control by developmental cell signaling. Genes Dev 2002;16:1167–1181. [DOI] [PubMed] [Google Scholar]
- 3. Manson MM. Cancer prevention – the potential for diet to modulate molecular signaling. Trends Mol Med 2003;9:11–18. [DOI] [PubMed] [Google Scholar]
- 4. Milenkovic D, Vanden Berghe W, Boby C, Leroux C, Declerck K, Szarc vel Szic K, Heyninck K, Laukens K, Bizet M, Defrance M, et al. Dietary flavanols modulate the transcription of genes associated with cardiovascular pathology without changes in their DNA methylation state. PLoS One 2014;9:e95527–e95521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scott MS, Calafell SJ, Thomas DY, Hallett MT. Refining protein subcellular localization. PLoS Comput Biol 2005;1:e66–e518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nair R, Rost B. Protein subcellular localization prediction using artificial intelligence technology. Methods Mol Biol 2008;484:435–463. [DOI] [PubMed] [Google Scholar]
- 7. Hung M‐C, Link W. Protein localization in disease and therapy. J Cell Sci 2011;124:3381–3392. [DOI] [PubMed] [Google Scholar]
- 8. Binder JX, Pletscher‐Frankild S, Tsafou K, Stolte C, O'Donoghue SI, Schneider R, Jensen LJ. COMPARTMENTS: Unification and visualization of protein subcellular localization evidence. Database 2014;2014:bau012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oliver JD, Roderick HL, Llewellyn DH, High S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell 1999;10:2573–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kimura T, Imaishi K, Hagiwara Y, Horibe T, Hayano T, Takahashi N, Urade R, Kato K, Kikuchi M. ERp57 bind competitively to protein disulfide isomerase and calreticulin. Biochem Biophys Res Commun 2005;331:224–230. [DOI] [PubMed] [Google Scholar]
- 11. Grillo C, D'Ambrosio C, Consalvi V, Chiaraluce R, Scaloni A, Maceroni M, Eufemi M, Altieri F. DNA‐binding activity of the ERp57 C‐terminal domain is related to a redox‐dependent conformational change. J Biol Chem 2007;282:10299–10310. [DOI] [PubMed] [Google Scholar]
- 12. Kim‐Han JS, O'Malley KL. Cell stress induced by the Parkinsonian mimetic, 6‐hydroxydopamine, is concurrent with oxidation of the chaperone, ERp57, and aggresome formation. Antioxid Redox Signal 2007;9:2255–2264. [DOI] [PubMed] [Google Scholar]
- 13. Grindel BJ, Rohe B, Safford SE, Bennett JJ, Farach‐Carson MC. Tumor necrosis factor α treatment of HepG2 cells mobilizes a cytoplasmic pool of ERp57/1,25D3‐MARRS to the nucleus. J Cell Biochem 2011;112:2606–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Brate A, Gianakakou P. The importance of p53 location: Nuclear or cytoplasmic zip code? Drug Resist Update 2003;6:313–322. [DOI] [PubMed] [Google Scholar]
- 15. Sun SC, Ley SC. New insights into NF‐κB regulation and function. Trends Immunol 2008;29:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iwaya K, Tsuda H, Hiraide H, Tamaki K, Tamakuma S, Fukutomi T, Mukai K, Hirohashi S. Nuclear p53 immunoreaction associated with poor prognosis of breast cancer. Jpn J Cancer Res 1991;82:835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quinn DI, Henshall SM, Head DR, Golovsky D, Wilson JD, Brenner PC, Turner JJ, Delprado W, Finlayson JF, Stricker PD, et al. Prognostic significance of p53 nuclear accumulation in localized prostate cancer treated with radical prostatectomy. Cancer Res 2000;60:1585–1594. [PubMed] [Google Scholar]
- 18. Gonzalez‐Angulo AM, Sneige N, Buzdar AU, Valero V, Kau SW, Broglio K, Yamamura Y, Hortobagyi GN, Cristofanilli M. p53 expression as a prognostic marker in inflammatory breast cancer. Clin Cancer Res 2004;10:6215–6221. [DOI] [PubMed] [Google Scholar]
- 19. Rayet B, Gélinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene 1999;18:6938–6947. [DOI] [PubMed] [Google Scholar]
