

Abstract
Topoisomerases are enzymes involved in maintaining the topological state of cellular DNA. Despite many structural, biophysical, and biochemical studies, their dynamic characteristics remain poorly understood. Recent single molecule experiments revealed that an important feature of the type IA topoisomerase mechanism is the presence of pauses between relaxation events. However, these experiments cannot determine whether the protein remains DNA bound during the pauses or the relationship between domain movements in the protein and topological changes in the DNA. By combining two orthogonal single molecule techniques, we observed that topoisomerase IA is constantly changing conformation and attempting to modify the topology of DNA, but only succeeds in a fraction of the attempts. Thus, its mechanism can be described as a series of DNA strand passage attempts that culminate in a successful relaxation event.
Introduction
Topoisomerases are ubiquitous enzymes involved in maintaining the supercoiled state of DNA in the cell1. Previous studies have provided an overall view of the mechanisms of DNA transformation by these enzymes, but many aspects, particularly their dynamic characteristics, remain poorly understood. Type IA topoisomerases change the topology of DNA using an enzyme-bridged strand passage mechanism, where one DNA strand is passed through a transient break in the second strand2. Escherichia coli topoisomerase I (EcTopoI), a type IA topoisomerase, can relax negatively supercoiled DNA and also decatenate DNA molecules. It requires both single stranded DNA (ssDNA) regions and magnesium for activity, but not ATP, instead utilizing the torsional stress stored in supercoiled DNA as an energy source2. Structural studies have shown that type IA topoisomerases are formed by a conserved toroidal shaped strand-passage module formed by four domains (I–IV)3. The strand-passage module contains the active site at the interface of domains I and III and binds the cleaved ssDNA in a groove formed by domains I, III, and IV (Fig. 1a)4. Some type IA enzymes contain additional domains at the N-terminus. The C-terminal region also varies in different type IA enzymes, but they all appear to be involved in DNA binding. For example, EcTopoI has C-terminal zinc-finger domains (V-IX) that bind to the uncleaved ssDNA opposite the strand bound in the strand-passage module (Fig. 1a)5. Although these other domains are important and modulate activity, the strand-passage module is the only conserved area and is central for activity.
Figure 1. Combined MT-TIRF experimental setup.
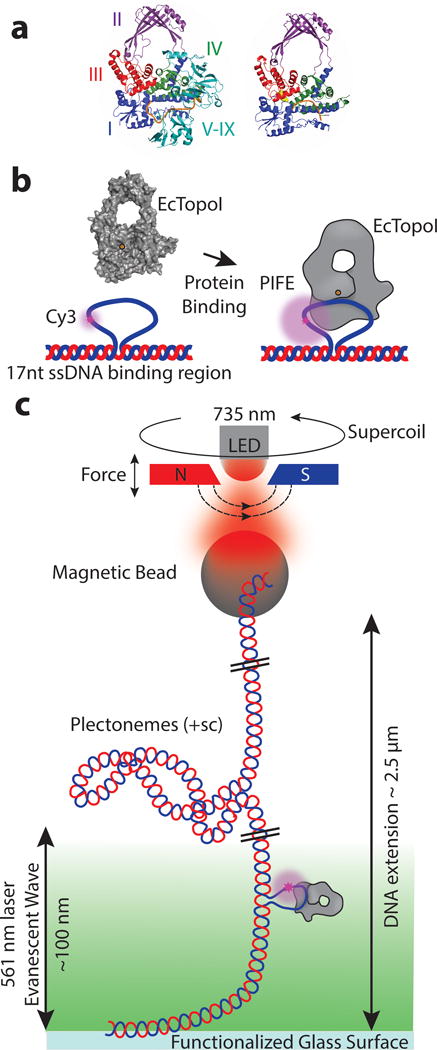
a) Left) Structure of full length EcTopoI with the C-terminal zinc-finger domains (V-IX) bound to ssDNA5. Right) The structure of the EcTopoI 67 kD N-terminal strand-passage module with ssDNA bound in the active site4 illustrates the four domain architecture of the conserved common region for all type IA topoisomerases. For clarity, only the conserved strand passage module is shown in subsequent figures. EcTopoI binds the cleaved ssDNA (orange) between domains I, III, and IV, with the active site tyrosine located in domain III (yellow). Domain III can separate from domains I and IV allowing for DNA strand passage, which leads to relaxation. b) When EcTopoI binds to the engineered 22nt Cy3 ssDNA bulge (with a 17nt binding region), it causes a distance-dependent increase in fluorescence intensity known as PIFE9,10. c) Schematic of the experimental set-up. Using the magnets, force can be applied to a tethered DNA molecule containing the 22nt Cy3 ssDNA bulge. Introduction of positive supercoils (+sc) does not produce single stranded regions for EcTopoI to bind to (except for the bulge) and generates plectonemes that shorten the DNA extension18. The ssDNA bulge serves as a binding site that targets the protein into the evanescent wave, where PIFE can be monitored. Bead extension, which reports on DNA topology, is monitored using light from a 735 nm LED, while PIFE, excited using a 561 nm laser, reports on protein/DNA interactions and domain movements. Both signals are measured concurrently (Supplementary Fig. 1).
Previously, an enzyme-bridged strand passage mechanism for type IA topoisomerases was proposed that involves several steps and conformational states3,4,6. Briefly, after ssDNA is bound across domain I, a transient break is formed in one DNA strand through a phosphotyrosine bond with the active site tyrosine in domain III, followed by movement of domain III away from domain I, creating an opening (or ‘gate’) in the protein and expanding the ssDNA break to allow passage of the complementary strand through the break and into the central hole of the protein. Reversal of the conformational changes allows religation of the phosphodiester backbone and subsequent reopening of the protein gate allows for the trapped DNA strand to exit, leading to strand passage and DNA relaxation.
Although this proposed mechanism is supported by many structural and biophysical observations3,4,6–8, the nature of the conformational changes and the details of the mechanism remain largely hypothetical. In order to study the correlation between conformational changes in the protein and topological changes in the DNA, we designed a novel single molecule experiment that combines two orthogonal single molecule techniques, magnetic tweezers (MT) and total internal reflection fluorescence (TIRF) microscopy. These orthogonal techniques allow us to follow the changes in overall DNA topology while at the same time observing the conformation of the protein. For the experiments, we took advantage of an inherent property of the Cyanine 3 fluorophore (Cy3). When Cy3 is covalently attached to DNA it displays a higher fluorescence intensity that depends on the proximity of the fluorophore to a nearby bound protein, a phenomenon termed protein-induced fluorescence enhancement (PIFE)9,10 (Fig. 1b). Using a home-built MT-TIRF microscope, we were able to use PIFE to monitor protein binding and conformational changes while simultaneously monitoring the DNA topology with the MT (Supplementary Fig. 1). Other combinations of single molecule manipulation and fluorescence have been utilized before, for example combining optical tweezers and fluorescence11–13, and combining MT and single molecule fluorescence14,15 or single molecule Förster Resonance Energy Transfer (smFRET)16, but this is the first time a combination of MT and PIFE has been used to study conformational changes in macromolecules.
