

Abstract
Liver diseases are associated with complex changes in the hemostatic system and elevated levels of the platelet-adhesive protein Von Willebrand factor (VWF) are reported in patients with acute and chronic liver damage. Although elevated levels of VWF are associated with fibrosis in the general population, the role of VWF in acute and chronic liver injury has not been examined in depth in experimental settings. We tested the hypothesis that VWF deficiency inhibits experimental liver injury and fibrosis. Wild-type (WT) and VWF-deficient mice were challenged with carbon tetrachloride (CCl4) and the impact of VWF deficiency on acute liver injury and chronic liver fibrosis was determined. VWF deficiency did not significantly affect acute CCl4-induced hepatocellular necrosis in mice. Chronic CCl4 challenge, twice weekly for 6 weeks, significantly increased hepatic stellate cell activation and collagen deposition in livers of WT mice. Interestingly, hepatic induction of several profibrogenic and stellate cell activation genes was attenuated in VWF-deficient mice. Moreover, birefringent sirius red staining (indicating type I and III collagens) and type I collagen immunofluorescence indicated a reduction in hepatic collagen deposition in CCl4-exposed VWF-deficient mice compared to CCl4-exposed WT mice. The results indicate that VWF deficiency attenuates chronic CCl4-induced liver fibrosis without affecting acute hepatocellular necrosis. The results are the first to demonstrate that VWF deficiency reduces the progression of liver fibrosis, suggesting a mechanistic role of elevated plasma VWF levels in cirrhosis.
Keywords: Liver cirrhosis, thrombosis, hemostatic disorders
Introduction
Complex changes in the hemostatic system are evident in patients with acute and chronic liver injury (Lisman et al., 2010; Tripodi and Mannucci, 2011). Chronic liver disease can result in replacement of functional liver mass with extracellular matrix, primarily collagens. The outcome of this process is cirrhosis, which can predispose patients to thrombosis, and may also contribute to progression of liver disease itself (Jairath and Burroughs, 2013). Among the hemostatic molecules disturbed in both acute and chronic liver disease is Von Willebrand factor (VWF), a glycoprotein that functions to bridge platelets with an exposed collagen surface. Several studies report that plasma levels of VWF (Reuken et al., 2015) are increased in patients with acute liver failure, as well as cirrhosis, with high levels predictive of poor outcome in this patient population (La Mura et al., 2011; Ferlitsch et al., 2012; Hugenholtz et al., 2013; Maieron et al., 2014; Kalambokis et al., 2016). Suggesting a functional connection, VWF levels are associated with the severity of liver fibrosis in the general population (Plompen et al., 2015). Thus, there is strong evidence in patients linking elevated VWF levels with both acute liver injury and with consequences of chronic liver disease such as cirrhosis.
Despite the association between VWF levels and cirrhosis in patients, the precise nature of this association is not understood. Increased plasma VWF levels in cirrhosis are most likely ascribed to endothelial cell activation, induction of hepatic expression (Ferro et al., 1996; Hollestelle et al., 2004), or perhaps reduced liver-mediated clearance (Lisman et al., 2006). That is, elevated plasma VWF levels may be a biomarker of elevated hepatic expression, driven by liver disease itself. However, strong evidence indicates that components of the hemostatic system actually drive the activation of hepatic stellate cells in experimental liver fibrosis (Anstee et al., 2011; Tripodi et al., 2011). Indeed, several studies illustrate the potential for coagulation proteases, both by activation of intracellular signaling and formation of intrahepatic microthrombi, to directly exacerbate liver fibrosis (Anstee et al., 2008; Rullier et al., 2008; Sullivan et al., 2010). Importantly, the association of VWF with cirrhosis, while inferred by clinical association studies, has not been examined in experimental studies to determine whether VWF plays a direct mechanistic role in liver fibrosis.
To address this critical gap in the literature, we tested the hypothesis that VWF directly contributes to experimental liver fibrosis by employing an established mouse model of genetic VWF deficiency. Using this tool, we determined the effect of VWF deficiency on acute carbon tetrachloride (CCl4)-induced liver injury and on liver fibrosis driven by chronic CCl4 challenge. Collectively, these studies represent the first published report describing the impact of VWF deficiency in experimental settings of acute and chronic liver injury.
