

Abstract
Nucleotide excision repair (NER) is a generalized DNA repair mechanism that is capable of removing a wide variety of DNA lesions induced by physical or chemical insults. UvrD, a member of the helicase SF1 superfamily and plays an essential role in the bacterial NER pathway by unwinding the duplex DNA in a 3′ to 5′ direction to displace the lesion-containing strand. In order to achieve conditional control over NER, we report the first generation of a light-activated DNA helicase. This was achieved through a site-specific insertion of a genetically encoded lysine at a crucial position in the ATP binding pocket of UvrD. The resulting caged enzyme was completely inactive in several functional assays. Moreover, enzymatic activity of the optically triggered UvrD was comparable to that of the wild-type protein, demonstrating excellent OFF to ON switching of the helicase. The developed approach provides optical control of NER, thereby laying a foundation for the regulation of ATP-dependent helicase functions in higher organisms. In addition, this methodology may be generally applicable to the light-activation of a wide range of other ATPases.
TOC image
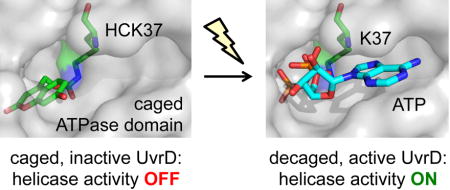
Using genetic code expansion, an optical switch was engineered into the DNA helicase UvrD. A hydroxycoumarin-caged lysine (HCK) was inserted into the active site of the UvrD ATPase domain, rendering the enzyme inactive, until irradiation removes the caging group and generates an active, wild-type helicase.
Manuscript
UvrD is a DNA-dependent ATPase and helicase that belongs to the helicase SF1 superfamily and catalyzes the unwinding of duplex DNA in a 3′ to 5′ direction.[1] UvrD plays essential roles in both methyl-directed mismatch repair and nucleotide excision repair (NER) in bacteria[2] and corresponding functions show a high degree of conservation in yeast, and human cells. NER is a generalized DNA repair mechanism that is capable of removing a wide variety of DNA lesions resulting from physical agents, such as sunlight or chemicals, such as cisplatin or polycyclic hydrocarbons. In bacteria, UvrA and UvrB recognize and bind the DNA lesion. Once UvrB is loaded at the site of damage, it helps to recruit UvrC to perform dual incisions on both sides of the lesion, leaving a UvrBC-DNA post-incision complex. UvrD and DNA polymerase 1 work in concert to turnover the UvrBC-DNA complex and UvrD unwinds the doubly nicked duplex DNA and displaces the lesion-containing strand (Figure 1).[3] DNA polymerase I is then able to undergo repair synthesis to generate a repair patch that is ligated during the final step.