Results
Combined magnetic tweezers and TIRF microscopy
In a typical experiment, we used a specifically designed DNA molecule with a 22 nucleotide (nt) ssDNA bulge containing a Cy3 label (Online Methods). The 22nt bulge consists of a 5nt region on the 3′ end followed by the Cy3 and a 17nt EcTopoI binding region, placing the Cy3 on the DNA strand that will be transiently attached to domain III by the phosphotyrosine linkage. When the DNA is positively supercoiled (+sc), there are no ssDNA regions for EcTopoI to bind to except for the ssDNA bulge8,17. In addition, the DNA construct located the fluorophore in the TIRF field, at ≈50 nm above the glass surface, to ensure that we could target a single EcTopoI molecule to the DNA and generate the PIFE signal (Fig. 1c). Once a suitable DNA molecule was found, the TIRF illumination was turned on and the DNA was supercoiled using the MT while the fluorescence signal of the supercoiled substrate was measured. As supercoiling occurs, plectonemes begin to form, shortening the extension of the DNA, which can be observed by monitoring the height of the tethered bead18. After the protein was flowed in, on average a 40% increase in fluorescence was observed due to PIFE. Real-time monitoring of the bead position tracked the topological state of the DNA while concurrently the fluorescence signal was measured.
Analysis of the fluorescence data in the absence of protein showed a constant signal with a Gaussian distribution of intensities (Fig. 2a). Addition of the protein showed two things: the intensity of the signal increased due to PIFE and the intensity distribution changed to a broader one, not described by a single Gaussian distribution and with a clear shoulder on the high intensity side (Fig. 2b). Possible explanations for the broader and asymmetric intensity distribution could be sliding of the protein in the ssDNA bulge or movement of protein domains, such as the C-terminal zinc-fingers or domain III.
Figure 2. PIFE is observed upon EcTopoI binding to DNA.
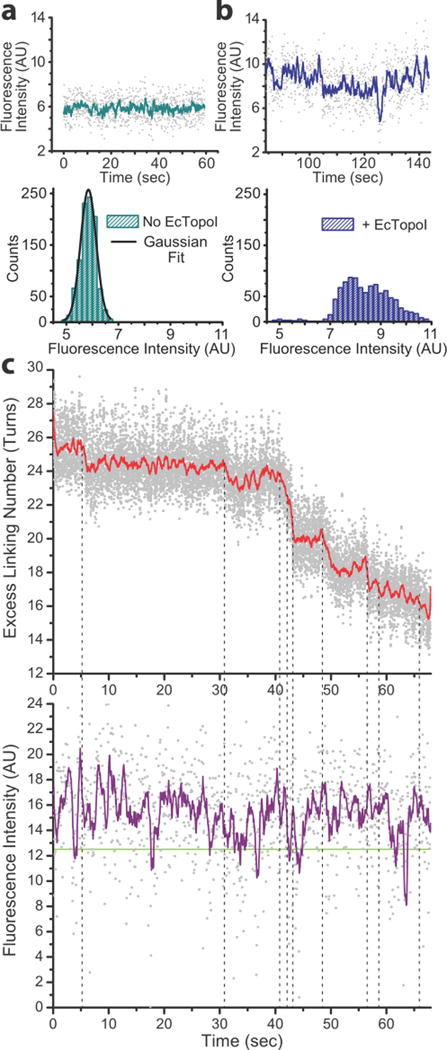
a) The fluorescence intensity signal of the Cy3 molecule attached to the 22nt bulge in the DNA displays a Gaussian distribution of values. b) Adding buffer with 40 nM of EcTopoI to the fluorescent substrate in a, leads to an increase in fluorescence intensity and broadening of the distribution. Additionally, a deviation from a single Gaussian distribution of intensity values occurs and a shoulder appears at higher intensities. The distribution could be modelled by several different Gaussians representing different states, but the lack of distinct states precludes a definitive fit. c) Concurrent fluorescence and DNA extension traces were aligned in time (n=9 DNA molecules). MT traces were converted from extension to excess linking number and the raw data (gray) were smoothed using a 100-point boxcar average (red). Fluorescence traces were smoothed with a 10-point boxcar average (purple). Note that the representative data trace in c) comes from a different fluorophore than in plots a and b –and hence the intensity values are not directly comparable. The plots in a and b serve to illustrate the difference is shape for the intensity distributions. The green line at ≈12.4 AU fluorescence intensity marks the center of the Gaussian distribution when no protein was present (Supplementary Fig. 4). Intensities just above or below this line represent the no-PIFE (protein unbound) state. While there are transient excursions to the unbound state, the majority of the time PIFE is observed, including during pauses in relaxation. Vertical dotted lines mark the relaxation events; these were found to be correlated with local maxima in the fluorescence trace (PIFE spikes) (Supplementary Fig. 5).
A dependence of the fluorescence intensity on the topological state of the DNA was generally observed, both in the presence or absence of EcTopoI, and is likely due to the movement of the fluorophore in the TIRF field. Using control DNA supercoiling data, we were able to determine a useable range in which to make our measurements that is not affected by the supercoiled state of the DNA. It is important to note that the exact fluorescence intensity values varied from molecule to molecule. For example, the baseline (no protein) intensity value and the increase in fluorescence upon protein binding vary for each different DNA molecule studied. However, the characteristics of the fluorescence signals (i.e. fluorescence increase with protein binding) analyzed here were consistent for all molecules.
We collected concurrent MT-TIRF data from 33 different DNA molecules, but only 9 produced good data from both the MT and PIFE channels, producing 93 independent relaxation events (Fig. 2, Supplementary Fig. 2, and Supplementary Table 1). In a few cases, unbinding/binding events were also observed, characterized by the fluorescence signal returning to the baseline value momentarily and then recovering to the PIFE state (Cy3 blinking events were easily distinguishable as well). We also observed fluctuations in fluorescence intensity, which could be due to domain movements or blinking of the Cy3 faster than the 60 ms imaging. Unbinding events were not common under our experimental conditions, indicating that the protein remains attached to the DNA most of the time, including during DNA relaxation pauses. This indicates that the pauses between relaxation events are generally not due to prolonged unbinding events, a possibility that could have explained the pausing behavior. Additional experiments were conducted with wild type EcTopoI fluorescently labeled with Alexa Fluor 555 to test directly for the presence of the protein during relaxation events. Alignment of the MT and protein fluorescence data in time directly confirmed that the protein is bound during DNA relaxation, including during the pauses (Supplementary Fig. 3).
Local fluorescent spikes correlated to relaxation events
Analysis of the fluorescence signal in the presence of protein showed the existence of local fluorescent spikes, above the average PIFE intensity (PIFE spikes) (Fig. 2c and Supplementary Fig. 2). To identify these PIFE spikes we used local maxima found in the fluorescence trace above a cut-off value determined from the control distribution without protein (Supplementary Fig. 4) (Origin (OriginLab, Northampton, MA). These spikes were found throughout the traces while the protein was bound, and many of them were associated with relaxation events. An illustration of the way the data in Figure 2c were analyzed, including PIFE spikes, is shown in Supplementary Figures 4 and 5.
When the local maxima and relaxation events were within 600 ms, they were considered correlated (due to the 10 pt smoothing of the 60 ms fluorescence trace). Comparison of the PIFE spikes and the relaxation events monitored by MT showed a correlation between the beginning of a relaxation event and a PIFE spike in 90% of the events (Nrelaxation=93, Ncorr=84). Visual inspection shows that while the relaxation events do not always align exactly with the local maxima, they fall within the spike of fluorescence associated with the local maxima. This could be due to the inexact identification of the start of a relaxation event, Cy3 blinking, or the smoothing used on the trace. However, averaging of these correlated PIFE spikes shows that they are centered on relaxation events (Fig. 3c).
Figure 3. PIFE spikes occur at relaxation events and during relaxation pauses.