- 20. Lessard L, Mes‐Masson AM, Lamarre L, Wall L, Lattouf JB, Saad F. NF‐κB nuclear localization and its prognostic significance in prostate cancer. BJU Int 2003;91:417–420. [DOI] [PubMed] [Google Scholar]
- 21. Cortés Sempere M, Rodríguez Fanjul V, Sánchez Pérez I, Perona R. The role of the NFkappaB signalling pathway in cancer. Clin Transl Oncol 2008;10:143–147. [DOI] [PubMed] [Google Scholar]
- 22. Hodge G, Hodge S, Han P. Increased levels of apoptosis of leukocyte subsets in cultured PBMCs compared to whole blood as shown by annexin V binding: Relevance to cytokine production. Cytokine 2000;12:1763–1768. [DOI] [PubMed] [Google Scholar]
- 23. Appay V, Reynard S, Voelter V, Romero P, Speiser DE, Leyvraz S. Immuno‐monitoring of CD8+ T cells in whole blood versus PBMC samples. J Immunol Methods 2006;309:192–199. [DOI] [PubMed] [Google Scholar]
- 24. Atkuri KR, Herzenberg LA, Niemi AK, Cowan T, Herzenberg LA. Importance of culturing primary lymphocytes at physiological oxygen levels. pnas 2007;104:4547–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silberer J, Ihorst G, Kopp MV. Cytokine levels in supernatants of whole blood and mononuclear cell cultures in adults and neonates reveal significant differences with respect to interleukin‐13 and interferon‐gamma. Pediatr Allergy Immunol 2007;19:140–147. [DOI] [PubMed] [Google Scholar]
- 26. Beliakova‐Bethell N, Massanella M, White C, Lada SM, Du P, Vaida F, Blanco J, Spina CA, Woelk CH. The effect of cell subset isolation method on gene expression in leukocytes. Cytometry A 2014;85A:94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wegner J, Hackenberg S, Scholz CJ, Chuvpilo S, Tyrsin D, Matskevich AA, Grigoleit GU, Stevanović S, Hünig T. High‐density preculture of PBMCs restores defective sensitivity of circulating CD8 T cells to virus‐ and tumor‐derived antigens. Blood 2015;126:185–194. [DOI] [PubMed] [Google Scholar]
- 28. Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, Tanner SD. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time‐of‐flight mass spectrometry. Anal Chem 2009;81:6813–6822. [DOI] [PubMed] [Google Scholar]
- 29. Nardozzi JD, Lott K, Cingolani G. Phosphorylation meets nuclear import: A review. Cell Commun Signal 2010;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trilling M, Le VTK, Rashidi‐Alavijeh J, Katschinski B, Scheller J, Rose‐Jone S, Androsiac GE, Jonjić S, Poli V, Pfeffer K, et al. “Activated” STAT proteins: A paradoxical consequence of inhibited JAK‐STAT signaling in cytomegalovirus‐infected cells. J Immunol 2014;192:447–458. [DOI] [PubMed] [Google Scholar]
- 31. Harir N, Pecquet C, Kerenyi M, Sonneck K, Kovacic B, Nyga R, Brevet M, Dhennin I, Gouilleux‐Gruart V, Beug H, et al. Constitutive activation of Stat5 promotes its cytoplasmic localization and association with PI3‐kinase in myeloid leukemias. Blood 2007;109:1678–1686. [DOI] [PubMed] [Google Scholar]
- 32. Bunting KD, Xie XY, Warshawsky I, His ED. Cytoplasmic localization of phosphorylated STAT5 in human acute myeloid leukemia is inversely correlated with Flt3‐ITD. Blood 2007;110:2775–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maguire O, O'Loughlin K, Minderman H. Simultaneous assessment of NF‐κB/p65 phosphorylation and nuclear localization using imaging flow cytometry. J Immunol Methods 2015;423:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pinidiyaarachchi A, Zieba A, Allalou A, Pardali K, Wählby C. A detailed analysis of 3D subcellular signal localization. Cytometry A 2009;75A:319–328. [DOI] [PubMed] [Google Scholar]