Materials and Methods
Mice
Congenic C57Bl/6J VWF−/− mice were obtained from Jackson Laboratory (Bar Harbor, ME), were maintained by homozygous breeding, and have been described previously (Denis et al., 1998). Age- and sex-matched wild-type C57Bl/6J mice were purchased from Jackson Laboratory and allowed to acclimate in the same facility for at least 3 weeks prior to experimental challenge. Male and female mice used for these studies were between the ages of 8–14 weeks. Mice were housed in an AAALAC-accredited facility and allowed ad libitum access to purified drinking water and rodent chow. All animal procedures were approved by the Michigan State University IACUC.
Acute and chronic CCl4 challenge
10% CCl4 in corn oil vehicle was administered to mice by intraperitoneal injection (10 ml/kg). For acute liver injury studies, samples were collected 48 or 72 hours after CCl4 challenge. Chronic CCl4-induced liver fibrosis was induced by administration of CCl4 every 3–4 days (i.e., every Tuesday and Friday) for 6 weeks. Blood and liver samples were collected three days after the last injection of CCl4.
Histopathology, immunohistochemistry and immunofluorescence
Hematoxylin and eosin (H&E), sirius red, alpha-smooth muscle actin (α-SMA), proliferating cell nuclear antigen (PCNA) and type I collagen stains were performed and analyzed as described previously (Sullivan et al., 2010; Joshi et al., 2016; Kopec et al., 2016). Necrosis area was calculated as described previously (Joshi et al., 2016).
Determination of VWF, lipid peroxidation, and serum ALT
Plasma VWF antigen levels were determined by ELISA as described previously (Groeneveld et al., 2016) and pooled normal human plasma was utilized to develop a standard curve. Thiobarbituric acid reactive substances (TBARS) were determined using a commercially available kit (Cayman Chemical, Ann Arbor, MI). Serum alanine aminotransferase (ALT) activity was determined using commercial reagents (Thermo Fisher, Waltham, MA).
RNA isolation, cDNA synthesis, and quantitative real-time PCR (qPCR)
Detection and quantification of select mRNAs by SYBR Green qPCR was performed using primers described previously (Joshi et al., 2016).
Statistical analyses
Comparison of two groups was performed using Student’s t-test. Comparison of three or more groups was performed using one- or two-way analysis of variance (ANOVA), as appropriate, and Student-Newman-Keul’s post hoc test. The criterion for statistical significance was P<0.05.
Results
Effect of VWF deficiency on acute CCl4-induced liver injury
Plasma VWF levels were significantly increased in CCl4-exposed wild-type mice at 48 and 72 hours (Fig. 1A). Complete VWF deficiency did not affect lipid peroxidation at 48 hours, a surrogate marker of CCl4 metabolism (Fig. 1B). Both male and female mice were challenged with CCl4 and there was no apparent sex-dependent difference in liver injury, in agreement with prior studies (Bhathal et al., 1983). Serum ALT activity, an indicator of hepatocellular injury, was significantly increased in CCl4-exposed male and female VWF−/− mice compared to wild-type mice at 48 hours (Fig. 1C). However, this did not correspond to a significant increase in centrilobular liver necrosis in CCl4-exposed VWF−/− mice (Fig. 1D–E). Serum ALT activity and hepatocellular necrosis were reduced at 72 hours, regardless of genotype (Fig. 1C–E). Moreover, the number of PCNA-positive hepatocytes at both 48 and 72 hours was not significantly affected by VWF deficiency (Fig. 1F–G).
Figure 1.

Effect of VWF deficiency on acute CCl4-induced liver injury. Male and female wild-type (WT) and VWF−/− mice were given carbon tetrachloride (CCl4) dissolved to 10% in corn oil (10 ml/kg, ip) or vehicle (corn oil) and (A) plasma VWF concentration, (B) hepatic malondialdehyde (MDA) levels, and (C) serum alanine aminotransferase (ALT) activity were assessed 48 and 72 hours after CCl4 challenge. (D) Representative photomicrographs (original magnification × 100) show areas of hepatocellular necrosis (stars) in H&E-stained liver sections. (E) Quantification of hepatocellular necrosis area as described in materials and methods. (F) Representative photomicrographs depict immunohistochemical labeling of proliferating cell nuclear antigen (PCNA)-positive hepatocytes (brown), which were (G) quantified as described in materials and methods. Data are expressed as mean + SEM. N=6–11 mice per group. *Significantly different from vehicle-treated mice. #Significantly different from CCl4-challenged WT mice at that time. P<0.05.