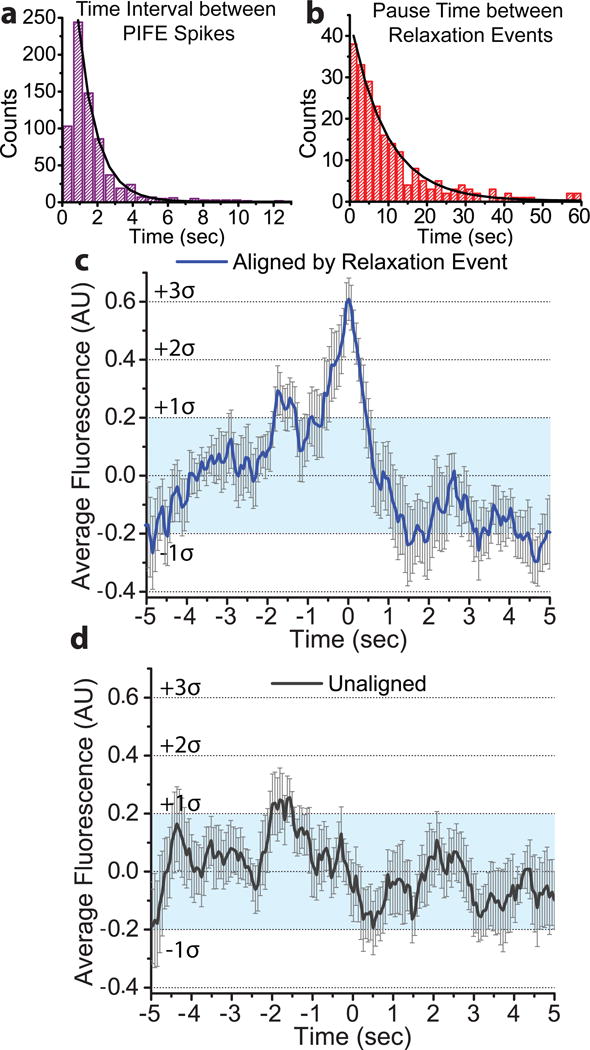
a) PIFE spikes were identified by local maxima and the time between spikes was binned in 600 ms intervals. An exponential decay function was fitted starting at the 600–1200 ms bin, due to the inability to distinguish between spikes that occur at shorter times. The mean lifetime of the time interval between PIFE spikes was found to be 1.08 ± 0.02 sec (N=734). b) The pause times between relaxation events from the MT data were binned in 2 sec intervals and fitted with an exponential decay function, giving a mean lifetime for the pauses of 9.3 ± 0.2 sec (N=233). c) Fluorescence traces of 73 separate relaxation events were normalized using the Z-score and the relaxation time (identified from the MT trace) was set to 0. These aligned traces were averaged (blue) and show a marked fluorescent spike (3σ, σ=0.20 (s.d.)) coincident with the relaxation event. The light blue shaded region indicates one standard deviation from the mean, the bars corresponds to error estimates (s.e.m.). For information on the error estimate analysis see Supplementary Note 1. A significant asymmetry of 0.24 ± 0.06 (s.e.m.) (where 0 is no asymmetry) was also found for the aligned trace, comparing the 2.5 seconds before and after the start of the relaxation event. d) 73 unaligned and averaged traces (black) did not display a similar peak, remaining primarily within the light blue shaded, one standard deviation region (error bars represent error estimates (s.e.m.)).
In order to confirm the observed correlation between a relaxation event and a PIFE spike, a group of 73 MT relaxation events were used to align the associated fluorescence traces. Fluorescence intensity data 5 seconds on either side of a DNA relaxation event were extracted and normalized to a mean of 0 and a standard deviation of 1, facilitating comparison between different traces (Supplementary Note 1). When the traces were averaged, a clear fluorescence spike at 0 seconds (the start of a relaxation event) and three standard deviations above the mean is evident (σ = 0.20 (s.d.)) (Fig. 3c). As a control, 73 unaligned 10 second traces were normalized and averaged, and no significant peak was observed (Fig. 3d). The averaged traces confirm that there is a strong correlation between a PIFE spike and a relaxation event.
Additionally, a significant asymmetry was observed in the aligned trace over time intervals of about 2 seconds before and after the relaxation event. For this analysis, 0 would indicate perfect symmetry of the peak on either side of 0 sec (the time of the relaxation event). For the window of 2.5 sec on either side of 0, we found a significant asymmetry of 0.240 ± 0.063 (s.e.m.), with the time region before the peak showing significantly greater fluorescence intensity than the time region after the peak (Supplementary Note 1). This indicates a difference in the Cy3 environment, with higher fluorescence intensity displayed from −2.5 to 0 sec compared to the intensity from 0 to 2.5 sec. This asymmetry likely reflects further domain movements during the relaxation process, in particular it could represent differences between the cleavage and religation states of the bound ssDNA.
While PIFE spikes are clearly correlated with relaxation events, we observed many more PIFE spikes than relaxation events, as correlated events account for only 11.4% of all the spikes observed (Ntot=734, Ncorr=84). This indicates that the majority of the PIFE spikes are occurring during pauses in relaxation. Analysis of the PIFE spikes showed the mean time interval between them to be 1.08 ± 0.02 seconds (i.e. the time elapsed between one local maximum and the next) (Fig. 3a). Similar analysis of the relaxation pauses in the MT data shows a mean duration of 9.3 ± 0.2 seconds (Fig. 3b). These mean lifetimes predict that ≈10% of the PIFE spikes should be associated with successful relaxation, which is in good agreement with the results of the correlation between relaxation events and PIFE spikes.
Magnesium is essential for the presence of PIFE spikes
By performing MT-TIRF experiments in the absence of magnesium, due to the presence of a chelator, we were able to isolate the role of magnesium in the reaction and to test whether binding of the protein alone or DNA cleavage and catalysis were necessary for the presence of PIFE spikes. As expected, whereas EcTopoI was able to bind to the ssDNA substrate in the absence of magnesium, no relaxation events were observed (data not shown). Additionally, for the traces that were analyzed in both the presence and absence of magnesium, no nicking events were observed (n=9 molecules).
Analysis of the protein bound fluorescence data in the absence of magnesium showed no asymmetry in the histograms of the fluorescence intensities (n=9 molecules), which fit well to a single Gaussian distribution (Fig. 4). In particular, no high intensity shoulder, which is characteristic of PIFE spikes, was observed without magnesium, implying that the PIFE spikes are the result of a magnesium dependent or magnesium stimulated event, such as cleavage or active site rearrangement19,20. However, upon addition of magnesium to the experiment the broader intensity distributions with high intensity fluorescence values were recovered (Fig. 4). This shows that the events that lead to the production of PIFE spikes in the presence of magnesium are not occurring in the absence of magnesium. This also indicates that the asymmetric intensity distributions observed in the presence of magnesium are not a result of protein sliding in the ssDNA bulge, since sliding should be possible when no magnesium is present.
Figure 4. PIFE spikes are not observed in the absence of magnesium.