Figure 1.
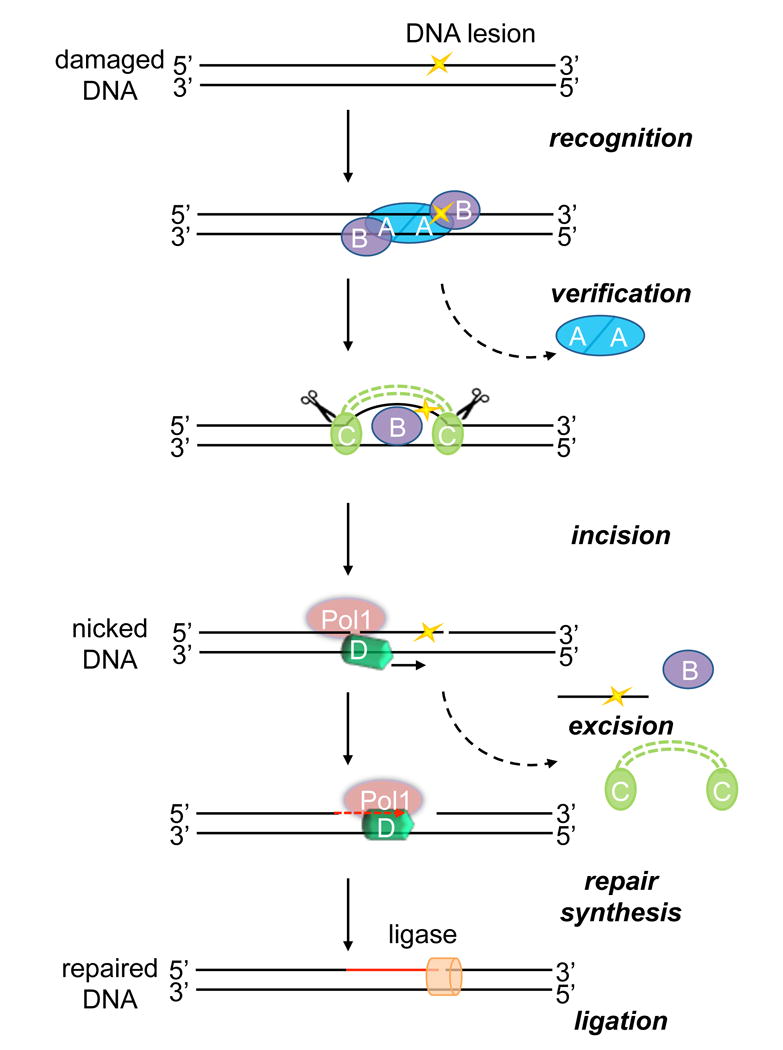
Schematic showing the role of UvrD in bacterial NER. During damage detection, UvrA and UvrB proteins bind at the site of damaged DNA. Once UvrB is loaded at the site of damage during the verification step, UvrA dissociates and UvrC is recruited and produces two incisions on the damaged strand both 5′ and 3′to the damaged nucleotide. The dual action of UvrD and Pol1 are necessary to remove the damaged oligonucleotide and carry out repair synthesis using the complementary strand as a template. DNA ligase seals the newly made repair patch.
Helicase defects have been linked to several human diseases including cancer and genetic disorders.[4] However, determining the molecular basis of helicase function defects that cause the pathophysiology associated with human diseases has been challenging, due to the involvement of a complex network of interconnected roles of DNA helicases and their protein interactions.[4] For example, XPD and XPB are key helicases that play important roles in human NER and are part of the seven-member TFIIH core complex.[5] Since many disease-causing mutations in either XPD or XPB affect the stability of TFIIH,[5] having the ability to directly initiate helicase activity of either protein within the TFIIH complex would be most desirable. Therefore, as proof of principle, we aimed to develop a strategy for conditional control of helicase function. In order to provide a general approach for the optical activation of helicase function, we photocaged a key amino acid residue that is conserved throughout the helicase protein family. The precise temporal control provided through photocaging[6] may allow assessment of heretofore intractable problems, such as how helicase-dependent DNA repair pathways are coordinated.
To engineer a light-activated helicase, we utilized the Methanosarcina barkeri pyrrolysyl tRNA synthetase/tRNACUA pair,[7] which we have previously applied to the optical control of transcription, nuclear translocation, protein folding, DNA recombination, genome editing, and kinase function.[8] The BHCKRS/tRNACUA pair directs the incorporation of hydroxycoumarin lysine (HCK, Figure 2A) in response to an amber stop codon, TAG.[8c] The genetically encoded coumarin lysine has been successfully applied as both a fluorescent cellular probe and a light-activated caging group for the optical activation of protein function. Here we show, for the first time, that site-specific incorporation of HCK at a key lysine residue allows for light-activated DNA helicase function through optical control of ATPase activity.
Figure 2.