VWF deficiency reduces α-smooth muscle actin and profibrogenic gene induction in liver after chronic CCl4 challenge
Plasma VWF levels were significantly increased in wild-type mice after chronic (6 weeks) CCl4 challenge (Fig. 2A). Serum ALT activity was increased modestly in wild-type mice challenged with chronic CCl4 and this was unaffected by VWF deficiency (Fig. 2B). As anticipated, chronic CCl4 challenge in wild-type mice was associated with increased hepatic stellate cell transition to myofibroblasts, marked by significant increases in hepatic Acta2 and corresponding α-SMA protein expression (Fig. 2C–E). Interestingly, induction of Acta2 and α-SMA protein was significantly reduced in VWF−/− mice (Fig. 2C–E). In agreement with these measures suggesting reduced stellate cell activation, induction of additional profibrogenic genes, Tgfb1, Tgfb2 and Col1a1, was also significantly reduced in CCl4-exposed VWF−/− mice compared to CCl4-exposed wild-type mice (Fig. 2F–H).
Figure 2.
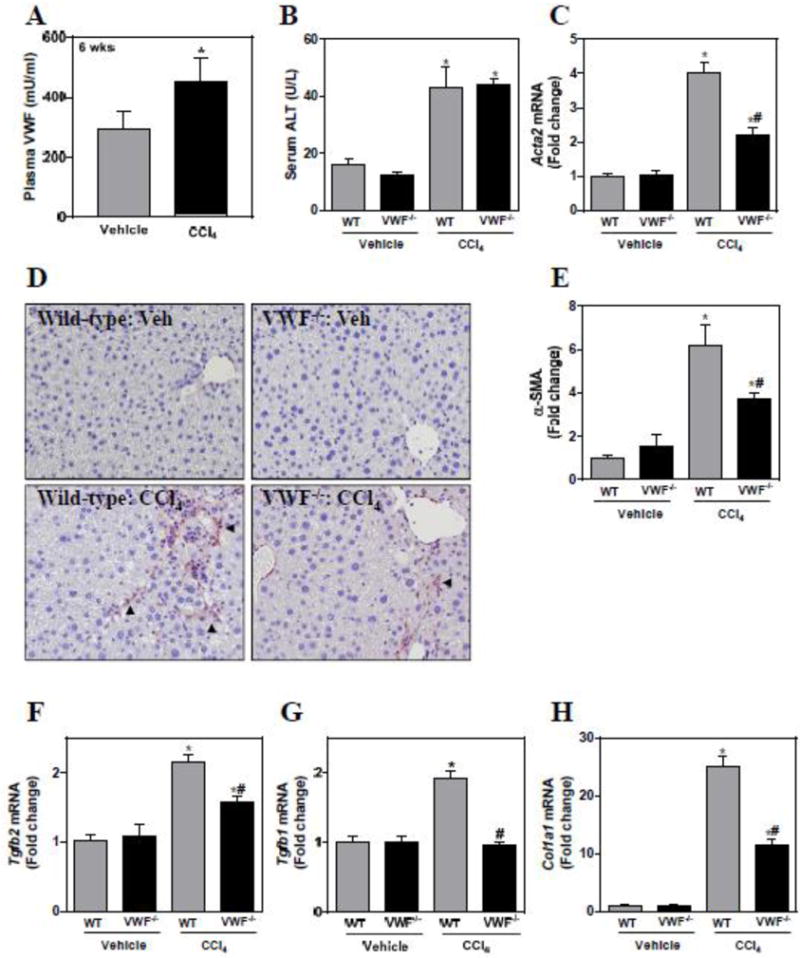
Effect of VWF deficiency on profibrogenic changes in liver after chronic CCl4 challenge. Female wild-type (WT) and VWF−/− mice were given carbon tetrachloride (CCl4) dissolved to 10% in corn oil (10 ml/kg, ip) twice weekly for 6 weeks. (A) Plasma VWF concentration. (B) Serum alanine aminotransferase (ALT) activity. Hepatic levels of profibrogenic genes (C) Acta2, (F) Tgfb2, (G) Tgfb1, and (H) Col1a1 were determined as described in materials and methods and expressed as fold-change vs. vehicle-treated mice of the same genotype. (D) Representative photomicrographs depict immunohistochemical labeling of alpha-smooth muscle actin (α-SMA) (arrowheads, brown) in liver sections. (E) The area of α-SMA staining was quantified as described in materials and methods. Data are expressed as mean + SEM. N=5–8 mice per group for vehicle-treated mice and 16–20 for CCl4-challenged mice. *Significantly different from vehicle-treated mice of the same genotype. #Significantly different from CCl4-challenged WT mice. P<0.05.