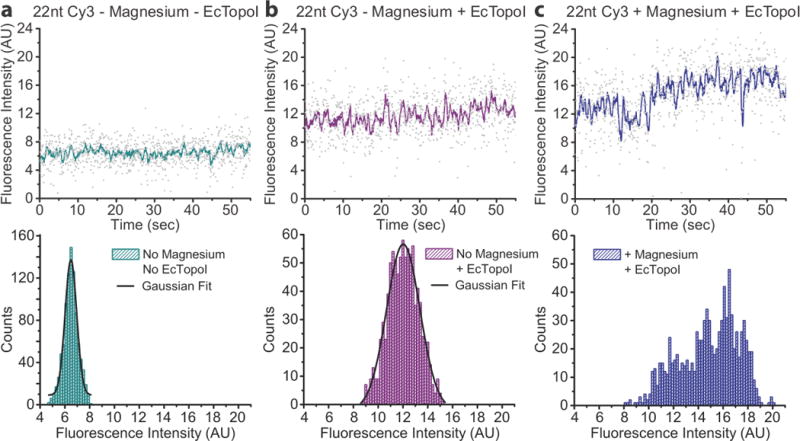
a) The fluorescence intensity of 22nt Cy3 bulge in a magnesium free buffer displays a Gaussian distribution. b) After addition of EcTopoI in magnesium free buffer to the same substrate as shown in a, an increase in fluorescence, characteristic of PIFE, is observed. The distribution of fluorescence intensity broadens, but no asymmetry is observed, and the histogram still fits well to a single Gaussian distribution. c) Addition of both magnesium buffer and EcTopoI to the same substrate in a and b shows a marked change in fluorescence intensity distribution (n=9 DNA molecules). The histogram no longer fits a single Gaussian distribution and is asymmetric, particularly with the appearance of high intensity fluorescence, which is characteristic of PIFE spikes.
Additionally, we observed binding and unbinding events when no magnesium was present, very different from the transient unbinding events observed when magnesium is present, suggesting a direct or indirect role of magnesium in sustained protein binding (Supplementary Fig. 6).
Discussion
PIFE is highly sensitive to the distance between the protein and the fluorophore9 and the spikes seen in the fluorescence data must reflect a change in the microscopic environment of the Cy3 molecule. This could be due to either the movement of the entire protein towards the fluorophore or conformational changes in the protein that move one or several protein domains closer to the location of the Cy3 on the DNA. The latter option is particularly attractive, as it is known that as part of the DNA relaxation cycle type IA topoisomerases change conformation to open and close a gate to accept ssDNA into the protein7. In particular, domain III (Fig. 1a) has to move to create the gate, but the nature of the movement is not known. Since the Cy3 molecule is found at the 3′ end of the ssDNA substrate, it remains attached to domain III during the relaxation cycle. Previous models, which proposed that domain III moves away from domain I2, would not necessarily account for an increase in PIFE as the Cy3 molecule would move further away from the protein. Instead, it is likely that domain III moves closer to the main body of the protein, possibly on the side of the molecule, and in this way creates an opening that allows the DNA to enter the protein (Fig. 5). This type of movement would not only account for the increase in PIFE, as it brings the protein domains closer to the fluorophore, it would also explain how to create a gate that seeks to capture the intact DNA strand (Supplementary Fig. 7).
Figure 5. Revised relaxation mechanism for type IA topoisomerases.
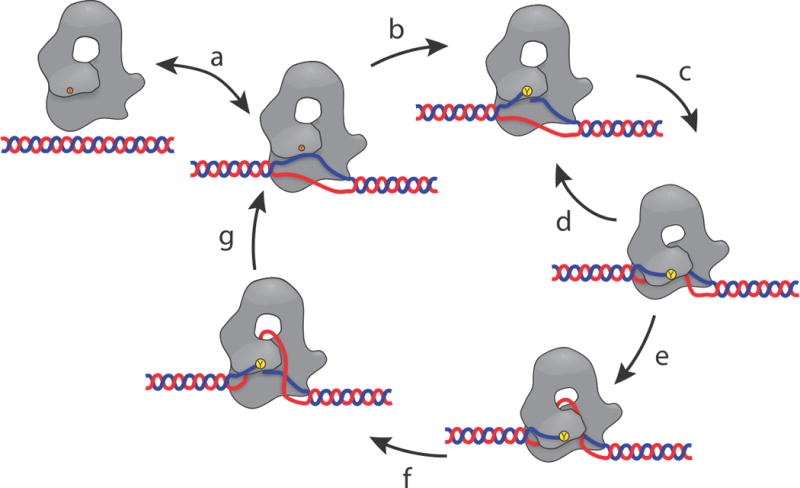
a) The enzyme binds to a ssDNA region, normally created by negatively supercoiled DNA. b) The protein forms a transient 5′-phosphotyrosine linkage with one of the DNA strands, creating a single stranded break in the DNA backbone. c) Domain III moves to attempt capture the intact ssDNA for passage through the transient DNA break. d) In many cases the enzyme fails to capture the intact ssDNA and domain III returns to its previous position. Many cycles of attempted capture may occur while waiting for the DNA to be in the correct orientation. This secondary cycle, (c – d) represents the multiple attempts at DNA relaxation that are the source of the pauses between relaxation events. e) ssDNA is successfully captured by domain III, allowing the intact DNA strand to pass through the DNA break and into the central hole in the protein. f) Domain III returns to its closed position and re-ligates the ssDNA break. g) The captured ssDNA is released, and the DNA either stays bound to the protein or unbinds. The overall cycle leads to a change in linking number by one. The revised aspects of this mechanism are the sliding of domain III past domain I and the sub-cycle of attempted capture of the ssDNA (c – d).
Type IA topoisomerases are torque-driven and non-reversible molecular machines, a fact which predicts a difference in the protein conformation before and after a relaxation event. The clear fluorescence asymmetry seen in the averaged trace suggests an asymmetry in the protein conformation on either side of a strand passage event (Fig. 3c), for instance a conformational difference between the cleavage and religation states, but further experiments are necessary to support this. Overall, based on the observations described here we can propose that DNA relaxation is described best as sliding of the domains past each other to create a gate, capture the second DNA strand, and trap it inside the protein.
This proposed mechanism is supported further by experiments performed in the absence of magnesium, which is a necessary cofactor for type IA topoisomerase activity and plays a role in stimulating DNA cleavage, active site conformational changes, and re-ligation19,20. Notably, no nicking events were observed on the DNA when magnesium was absent. This is significant, since magnesium has previously been shown to be unnecessary for cleavage, but necessary for re-ligation19. In the experimental conditions without magnesium, if the DNA had been cleaved, religation of the cleaved substrate would not have been possible, resulting in nicked DNA upon EcTopoI dissociation. In MT experiments nicking of DNA causes immediate relaxation and renders the DNA molecule impervious to supercoiling. This was not seen, as the analyzed DNA substrates were capable of being supercoiled in subsequent experiments with magnesium. Therefore, we did not observe cleavage in the absence of magnesium (nicking), even though protein binding and unbinding was observed.
In the absence of magnesium, unimodal fluorescence distributions were observed, which contrast with the broader, multiple Gaussian distributions seen in the presence of magnesium. When magnesium was added to the sample, the multiple distributions, including high intensity fluorescence, were recovered, indicating that magnesium is the triggering factor for the observed fluorescence spikes. This leads us to conclude that the events leading to PIFE spikes do not occur in the absence of magnesium, and that the spikes are not the result of protein binding to the DNA alone, but instead are the consequence of events that necessitate the presence of magnesium, potentially DNA cleavage or active site conformational changes. The Gaussian fluorescence distributions in the absence of magnesium further support the conclusion that the fluctuations in fluorescence intensity observed when magnesium is present are a result of domain movements, since protein sliding in the ssDNA bulge would be possible without magnesium, but was not observed. With regards to the frequent binding/unbinding events in the presence of magnesium, one possible explanation is that the absence of magnesium could affect the conformation of the ssDNA bulge, making it difficult for the protein to remain bound to it. Alternatively, it could indicate that cleavage/religation of DNA or active site conformation is closely associated with stable DNA binding.