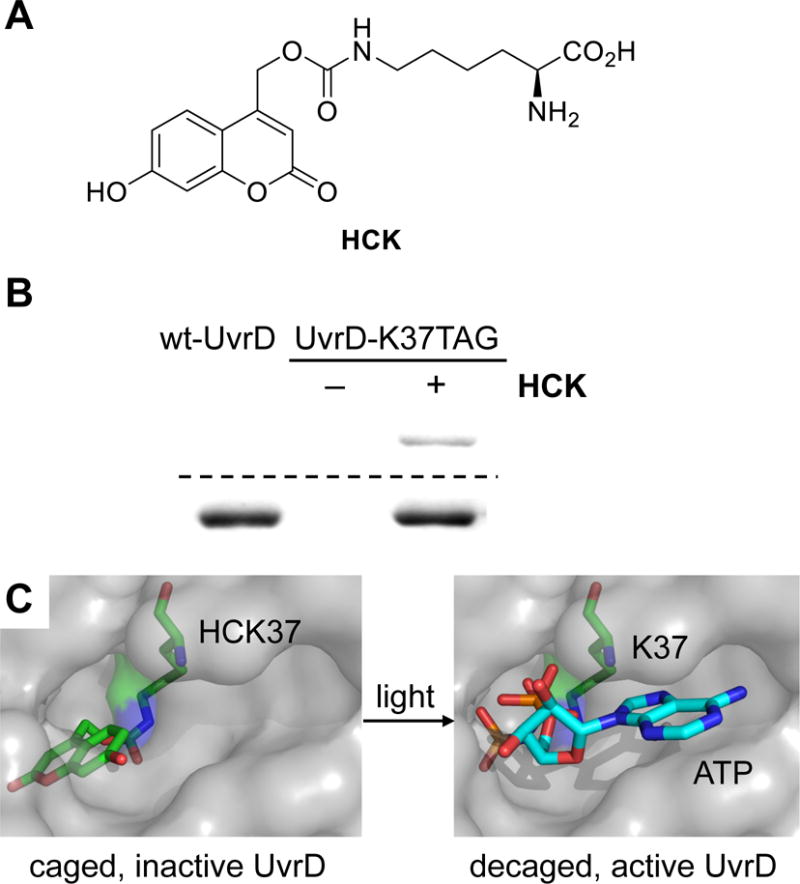
Site-specific incorporation of coumarin lysine into UvrD at residue K37. A) Structure of photocaged lysine HCK. B) SDS-PAGE analysis of wild-type UvrD and photocaged UvrD (UvrD-HCK) expressed in E. coli. The gel was stained with Coomassie blue (bottom), and the in-gel fluorescence was imaged via excitation at 365 nm (top). C) HCK modeled into the active site of UvrD (PDB 4JA8) obstructs the ATP binding site until photochemically removed through irradiation, delivering the wild-type enzyme.
Results and Discussion
We selected the lysine residue at position 37 as a target for hydroxycoumarin caging group introduction (Figure 2C), since it is highly conserved in the helicase superfamily and is essential for ATP binding and hydrolysis.[9] K37 is localized in the nucleotide binding motif I and is one of the four basic residues that are responsible for coordination of the triphosphate moiety, in particular the γ-phosphate.[9] Herein, we reasoned that photocaging K37 in UvrD (K37→HCK) prevents ATP-dependent DNA unwinding and thus renders the protein inactive. UvrD-HCK could be temporally controlled through activation by light-induced decaging (Figure 2B), which in turn generates wild-type UvrD.
A 6×HIS-tagged UvrD was generated through amplification of the uvrD gene from pETM11-UvrD and cloned into the pBAD-PylT backbone, creating pBAD-UvrD-PylT. A TAG codon or an alanine codon were introduced at position 37 for the mutated UvrD-K37TAG (pETM11-UvrD-K37TAG) or UvrD-K37A (pETM11-UvrD-K37A), respectively. These constructs were cloned into the pBAD-PylT expression vector, creating the pBAD-UvrD-K37TAG-PylT and pBAD-UvrD-K37A-PylT plasmids, respectively. Expression of full-length UvrD from E. coli bearing UvrD-K37TAG and BHCKRS/tRNACUA pair was dependent on the presence of HCK. The purified proteins were analyzed by SDS-PAGE (Figure 2B) and confirmed by MS/MS sequencing (Supporting Information Figure S1).
In order to evaluate the optical control of UvrD function through introduction of the K37HCK mutation, we applied several fluorescence-based functional assays due to their high sensitivity and ease of quantification. Fluorophores with emission wavelengths >450 nm were selected to prevent absorbance by the coumarin caging group. UvrD is a DNA-dependent ATPase,[10] and its ATPase activity was characterized and quantified using a coupled assay that converts one molecule of NADH to NAD+ for every ATP molecule consumed (Figure 3A). This coupled reaction relies on an ATP regenerating system through pyruvate kinase and phosphoenolpyruvate and the subsequent pyruvate to lactate reaction by lactate dehydrogenase which oxidizes NADH to NAD. The ATPase activities of wild-type, photocaged, decaged, and inactive UvrD were determined by time-course measurement of the fluorescence signal generated by NADH, as it is directly linked to the formation of ADP by UvrD (Figure 3B). The photocaged UvrD (UvrD-HCK) was enzymatically inactive (Figure 3B, red), as evident when compared to the UvrD-K37A negative control mutant (Figure 3B, blue), indicating that the introduction of the caging group into the ATPase active site inhibited its function. A rapid drop of NADH signal within minutes of ATP addition to the light-activated UvrD was observed (Figure 3B, purple), indicating a dramatic increase in ATP consumption rate due to decaging of the ATPase domain. Further, complete optical activation of ATPase function was achieved, since wild-type UvrD showed an identical kinetic profile (Figure 3B, green). ATPase activities of wild-type UvrD and light-activated UvrD were calculated at 46.0 ± 20.9 s−1 and 41.2 ± 14.6 s−1, respectively (see Supporting Information Table S3). Because ATP hydrolysis is a shared feature of all cellular ATPases and other ATP-dependent enzymes, this may represent a general approach to the optical control of many ATP-dependent enzymes.