VWF deficiency reduces liver fibrosis after chronic CCl4 challenge
Hepatic stellate cells are well-recognized as the primary cellular source of extracellular matrix (e.g., collagens) in liver fibrosis; thus, we utilized multiple approaches to examine the effect of VWF deficiency on liver fibrosis. First, we quantified hepatic collagen deposition by examining sirius red-stained sections under polarized light, an approach that isolates the birefringent property of type I and III collagen (Junqueira et al., 1978; Street et al., 2014). Collagen deposition significantly increased in CCl4-challenged wild-type mice, but this increase was significantly reduced in CCl4-challenged VWF−/− mice (Fig. 3A–B). To validate the reduction in collagen deposition in VWF−/− mice, we evaluated type I collagen deposits by a direct immunolabeling approach. By this method, type I collagen deposits significantly increased in livers of wild-type CCl4-challenged mice, and this was significantly attenuated in CCl4-challenged VWF−/− mice (Fig. 3C–D). Altogether, the results indicate that VWF deficiency significantly reduces collagen deposition in a well-validated experimental setting of liver fibrosis.
Figure 3.
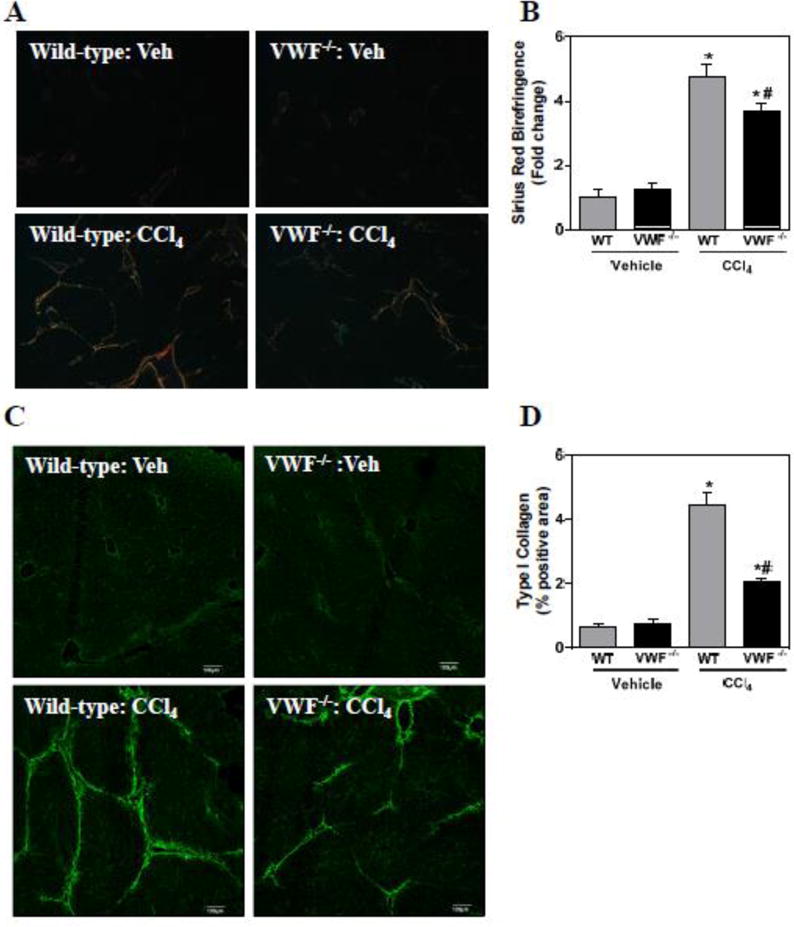
Effect of VWF deficiency on liver fibrosis after chronic CCl4 challenge. Female wild-type (WT) and VWF−/− mice were given carbon tetrachloride (CCl4) dissolved to 10% in corn oil (10 ml/kg, ip) twice weekly for 6 weeks. (A–B) Representative photomicrographs and quantification of sirius red birefringence in liver sections visualized under polarized light. (C–D) Representative photomicrographs and quantification of type I collagen immunofluorescent staining of frozen liver sections. Data are expressed as mean + SEM. For panel B, N=5–8 mice per group for vehicle-treated mice and 16–20 for CCl4-challenged mice. N=3 mice per group for panel D. *Significantly different from vehicle-treated mice of the same genotype. #Significantly different from CCl4-challenged WT mice. P<0.05.