PIFE spikes occur even during the pauses between relaxation events, when the protein is bound but DNA relaxation is not occurring, and depend on the presence of magnesium, with only 11% of spikes correlated to relaxation events. This suggests that the uncorrelated spikes represent conformational changes triggered by the presence of magnesium, but in these cases the protein fails to pass the intact strand into its interior, i.e., the spikes correspond to attempts to capture the second DNA strand for passage through the transient DNA break. When the intact DNA strand is successfully captured, the catalytic cycle continues and relaxation occurs. In the unsuccessful cases, cleavage occurs and the domains move, but no strand is captured and relaxation does not occur (Fig. 5). The data also indicate that successful events are a minority of all events and occur randomly, which is reflected in the variable length of the pauses between successful relaxation events. Whereas relaxation pauses for the optimal substrate, negatively supercoiled DNA, are less frequent and have a shorter mean lifetime of 5.6 ± 1.3 sec, on other substrates EcTopoI may require more attempts to achieve a successful relaxation event, leading to a lengthening of the pausing time, as has been observed before21 (Supplementary Table 2).
The relative orientation of the protein and DNA may be crucial for successful relaxation and the unsuccessful strand passage attempts may be the consequence of the DNA adopting conformations not optimal for capture and passage. This offers an explanation for why multiple relaxation cycles can occur in quick succession, as after a successful strand passage cycle the DNA may be well positioned for further events. In this scenario pauses, which occur due to unsuccessful strand capture events, are a consequence of suboptimal relative positioning of the enzyme and the DNA.
Our observations indicate a mechanism of strand passage for type IA topoisomerases where the domains move past each other, not away from each other. It is possible that creation of the transient phosphotyrosine bond triggers the domain movements and implies a close synergy between DNA cleavage, domain movement, and strand passage. Furthermore, the experiments show that a dominant feature of the relaxation cycle is a sub-cycle where the protein attempts to capture the intact DNA strand many times before succeeding (Fig. 5).
The combination of two orthogonal techniques, MT and PIFE/TIRF, allowed us to study a complex reaction catalyzed by a molecular machine at the single molecule level and uncover key dynamical features that could not be learned from either bulk experiments or single molecule experiments using one reporting channel. The experiments provide direct visualization of a molecular machine making multiple attempts before achieving successful catalysis, since every attempt does not lead to a successful event. This indicates that the dynamics of the reactions are the result of a complex interplay between successful and unsuccessful strand passage events. These observations are likely to extend to type II topoisomerases, which also employ an enzyme-bridge strand passage mechanism, require the presence of magnesium, and where domain movements in the absence of catalysis have been observed22–24. Furthermore, other molecular machines that exhibit pausing behavior may employ a similar strategy, particularly in the cases where a mechanical event is not tightly coupled to a chemical event. For type IA topoisomerases, successful strand passage only happens after several unsuccessful coupled DNA strand cleavage and protein conformational changes. Strand passage is the result of both a successful chemical (DNA cleavage) and mechanical (strand passage) step, which occurs following several chemical steps that do not lead to a mechanical step.
Online Methods
Combined MT-TIRF instrument
The MT-TIRF microscope was built by mounting a compact MT setup25 on top of an objective-type TIRF microscope (IX81, Olympus, Tokyo, Japan) using an oil-immersion 100X, 1.4 NA objective (Supplementary Fig. 1). Fine control of objective height was achieved with a piezo objective positioner (NV40/1CLE, Piezosystem Jena, Hopedale, MA) and temperature was kept at 37°C with an objective heater (OH-XX, 20/20 Technology, Inc.). To illuminate the flow cell and image the magnetic beads, the MT utilizes a near infrared 735 nm LED (JET-735-05, Roithner LaserTechnik GmbH, Vienna, Austria) located just above the magnets and attached to the magnet holder. The magnets can be rotated to introduce supercoils and translated vertically to vary the force exerted on the magnetic beads by using a dual motion linear and rotational actuator (LR43HH4AC-05-001, Haydon Kerk, Waterbury, CT). The motor was built with a custom shaft for mounting the magnet/LED holder, which was machined in house. Cylindrical N52 neodymium magnets (1 inch diameter × 1/2 inch height) were mounted with opposing polarity on either side of a 3 mm diameter hole, which allows light from the LED to reach the sample. The magnetic field was further enhanced by use of 0.05 inch thick moly-permalloy disks on top of the magnets26. The motor and magnet/LED holder were mounted on a custom machined base plate and attached to the microscope using 4 posts. Power was supplied to the LED via a slip-ring (503-0200, Orbex Group, Fremont, CA). Two stepper motor controllers (TIMS 201, Jova Solutions, Berkeley, CA) run the motors and an LED evaluation board (LM3404EVAL/NOPB, Texas Instruments, Dallas, TX), modified with a 0.47 ohm resistor, provided power to the LED.
A 561 nm laser (85-YCA, 75 mW, Melles Griot, Rochester, NY) at the TIR angle was used to generate an evanescent field to illuminate the fluorescently labeled DNA substrate located < 100 nm above the coverslip surface27. The light from the LED and the laser were separated using a 721 nm short pass dichroic mirror (FF721-SDi01, Semrock, Rochester, NY) mounted in a dual camera port (U-DPCAD, Olympus). An ImagEM EMCCD (C9100-13, Hamamatsu Photonics, Japan) was used to measure the fluorescence signal at 16.6 frames per second and a CMOS monochrome camera (PL-B761U, PixeLink, Ottawa, Canada) was used to track the bead position at 166 frames per second. Light cross-contamination between the two cameras was prevented by using a 710 nm long-pass filter (XVL0710, Asahi Spectra, Torrance, CA) placed just after the LED and a 694 nm short-pass filter (FF01-694/SP-25, Semrock) located after the dichroic mirror (Supplementary Fig. 1). The two instruments were controlled separately by purpose-written software running on the same computer. Fluorescent beads were used to establish the spatial relation between the two cameras, so that the location of the beads in the MT channel can be found in the TIRF channel.
Protein purification
E. coli Topoisomerase I (EcTopoI) was purified as previously described28 and stored at −20°C at 2 mg/mL in storage buffer (50 mM Tris-HCl pH8, 1mM DTT, 50 mM KCl, 50% glycerol). To create fluorescently labeled EcTopoI, the purified protein was dialyzed into de-gassed labeling buffer (50 mM Tris-HCl pH 7, 150 mM KCl, 1 mM EDTA). Protein was then incubated overnight at 4°C with 10-fold molar excess of Alexa Fluor 555 maleimide (A20346, Thermo Fisher, Waltham, MA). After incubation, protein and dye were purified over a Sephacryl S-100 resin (17-1194-01, GE Healthcare Life Sciences) to remove excess dye. After purification mass spectrometry confirmed primarily single labeling of the protein, although the location of the label is unknown. Bulk activity assays confirmed relaxation activity for Alexa 555 labeled EcTopoI, although at approximately five times lower rate than wild type EcTopoI. For the experiments, EcTopoI was first diluted 20-fold in buffer (50 mM Tris-HCl pH8, 150 mM KCl, 1mM EDTA) and stored on ice until needed. Immediately prior to flowing protein into the flow cell, EcTopoI was further diluted to 40 nM in the single molecule fluorescence oxygen scavenging buffer, described below. For experiments with Alexa 555 labeled EcTopoI, the protein was diluted to 2 nM.