Figure 3.
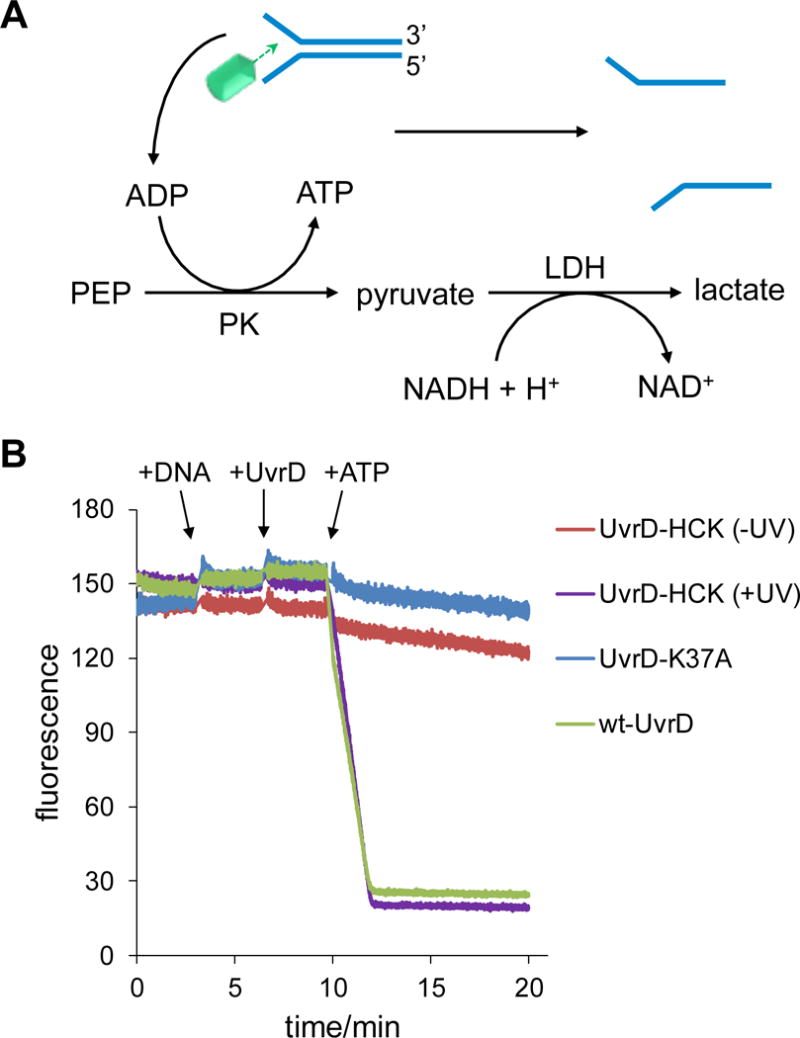
Fluorescence-based coupled ATPase activity assay. A) Mechanism of the coupled ATPase activity assay. B) Time-course fluorescence measurement of NADH consumption driven by UvrD ATPase. This trace is representative of three independent experiments (see Supporting Information Table S3).
Since the ATPase activity of UvrD-HCK could be restored through UV exposure, actual helicase activities were characterized by using a fluorescence-quenched Y-shaped double-stranded DNA substrate, containing a 5′ FAM (fluorescein) labeled top strand and a 3′ DAB (dimethylamino phenylazophenyl) labeled bottom strand. FAM fluorescence is quenched by DAB until the strands are separated by UvrD helicase activity (Figure 4A). The DNA substrate and wild-type enzyme were combined and, after initial stabilization of the fluorescence readout, ATP was added to initiate the reaction. A rapid increase in fluorescence was observed, suggesting the separation of the fluorescein-labeled top strand from the DAB-attached bottom strand as a result of unwinding. The absence of the DAB quencher molecule in the vicinity of the FAM label on the single-stranded DNA led to activation of FAM fluorescence (λex = 485 nm, λem = 520 nm). After reaching maximum fluorescence intensity, fluorescence intensity began to decrease due to the depletion of ATP (data not shown) and annealing of the fluorescent top strand back to the quencher bottom strand (Figure 3B). Following validation of the assay for the wild-type helicase, the experiment was repeated with the same concentration of photocaged UvrD-HCK that was either kept in the dark or irradiated at 365 nm. Only in the presence of ATP, and following UV exposure of the protein, was an increase in total fluorescence intensity detected, revealing light-activation of the UvrD-HCK helicase activity comparable to that of wild-type UvrD (Figure 3C). As expected, three negative control experiments showed that only minimal fluorescence was observed in the cases of the photocaged, non-irradiated UvrD system in the presence or absence of ATP and the light-activated UvrD system without ATP (Figure 3C). Therefore, optical control of UvrD helicase was achieved as determined by the Y-shaped double-stranded DNA substrate assay.