Discussion
Increased plasma VWF levels are closely associated with poor outcome in patients with cirrhosis, and VWF levels are associated with fibrosis in the general population (La Mura et al., 2011; Ferlitsch et al., 2012; Maieron et al., 2014; Plompen et al., 2015; Kalambokis et al., 2016). Building on this clinical association, we provide the first experimental evidence that VWF directly contributes to liver fibrosis. Similar to patients with liver disease, we observed that both acute and chronic experimental liver injury elevated plasma VWF levels. Whereas VWF deficiency had minimal effect on acute CCl4 hepatotoxicity, activation of hepatic stellate cells, suggested by α-SMA expression, and hepatic collagen deposition triggered by chronic CCl4 challenge were reduced in VWF-deficient mice. These studies demonstrate a causal link between VWF and fibrosis, an important observation building on the clinical association between VWF and cirrhosis in patients. This suggests that in addition to serving as a biomarker of cirrhosis severity, elevated plasma VWF levels offer insight into how components of the hemostatic system contribute to fibrosis.
The mechanism whereby VWF contributes to experimental liver fibrosis is not completely understood. The hemostatic system is altered in patients with cirrhosis, predisposing patients to thrombosis (Tripodi, 2015). Experimental studies also indicate that components of the hemostatic system drive liver fibrosis (Anstee et al., 2011). Published clinical studies support this hypothesis (Villa et al., 2012; Poujol-Robert et al., 2014) and additional clinical trials are underway to examine the effect of anticoagulants in cirrhosis (e.g., NCT02271295, NCT02643212). Elevated VWF levels are one of the reported hemostatic alterations in patients with cirrhosis. VWF binding to the GPIb/IX/V complex, and to collagen, bridges platelets to exposed collagen surfaces (Ruggeri, 2003). VWF deficiency predisposes to bleeding in humans and also reduces thrombus formation in experimental models (Denis et al., 1998; Leebeek and Eikenboom, 2016). Thus, one mechanism whereby VWF deficiency could reduce fibrosis in experimental systems is by reducing platelet accumulation/function. Limitation in next level mechanistic studies examining this possibility is that interaction between VWF and platelets is rapid and may be missed without extensive time course investigation. It is also possible that VWF-platelet interaction drives liver fibrosis through formation of thrombi in extrahepatic vessels.
Anticoagulants have emerged as a candidate therapeutic class to reduce cirrhosis and have been shown to reduce disease progression in both experimental animal models as well as in humans (Ganey et al., 2007; Fujita et al., 2008; Anstee et al., 2011; Villa et al., 2012). Although currently available anticoagulants show promising results, the risk of bleeding is still a major drawback. Especially in patients with impaired liver function, which is associated with a complex coagulopathy, bleeding risk dramatically limits therapeutic potential of existing drugs (Lisman et al., 2013). Agents inhibiting VWF function by blocking the VWF-GPIbα or VWF-collagen interaction have been developed in the past decade (De Meyer et al., 2012). These inhibitors include monoclonal antibodies against VWF and DNA aptamers that act like chemical antibodies by binding to VWF with high affinity. Preclinical and clinical studies have already demonstrated the antithrombotic potential of such inhibitors (reviewed in (De Meyer et al., 2012). Moreover, clinical testing in animal models and healthy individuals points towards a profoundly lower bleeding risk with equal or even higher efficacy as compared to established anti-platelet drugs (Kageyama et al., 1997; Kageyama et al., 2002; Ulrichts et al., 2011). Our results indicate that VWF directly contributes to liver fibrosis. Thus, targeting VWF functionality with these monoclonal antibodies or DNA aptamers could therefore be a novel therapeutic approach in the treatment of chronic liver disease.