- 35. Barteneva NS, Fasler‐Kan E, Vorobjev IA. Imaging flow cytometry: Coping with heterogeneity in biological systems. J Histochem Cytochem 2012;60:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dominical V, Samsel L, McCoy JP. Masks in imaging flow cytometry. Methods 2017;112:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fei C, Lillico DM, Hall B, Rieger AM, Stafford JL. Connected component masking accurately identifies the ratio of phagocytosed and cell‐bound particles in individual cells by imaging flow cytometry. Cytometry A 2017; doi:10.1002/cyto.a.23050. [DOI] [PubMed] [Google Scholar]
- 38. Pozarowski P, Holden E, Darzynkiewicz Z. Laser scanning cytometry: Principles and applications‐An update. Methods Mol Biol 2013;931:187–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: Where they are and how they behave. Nat Rev Mol Cell Biol 2008;9:112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: Building blocks of nuclear architecture. Genes Dev 2002;16:533–547. [DOI] [PubMed] [Google Scholar]
- 41. Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 2008;22:832–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability signaling and therapeutic targeting. Biochem J 2011;434:181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burri L, Strahm Y, Hawkins CJ, Gentle IE, Puryer MA, Verhagen A, Callus B, Vaux D, Lithgow T. Mature DIABLO/Smac is produced by the IMP protease complex on the mitochondrial inner membrane. Mol Biol Cell 2005;16:2926–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alavian KN, Li H, Collis L, Bonanni L, Zen L, Silvio S, Lazrove E, Nabili P, Flaherty B, Graham M, et al. Bcl‐xL Regulates metabolic efficiency of neurons through interaction with the mitochondrial F1F0 ATP synthase. Nat Cell Biol 2011;13:1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JML, Rangell L, Bennett MJ, Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell 2005;16:1543–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: Functions beyond respiration. Nat Rev Mol Cell Biol 2008;9:532–542. [DOI] [PubMed] [Google Scholar]
- 47. Dupont G, Demaret J, Venet F, Malergue F, Malcus C, Poiteven‐Later F, Morel J, Monneret G. Comparative dose‐responses of recombinant human IL‐2 and IL‐7 on STAT5 phosphorylation in CD4+FOXP3‐ cells versus regulatory T cells: A whole blood perspective. Cytokine 2014;69:146–149. [DOI] [PubMed] [Google Scholar]
- 48. Dudek J, Rehling P, van der Laan M. Mitochondrial protein import: Common principles and physiological networks. BBA‐Mol Cell Res 2013;1833:274–285. [DOI] [PubMed] [Google Scholar]
- 49. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet 2009;43:95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Christian F, Smith EL, Carmody RJ, The regulation of NF. κB subunits by phosphorylation. Cells 2016;5:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu J, Nakano H, Sakuri H, Colburn NH. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF‐kappaB activation and transformation in resistant JB6 cells. Carcinogenesis 2004;25:1991–2003. [DOI] [PubMed] [Google Scholar]
- 52. Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, Kracht M, Schmitz ML. Transient and selective NF‐kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J Immunol 2004;172:6336–6344. [DOI] [PubMed] [Google Scholar]
- 53. Bird TA, Schooley K, Dower SK, Hagen H, Virca GD. Activation of nuclear transcription factor NF‐kappaB by interleukin‐1 is accompanied by casein kinase II‐mediated phosphorylation of the p65 subunit. J Biol Chem 1997;272:32606–32612. [DOI] [PubMed] [Google Scholar]
- 54. Wang D, Baldwin AS, Jr. Activation of nuclear factor‐kappaB‐dependent transcription by tumor necrosis factor‐alpha is mediated through phosphorylation of RelA/p65 on serine 529. J Biol Chem 1998;273:29411–29416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information