Figure 4.
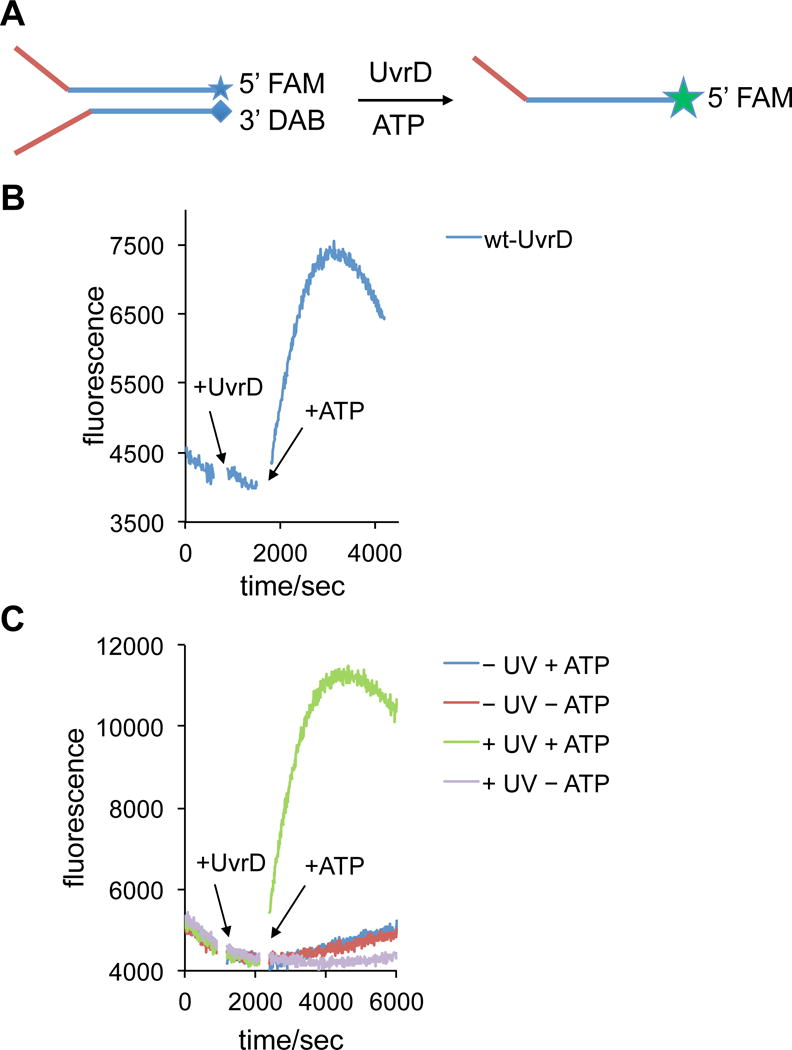
Fluorescence-based UvrD helicase activity assay using a quenched Y-shaped DNA duplex. A) Assay design based on fluorophore activation as a result of DNA duplex dehybridization. B) Time-course measurement of the helicase activity of wild-type UvrD, C) Time-course measurement of the helicase activities of photocaged UvrD-HCK, with or without light activation, in the absence or presence of ATP.
Finally, in order to further investigate light-activation of UvrD in a context that is more relevant to the NER process, an unwinding assay using a dual-nicked plasmid DNA substrate was performed. This gel-based helicase assay measures the release of an unligated and site-specific fluorescein-modified thymine (Fl-dT)-containing 16-mer that had been annealed to a gapped plasmid.[11] The substrate resembles the post-incision product and thus mimics an intermediate state of the NER process. Since this assay demonstrates the helicase function of UvrD in the context of the excision step of bacterial NER, it complements and lends support to the validity of the fluorescence-based helicase activity measurements. Displacement of the fluorescent strand through unwinding by the helicase was analyzed by polyacrylamide gel electrophoresis (Figure 5A).