In summary, we report that VWF deficiency significantly attenuates the cellular profibrogenic response and fibrosis in experimental chronic liver injury. The results solidify the role of VWF as a causal factor in liver fibrosis, building on strong clinical evidence of an association between elevated VWF levels with cirrhosis. Additional mechanistic studies defining the mechanism whereby VWF exacerbates experimental fibrosis are likely to reveal novel links between the hemostatic system and cirrhosis. Such studies have the potential to identify specific VWF functions as putative therapeutic targets for liver cirrhosis.
Highlights.
Plasma von Willebrand Factor (VWF) levels increase in experimental liver damage
VWF contributes to liver myofibroblast activation in experimental fibrosis
VWF deficiency reduces experimental fibrosis without affecting acute liver injury
Acknowledgments
This work was supported by the National Institutes of Health National Institute of Environmental Health Sciences [R01 ES017537]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anstee QM, Dhar A, Thursz MR. The role of hypercoagulability in liver fibrogenesis. Clinics and research in hepatology and gastroenterology. 2011;35:526–533. doi: 10.1016/j.clinre.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Anstee QM, Goldin RD, Wright M, Martinelli A, Cox R, Thursz MR. Coagulation status modulates murine hepatic fibrogenesis: implications for the development of novel therapies. Journal of thrombosis and haemostasis: JTH. 2008;6:1336–1343. doi: 10.1111/j.1538-7836.2008.03015.x. [DOI] [PubMed] [Google Scholar]
- Bhathal PS, Rose NR, Mackay IR, Whittingham S. Strain differences in mice in carbon tetrachloride-induced liver injury. British journal of experimental pathology. 1983;64:524–533. [PMC free article] [PubMed] [Google Scholar]
- De Meyer SF, Stoll G, Wagner DD, Kleinschnitz C. von Willebrand factor: an emerging target in stroke therapy. Stroke. 2012;43:599–606. doi: 10.1161/STROKEAHA.111.628867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Cullere M, Hynes RO, Wagner DD. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9524–9529. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlitsch M, Reiberger T, Hoke M, Salzl P, Schwengerer B, Ulbrich G, Payer BA, Trauner M, Peck-Radosavljevic M, Ferlitsch A. von Willebrand factor as new noninvasive predictor of portal hypertension, decompensation and mortality in patients with liver cirrhosis. Hepatology. 2012;56:1439–1447. doi: 10.1002/hep.25806. [DOI] [PubMed] [Google Scholar]
- Ferro D, Quintarelli C, Lattuada A, Leo R, Alessandroni M, Mannucci PM, Violi F. High plasma levels of von Willebrand factor as a marker of endothelial perturbation in cirrhosis: relationship to endotoxemia. Hepatology. 1996;23:1377–1383. doi: 10.1002/hep.510230613. [DOI] [PubMed] [Google Scholar]
- Fujita K, Nozaki Y, Wada K, Yoneda M, Endo H, Takahashi H, Iwasaki T, Inamori M, Abe Y, Kobayashi N, Kirikoshi H, Kubota K, Saito S, Nagashima Y, Nakajima A. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut. 2008;57:1583–1591. doi: 10.1136/gut.2007.144550. [DOI] [PubMed] [Google Scholar]
- Ganey PE, Luyendyk JP, Newport SW, Eagle TM, Maddox JF, Mackman N, Roth RA. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007;46:1177–1186. doi: 10.1002/hep.21779. [DOI] [PubMed] [Google Scholar]
- Groeneveld DJ, Alkozai EM, Adelmeijer J, Porte RJ, Lisman T. Balance between von Willebrand factor and ADAMTS13 following major partial hepatectomy. The British journal of surgery. 2016 doi: 10.1002/bjs.10107. [DOI] [PubMed] [Google Scholar]
- Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thrombosis and haemostasis. 2004;91:267–275. doi: 10.1160/TH03-05-0310. [DOI] [PubMed] [Google Scholar]