DNA substrate preparation
DNA substrates were created by modifying a previous procedure21 to create asymmetric DNA molecules labelled with digoxigenin-11-dUTP alkali stable (dig) and Biotin-16-dUTP (bio) on either end (11093088910 and 11093070910, Roche, Indianapolis, IN). In addition, a single stranded DNA (ssDNA) bulge was used for targeting of EcTopoI and was located near the dig-labelled end of the DNA molecule to ensure it was in close proximity to the coverslip (≈50 nm) and, hence within the TIRF field.
To reduce the variation in height from random incorporation of functionalized dUTPs, 250 base pair (bp) “handles” with 50% dUTP labeling were created using PCR. The ssDNA bulge was created by annealing two oligonucleotides (Integrated DNA Technologies, Coralville, IA) that shared 100% sequence identity, except for the overhanging ends and a 22nt bulge region in the middle of one of the oligonucleotides, which also included a Cy3 fluorophore 5nt from the 3′ end of the bulge, thus providing a 17nt binding region (Fig. 1b). Oligonucleotide with 22nt Cy3 bulge (phosphorylated): 5′ – /5Phos/GACAGATTTCGCCCACTACGTGATCGAATCGGCATTGCGCAAACCAAG /iCy3/ ACAGCCCAAGCTGAGCGTACGCTGCTTTAATGCGG – 3′ Oligonucleotide for opposite bulge (with XmaI and SacI overhangs): 5′ – CCGGCCGCATTAAAGCAGCGTACGCTCAGCTTGGCGATTCGATCACGTAGTGGGCGAAA TCTGTCAGCT – 3′
The optimal size of the bulge (22nt) was arrived at experimentally; a 27nt bulge (22nt binding region) was found to be too large to produce consistent PIFE and a 17nt bulge (12nt binding region) displayed no relaxation activity, although it showed protein binding (data not shown). The annealed bulge was ligated to a PCR-produced 162bp fragment of plasmid pMal-pIII digested with SacI (all enzymes from New England Biolabs, Ipswich, MA) and gel purified (QIAquick Gel Extraction Kit, Qiagen, Valencia, CA). The 162bp fragment was designed to act as a spacer between the dig handle, attached to the coverslip, and the Cy3 labeled bulge, placing it within the evanescent wave ≈50 nm away from the coverslip (162bp + 31bp of oligo). The combined 162bp/Cy3 bulge DNA and the dig handle were both digested with ApaI, ligated together, and then gel purified. Plasmid pMal-pIII was modified to extend its length from 6.7 kb to 8.9kb and then digested with NotI and XmaI. The bio handle was also digested with NotI. The dig handle/162bp/Cy3 bulge fragment was phosphorylated and ligated to the digested 8.9kb plasmid and the bio handle overnight at 16°C. The resulting 9.5 kb DNA was used for the experiments (Fig. 1c).
Bulk Fluorescence
EcTopoI and the annealed Cy3 bulge were tested in bulk using an ISS PC1 spectrofluorometer (ISS, Champaign, IL) to confirm the presence of PIFE for the different size bulges. Different size annealed DNA Cy3 bulges at 10 nM were mixed with 0 to 40 nM of EcTopoI in de-gassed buffer (50 mM HEPES pH 7, 50 mM KCl, 1 mM MgCl2, 10% glycerol, 0.1 mg/mL BSA) and placed in a 5 mm × 10 mm quartz cuvette. Spectra were excited at 520 nm and emission was collected from 550–700 nm at room temperature.
Flow cell construction
Flow cells were created by adapting a method described before29, but instead of using a syringe pump, we used a ≈100 μL reservoir at one inlet to allow for gentle capillary flow. The flow speed could be increased by using a paper tissue to absorb liquid at the outlet. Coverslips and glass slides were cleaned by sonication as described29, but using only 10% Alconox, 1 M KOH, and 100% Ethanol. A cleaned coverslip was coated with cleaned streptavidin coated magnetic beads (described below) that had been diluted 10-fold in 100% ethanol. Subsequently, the coverslip was heated on a 80°C heat plate for 15 seconds to adhere the beads to the coverslip for use as reference beads during the experiments30. Sheep anti-Digoxigenin (anti-dig) Fab fragments (11214667001, Roche) were non-covalently attached to the bead-coated coverslips by overnight incubation with 0.2 mg/mL anti-dig at 4°C. Prior to attachment of the DNA substrates, the flow cell was passivated with 10 flow-cell volumes (FCV) of 0.4 mg/mL Bovine Serum Albumin (BSA) and incubated for at least 5 minutes. For the experiments, 1 μm streptavidin magnetic beads (S1420S, New England Biolabs) were used due to their lack of intrinsic fluorescence at 561 nm. Before use, the 4 mg/mL streptavidin magnetic beads were cleaned by diluting 10-fold in phosphate buffered saline (PBS) and vortexing for at least 1 min. The beads were then pelleted with a magnet and the process repeated 3 times before finally resuspending in 6 times excess of 0.4 mg/mL BSA solution for a final bead concentration of 0.6 mg/mL and stored at 4°C.
Two tubes containing 2 μL of 0.5 ng/μL 9.5 kb DNA substrate (with incorporated 22nt Cy3 bulge) were incubated with 2 μL of 0.6 mg/mL cleaned beads for 5 minutes at room temperature. The two reactions were stopped by the addition of a total of 45 μL PBS to both, for a final combined volume of ≈55 μL (≈2 FCV). The diluted beads and DNA were flowed into the flow cell using a paper tissue to pull most of the sample through the flow cell. An additional 55 μL of PBS was put in the reservoir, to prevent drying. The DNA and beads were incubated in the flow cell for 10 minutes at RT before putting the flow cell on the microscope and lowering the magnet to remove any beads that were not attached to the surface via the DNA substrate. This process was repeated as necessary, rinsing the flow cell with 0.4 mg/mL BSA before applying a new sample of beads and DNA. To maintain a constant 37°C temperature in the flow cell during the experiment, the objective was kept at 37°C.
DNA selection
A prepared flow cell was placed in the MT-TIRF microscope and the beads were examined using only the MT illumination to minimize photobleaching of the fluorophore. Once a reference bead and a DNA-attached bead were located in close proximity, the magnet was turned to assess whether the DNA was intact (a single supercoilable tether) and also whether the length was within the expected length of ≈2.5 μm at 1 pN force. Once a suitable molecule was identified, a lookup table to calculate the height of the tethered bead above the cover slip, was collected as described before25.
Force calibration and DNA characterization
Force was calibrated by using the Brownian motion of the tethered bead as described before18,21,31. Subsequently, a linking number vs. extension plot was measured at different forces, where the magnet was turned and bead height recorded for each turn. The resulting curves show a typical shape for a single intact DNA tether18. For the experiments, a force of 1.0 pN was used. The linear portion of the linking number vs. extension plot at 1pN was later used to convert the collected data from extension (microns) into excess linking number (turns) for analysis.
Oxygen scavenging
To increase the lifetime of the fluorophores, an oxygen scavenging system (OSS) was used consisting of 0.3 mg/mL pyranose oxidase from Coriolus sp. (P4234, Sigma-Aldrich), 90 U/mL catalase purified from human erythrocytes (C3556, Sigma-Aldrich), 0.5% (w/v) D-glucose, and 143 mM beta-mercaptoethanol. Pyranose oxidase was used due to its pH stability32, since we found that glucose oxidase inhibited EcTopoI over time due to acidification. To ameliorate problems caused by nuclease contamination in commercial pyranose oxidase, similar to the contamination observed in glucose oxidase33, we further purified the pyranose oxidase by size exclusion chromatography using an S-300 resin (50nM Tris-HCl pH 7, 100 mM KCl). Catalase from human erythrocytes was used based on the report that it exhibits almost no nuclease activity relative to other commercial catalases33. The OSS was combined with EcTopoI single molecule buffer (50 mm Tris-HCl pH 7, 120 mM NaCl, 1 mM MgCl2, 1 mg/mL BSA), for a buffer we called single molecule fluorescence buffer. For experiments in the absence of magnesium, the buffer was altered by the removal of MgCl2 and the addition of 1 mM EDTA.