- Hugenholtz GC, Adelmeijer J, Meijers JC, Porte RJ, Stravitz RT, Lisman T. An unbalance between von Willebrand factor and ADAMTS13 in acute liver failure: implications for hemostasis and clinical outcome. Hepatology. 2013;58:752–761. doi: 10.1002/hep.26372. [DOI] [PubMed] [Google Scholar]
- Jairath V, Burroughs AK. Anticoagulation in patients with liver cirrhosis: complication or therapeutic opportunity? Gut. 2013;62:479–482. doi: 10.1136/gutjnl-2012-303088. [DOI] [PubMed] [Google Scholar]
- Joshi N, Kopec AK, Ray JL, Cline-Fedewa H, Nawabi A, Schmitt T, Nault R, Zacharewski TR, Rockwell CE, Flick MJ, Luyendyk JP. Fibrin deposition following bile duct injury limits fibrosis through an alphaMbeta2-dependent mechanism. Blood. 2016;127:2751–2762. doi: 10.1182/blood-2015-09-670703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junqueira LC, Cossermelli W, Brentani R. Differential staining of collagens type I, II and III by Sirius Red and polarization microscopy. Archivum histologicum Japonicum = Nihon soshikigaku kiroku. 1978;41:267–274. doi: 10.1679/aohc1950.41.267. [DOI] [PubMed] [Google Scholar]
- Kageyama S, Yamamoto H, Nagano M, Arisaka H, Kayahara T, Yoshimoto R. Anti-thrombotic effects and bleeding risk of AJvW-2, a monoclonal antibody against human von Willebrand factor. British journal of pharmacology. 1997;122:165–171. doi: 10.1038/sj.bjp.0701354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama S, Yamamoto H, Nakazawa H, Matsushita J, Kouyama T, Gonsho A, Ikeda Y, Yoshimoto R. Pharmacokinetics and pharmacodynamics of AJW200, a humanized monoclonal antibody to von Willebrand factor, in monkeys. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:187–192. doi: 10.1161/hq0102.101520. [DOI] [PubMed] [Google Scholar]
- Kalambokis GN, Oikonomou A, Christou L, Kolaitis NI, Tsianos EV, Christodoulou D, Baltayiannis G. von Willebrand factor and procoagulant imbalance predict outcome in patients with cirrhosis and thrombocytopenia. Journal of hepatology. 2016;65:921–928. doi: 10.1016/j.jhep.2016.06.002. [DOI] [PubMed] [Google Scholar]
- Kopec AK, Joshi N, Cline-Fedewa H, Wojcicki AV, Ray JL, Sullivan BP, Froehlich JE, Johnson BF, Flick MJ, Luyendyk JP. Fibri(nogen) drives repair after acetaminophen-induced liver injury via leukocyte alphaMbeta2 integrin-dependent upregulation of MMP12. Journal of hepatology. 2016 doi: 10.1016/j.jhep.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Mura V, Reverter JC, Flores-Arroyo A, Raffa S, Reverter E, Seijo S, Abraldes JG, Bosch J, Garcia-Pagan JC. Von Willebrand factor levels predict clinical outcome in patients with cirrhosis and portal hypertension. Gut. 2011;60:1133–1138. doi: 10.1136/gut.2010.235689. [DOI] [PubMed] [Google Scholar]
- Leebeek FW, Eikenboom JC. Von Willebrand’s Disease. The New England journal of medicine. 2016;375:2067–2080. doi: 10.1056/NEJMra1601561. [DOI] [PubMed] [Google Scholar]
- Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, Leebeek FW. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006;44:53–61. doi: 10.1002/hep.21231. [DOI] [PubMed] [Google Scholar]
- Lisman T, Caldwell SH, Burroughs AK, Northup PG, Senzolo M, Stravitz RT, Tripodi A, Trotter JF, Valla DC, Porte RJ, Coagulation in Liver Disease Study, G Hemostasis and thrombosis in patients with liver disease: the ups and downs. Journal of hepatology. 2010;53:362–371. doi: 10.1016/j.jhep.2010.01.042. [DOI] [PubMed] [Google Scholar]
- Lisman T, Kamphuisen PW, Northup PG, Porte RJ. Established and new-generation antithrombotic drugs in patients with cirrhosis - possibilities and caveats. Journal of hepatology. 2013;59:358–366. doi: 10.1016/j.jhep.2013.03.027. [DOI] [PubMed] [Google Scholar]