Data collection
After identification of a suitable DNA tether and collection of an extension vs. linking number curve in PBS, 4 FCV of single molecule fluorescence buffer were flowed in using capillary flow. In some cases a paper tissue was used to remove buffer that had pooled at the outlet. The flow cell was allowed to stabilize for 15 minutes before beginning data collection. Control data, with no protein present, were collected first, with the MT recording the extension of the DNA tether. The 561 nm laser was then turned on and TIRF data collection begun, which records movies that are later converted into integrated fluorescence intensity values using purpose-written software. While recording both data channels, 30 positive supercoils (+sc) were introduced by rotating the magnet. After waiting a brief time (≈15 sec), the magnet was turned in the opposite direction to bring the excess linking number back to 0. Data collection was stopped and the laser turned off to preserve fluorophore lifetime. EcTopoI was diluted to 40 nM with 4 FCV of single molecule fluorescence buffer and passed through the flow cell with gravity flow. After ≈5 min the magnetic bead had returned to its original position after deflection due to buffer flow. Data collection was begun again in the above order, followed by the introduction of ≈30 +sc. Data collection continued until the tether had been fully relaxed, the fluorophore was bleached, or the tether was nicked, therefore losing its ability to supercoil. Bleaching time for the Cy3 labeled DNA varied greatly, ranging from almost instantaneous bleaching to longer than 10 min. An example of a bleaching event that occurred after ≈2 min is shown in Supplementary Figure 2b. If the tether reached full relaxation, ≈30 +sc were introduced again. For magnesium-free experiments, the procedure above was modified to include a single molecule fluorescence oxygen scavenging buffer where MgCl2 was replaced by EDTA. This buffer was flowed into the flow cell and control data were collected as described above. Then the magnesium-free buffer was used to dilute EcTopoI to 40 nM and that mixture was flowed in and data collected. Finally, 40 nM EcTopoI in regular single molecule fluorescence oxygen scavenging buffer (with MgCl2 and no EDTA) was flowed in and data were collected. For experiments with fluorescently labeled EcTopoI, the Cy3 label on the 22nt bulge DNA was bleached after identifying the exact location of the bulge; this location was then monitored for the appearance of fluorescent protein. After flowing in 2nM of EcTopoI labeled with Alexa Fluor 555, the DNA was supercoiled and the presence of labeled EcTopoI at the DNA bulge was observed with the 561 nm laser. Data collection was continued until relaxation was observed.
Data analysis
After data collection, the MT and fluorescence data sets were combined using their recorded internal time stamps (Fig. 2c). The elapsed time between 233 relaxation events was extracted, binned in 2 second intervals, and fitted to a simple exponential decay function, giving a mean-lifetime of 9.3 ± 0.2 seconds (Fig. 3b). The total relaxation rates of 23 traces (n=12 molecules) were also fitted to exponential decay functions, giving a mean relaxation rate of 0.22 ± 0.07 linking numbers/second (Supplementary Table 2). To determine the pause lifetime and relaxation rate, relaxation traces without good fluorescence data were also included since these statistics are independent of the fluorescence data.
For the combined MT-PIFE experiments, data were collected from a total of 33 individual DNA molecules, with 9 used for data analysis (Supplementary Table 1). For the fluorescence data analysis, 10-point boxcar averages of the raw fluorescence data were used. The supercoiled fluorescence intensity prior to addition of EcTopoI was used to determine the base level of fluorescence and to set a cut-off fluorescence intensity (Supplementary Fig. 4 and Supplementary Note 1). Sustained fluorescence greater than the cut-off was determined to be PIFE, representing the protein bound state. As expected, when no EcTopoI is present the fluorescence signal displays a narrow Gaussian distribution of values, which is apparent in a histogram of the data (Fig. 2a). Upon addition of protein, we observed an expected increase in fluorescence intensity due to PIFE and an increase in the variance of the fluorescence signal, which is manifested as a broadening of the distribution9. We also saw a more asymmetrical distribution of intensities around the mean, particularly a shoulder appearing at higher fluorescence intensities, so that the signal no longer fit to a single Gaussian distribution (Fig. 2b).
Traces with fluorescently labeled EcTopoI were aligned in time to observe the correlation between the presence of protein and relaxation of the DNA (n=3 molecules) (Supplementary Fig. 3 and Supplementary Table 1).
When fluorescence data from DNA substrates in the presence of EcTopoI were measured, but without magnesium, no asymmetry or high fluorescence intensity spikes were observed (Fig. 4b). However, addition of protein with magnesium buffer shifted the fluorescence distribution back to the asymmetry typically observed (Fig. 4c). We also observed frequent unbinding events when magnesium was not present in the buffer, with both bound and unbound fluorescence intensities described by Gaussian distributions (Supplementary Fig. 6). The experiments with and without magnesium were conducted on 17 individual DNA molecules, with 9 used for data analysis (Supplementary Table 1 and Supplementary Note 1).
To recognize the PIFE spikes, a cut-off intensity value was calculated as described above and the local maxima were extracted using a 5-point window Origin (OriginLab, Northampton, MA). In total, 734 PIFE spikes were identified using this method. The elapsed times between PIFE spikes from the local maxima dataset were calculated, binned in 600 ms intervals, and fitted to a simple exponential decay function, giving a mean-lifetime of 1.08 ± 0.02 seconds (Fig. 3a). Out of 93 relaxation events, 84 PIFE spikes within the coincidence time window were identified or 90% of the total (Supplementary Fig. 5 and Supplementary Note 1). Only 9 relaxation events did not show a coincident PIFE spike or the spike was too weak or noisy to be confident of the assignment (Supplementary Fig. 2a, yellow triangle). Of the 734 PIFE spikes identified, only 84 corresponded to relaxation events or 11%.
Statistical Analysis
Statistical significance for averaging of the PIFE spikes was analyzed by standard deviation (σ = 0.20) and bootstrap aggregating of 73 events. For more details of the statistical analysis see Supplementary Note 1.
Data Availability
Source data for Figure 2 and Figure 3 are available with the paper online. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank members of the Mondragón and Marko laboratories for discussions and assistance and Ksenia Terekhova for early work on the project. Research was supported by the NIH (A.M., grant R01-GM051350 and J.F.M., grants R01-GM105847 and U54-CA193419 (CR-PS-OC) and subcontract to U54DK107980) and by the NSF (J.F.M., grant DMR-1206868). K.H.G. was supported by a Dr. John N. Nicholson Fellowship and a NRSA pre-doctoral training grant (T32-GM008382). We acknowledge the help from the Keck Biophysics Facility and the Northwestern University Instrument Shop.
Footnotes
Author Contributions:
K.H.G., J.F.M., and A.M. planned the experiments, designed the MT-TIRF microscope, analyzed data, and edited the manuscript. K.H.G. and A.M. constructed the MT-TIRF microscope and wrote the manuscript. A.M. wrote the MT-TIRF software. K.H.G. prepared reagents and collected data.
Competing Financial Interests:
The authors declare no competing financial interests.
References
- 1.Schoeffler AJ, Berger JM. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q Rev Biophys. 2008;41:41–101. doi: 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- 2.Baker NM, Rajan R, Mondragon A. Structural studies of type I topoisomerases. Nucleic Acids Res. 2009;37:693–701. doi: 10.1093/nar/gkn1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lima CD, Wang JC, Mondragon A. Three-dimensional structure of the 67K N-terminal fragment of E. coli DNA topoisomerase I. Nature. 1994;367:138–46. doi: 10.1038/367138a0. [DOI] [PubMed] [Google Scholar]
- 4.Perry K, Mondragon A. Structure of a complex between E. coli DNA topoisomerase I and single-stranded DNA. Structure. 2003;11:1349–1358. doi: 10.1016/j.str.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Tan K, et al. Structural basis for suppression of hypernegative DNA supercoiling by E. coli topoisomerase I. Nucleic Acids Res. 2015;43:11031–46. doi: 10.1093/nar/gkv1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Changela A, DiGate RJ, Mondragon A. Structural studies of E. coli topoisomerase III-DNA complexes reveal a novel type IA topoisomerase-DNA conformational intermediate. J Mol Biol. 2007;368:105–18. doi: 10.1016/j.jmb.2007.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Z, Mondragon A, DiGate RJ. The mechanism of type IA topoisomerase-mediated DNA topological transformations. Mol Cell. 2001;7:301–7. doi: 10.1016/s1097-2765(01)00178-2. [DOI] [PubMed] [Google Scholar]
- 8.Dekker NH, et al. The mechanism of type IA topoisomerases. Proc Natl Acad Sci U S A. 2002;99:12126–31. doi: 10.1073/pnas.132378799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang H, Kim H, Myong S. Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc Natl Acad Sci U S A. 2011;108:7414–8. doi: 10.1073/pnas.1017672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stennett EM, Ciuba MA, Lin S, Levitus M. Demystifying PIFE: The Photophysics Behind the Protein-Induced Fluorescence Enhancement Phenomenon in Cy3. J Phys Chem Lett. 2015;6:1819–23. doi: 10.1021/acs.jpclett.5b00613. [DOI] [PubMed] [Google Scholar]
- 11.Lang MJ, Fordyce PM, Engh AM, Neuman KC, Block SM. Simultaneous, coincident optical trapping and single-molecule fluorescence. Nat Methods. 2004;1:133–9. doi: 10.1038/nmeth714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comstock MJ, et al. Protein structure. Direct observation of structure-function relationship in a nucleic acid-processing enzyme. Science. 2015;348:352–4. doi: 10.1126/science.aaa0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordova JC, Das DK, Manning HW, Lang MJ. Combining single-molecule manipulation and single-molecule detection. Curr Opin Struct Biol. 2014;28:142–8. doi: 10.1016/j.sbi.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 14.Graves ET, et al. A dynamic DNA-repair complex observed by correlative single-molecule nanomanipulation and fluorescence. Nat Struct Mol Biol. 2015;22:452–7. doi: 10.1038/nsmb.3019. [DOI] [PubMed] [Google Scholar]
- 15.Fan J, Leroux-Coyau M, Savery NJ, Strick TR. Reconstruction of bacterial transcription-coupled repair at single-molecule resolution. Nature. 2016;536:234–7. doi: 10.1038/nature19080. [DOI] [PubMed] [Google Scholar]
- 16.Kemmerich FE, et al. Simultaneous Single-Molecule Force and Fluorescence Sampling of DNA Nanostructure Conformations Using Magnetic Tweezers. Nano Lett. 2016;16:381–6. doi: 10.1021/acs.nanolett.5b03956. [DOI] [PubMed] [Google Scholar]
- 17.Kirkegaard K, Wang JC. Bacterial DNA topoisomerase I can relax positively supercoiled DNA containing a single-stranded loop. J Mol Biol. 1985;185:625–37. doi: 10.1016/0022-2836(85)90075-0. [DOI] [PubMed] [Google Scholar]
- 18.Strick TR, Allemand JF, Bensimon D, Bensimon A, Croquette V. The elasticity of a single supercoiled DNA molecule. Science. 1996;271:1835–7. doi: 10.1126/science.271.5257.1835. [DOI] [PubMed] [Google Scholar]
- 19.Sissi C, Palumbo M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 2009;37:702–11. doi: 10.1093/nar/gkp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt BH, Burgin AB, Deweese JE, Osheroff N, Berger JM. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature. 2010;465:641–4. doi: 10.1038/nature08974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terekhova K, Gunn KH, Marko JF, Mondragon A. Bacterial topoisomerase I and topoisomerase III relax supercoiled DNA via distinct pathways. Nucleic Acids Res. 2012;40:10432–40. doi: 10.1093/nar/gks780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins TR, Hammes GG, Hsieh TS. Analysis of the eukaryotic topoisomerase II DNA gate: a single-molecule FRET and structural perspective. Nucleic Acids Res. 2009;37:712–20. doi: 10.1093/nar/gkn1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Garcia B, et al. Topoisomerase II minimizes DNA entanglements by proofreading DNA topology after DNA strand passage. Nucleic Acids Res. 2014;42:1821–30. doi: 10.1093/nar/gkt1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basu A, Parente AC, Bryant Z. Structural Dynamics and Mechanochemical Coupling in DNA Gyrase. J Mol Biol. 2016;428:1833–45. doi: 10.1016/j.jmb.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gosse C, Croquette V. Magnetic tweezers: micromanipulation and force measurement at the molecular level. Biophys J. 2002;82:3314–29. doi: 10.1016/S0006-3495(02)75672-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meeker DC. Finite Element Method Magnetics. Build. (4.2) 2013 Aug 25; ( http://www.femm.info 2013)
- 27.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003;361:1–33. doi: 10.1016/s0076-6879(03)61003-7. [DOI] [PubMed] [Google Scholar]
- 28.Perry K, Mondragon A. Biochemical characterization of an invariant histidine involved in Escherichia coli DNA topoisomerase I catalysis. J Biol Chem. 2002;277:13237–45. doi: 10.1074/jbc.M112019200. [DOI] [PubMed] [Google Scholar]
- 29.Joo C, Ha T. Preparing sample chambers for single-molecule FRET. Cold Spring Harb Protoc. 2012;2012:1104–8. doi: 10.1101/pdb.prot071530. [DOI] [PubMed] [Google Scholar]
- 30.Burnham DR, De Vlaminck I, Henighan T, Dekker C. Skewed brownian fluctuations in single-molecule magnetic tweezers. PLoS One. 2014;9:e108271. doi: 10.1371/journal.pone.0108271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skoko D, Wong B, Johnson RC, Marko JF. Micromechanical analysis of the binding of DNA-bending proteins HMGB1, NHP6A, and HU reveals their ability to form highly stable DNA-protein complexes. Biochemistry. 2004;43:13867–74. doi: 10.1021/bi048428o. [DOI] [PubMed] [Google Scholar]
- 32.Swoboda M, et al. Enzymatic oxygen scavenging for photostability without pH drop in single-molecule experiments. ACS Nano. 2012;6:6364–9. doi: 10.1021/nn301895c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Senavirathne G, et al. Widespread nuclease contamination in commonly used oxygen-scavenging systems. Nat Methods. 2015;12:901–2. doi: 10.1038/nmeth.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Figure 2 and Figure 3 are available with the paper online. The data that support the findings of this study are available from the corresponding author upon reasonable request.