- Maieron A, Salzl P, Peck-Radosavljevic M, Trauner M, Hametner S, Schofl R, Ferenci P, Ferlitsch M. Von Willebrand Factor as a new marker for non-invasive assessment of liver fibrosis and cirrhosis in patients with chronic hepatitis C. Alimentary pharmacology & therapeutics. 2014;39:331–338. doi: 10.1111/apt.12564. [DOI] [PubMed] [Google Scholar]
- Plompen EP, Darwish Murad S, Hansen BE, Loth DW, Schouten JN, Taimr P, Hofman A, Uitterlinden AG, Stricker BH, Janssen HL, Leebeek FW. Prothrombotic genetic risk factors are associated with an increased risk of liver fibrosis in the general population: The Rotterdam Study. Journal of hepatology. 2015;63:1459–1465. doi: 10.1016/j.jhep.2015.07.026. [DOI] [PubMed] [Google Scholar]
- Poujol-Robert A, Boelle PY, Conti F, Durand F, Duvoux C, Wendum D, Paradis V, Mackiewicz V, Chazouilleres O, Corpechot C, Poupon R. Aspirin may reduce liver fibrosis progression: Evidence from a multicenter retrospective study of recurrent hepatitis C after liver transplantation. Clinics and research in hepatology and gastroenterology. 2014;38:570–576. doi: 10.1016/j.clinre.2014.07.004. [DOI] [PubMed] [Google Scholar]
- Reuken PA, Kussmann A, Kiehntopf M, Budde U, Stallmach A, Claus RA, Bruns T. Imbalance of von Willebrand factor and its cleaving protease ADAMTS13 during systemic inflammation superimposed on advanced cirrhosis. Liver international: official journal of the International Association for the Study of the Liver. 2015;35:37–45. doi: 10.1111/liv.12657. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. Journal of thrombosis and haemostasis: JTH. 2003;1:1335–1342. doi: 10.1046/j.1538-7836.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- Rullier A, Gillibert-Duplantier J, Costet P, Cubel G, Haurie V, Petibois C, Taras D, Dugot-Senant N, Deleris G, Bioulac-Sage P, Rosenbaum J. Protease-activated receptor 1 knockout reduces experimentally induced liver fibrosis. American journal of physiology Gastrointestinal and liver physiology. 2008;294:G226–235. doi: 10.1152/ajpgi.00444.2007. [DOI] [PubMed] [Google Scholar]
- Street JM, Souza AC, Alvarez-Prats A, Horino T, Hu X, Yuen PS, Star RA. Automated quantification of renal fibrosis with Sirius Red and polarization contrast microscopy. Physiological reports. 2014;2 doi: 10.14814/phy2.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BP, Weinreb PH, Violette SM, Luyendyk JP. The coagulation system contributes to alphaVbeta6 integrin expression and liver fibrosis induced by cholestasis. The American journal of pathology. 2010;177:2837–2849. doi: 10.2353/ajpath.2010.100425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripodi A. Hemostasis abnormalities in cirrhosis. Current opinion in hematology. 2015;22:406–412. doi: 10.1097/MOH.0000000000000164. [DOI] [PubMed] [Google Scholar]
- Tripodi A, Anstee QM, Sogaard KK, Primignani M, Valla DC. Hypercoagulability in cirrhosis: causes and consequences. Journal of thrombosis and haemostasis: JTH. 2011;9:1713–1723. doi: 10.1111/j.1538-7836.2011.04429.x. [DOI] [PubMed] [Google Scholar]
- Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. The New England journal of medicine. 2011;365:147–156. doi: 10.1056/NEJMra1011170. [DOI] [PubMed] [Google Scholar]
- Ulrichts H, Silence K, Schoolmeester A, de Jaegere P, Rossenu S, Roodt J, Priem S, Lauwereys M, Casteels P, Van Bockstaele F, Verschueren K, Stanssens P, Baumeister J, Holz JB. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood. 2011;118:757–765. doi: 10.1182/blood-2010-11-317859. [DOI] [PubMed] [Google Scholar]
- Villa E, Camma C, Marietta M, Luongo M, Critelli R, Colopi S, Tata C, Zecchini R, Gitto S, Petta S, Lei B, Bernabucci V, Vukotic R, De Maria N, Schepis F, Karampatou A, Caporali C, Simoni L, Del Buono M, Zambotto B, Turola E, Fornaciari G, Schianchi S, Ferrari A, Valla D. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology. 2012;143:1253–1260 e1251. 1254. doi: 10.1053/j.gastro.2012.07.018. [DOI] [PubMed] [Google Scholar]