Figure 5.
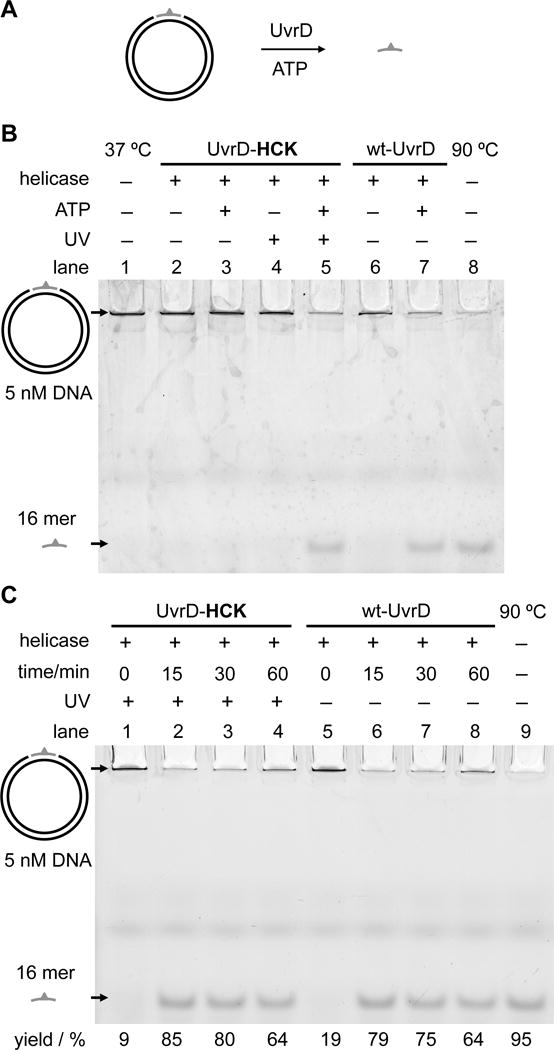
Analysis of light-activated helicase activity using a substrate resembling the post-incision intermediate of bacterial NER. A) Design of the nicked plasmid unwinding assay: released 16-mer ssDNA is shown in gray; the Fl-dT fluorophore is indicated by the green triangle. B) Unwinding reactions of wild-type and photocaged UvrD on the nicked plasmid DNA substrate. Unwound Fl-dT-containing 16-mer ssDNA was separated from the plasmid substrate by a 12% native PAGE. Top band: plasmid DNA containing the 16-mer ssDNA insert. Bottom band: unwound 16-mer ssDNA. C) Time-course of the nicked plasmid unwinding assays of wild-type and light-activated UvrD analyzed by native PAGE.
Negative controls showed that the 16-mer unwound ssDNA was not observed without UvrD (Figure 5B, lane 1) or with UvrD minus ATP (Figure 5B, lane 6). The incorporation of HCK into UvrD resulted in the complete inhibition of unwinding activity before UV irradiation, in the presence or absence of ATP (Figure 5B, lanes 2 and 3), comparable to the lack of activity in the negative controls (Figure 5B, lanes 1 and 6). As expected, UV irradiation of UvrD-HCK led to protein activation and observation of enzymatic activity only in the presence of ATP (Figure 5, lanes 4 and 5) consistent with wild-type protein (Figure 5B, lanes 6 and 7). In addition, time-course helicase assays were performed to evaluate the rates of unwinding for wild-type and light-activated UvrD proteins (Figure 5C). Based on the measured unwinding yields over time, light activation of UvrD-HCK was shown to significantly stimulate its helicase activity, in a time-dependent manner, reaching that of the wild-type UvrD levels (Figure 5C, Supporting Information Figure S5). Therefore, this gel-based helicase assay is a robust and reliable way to complement the fluorescence-based helicase assay. It demonstrates that UvrD helicase function can be optically controlled for repair of nicked DNA in the NER pathway.
In summary, we have engineered a genetically encoded, light-activated bacterial UvrD helicase. The activity of the enzyme can be stringently regulated through the use of a light-removable caging group installed directly on the essential lysine residue K37 in the ATP binding pocket of the active site. This was achieved by adding an engineered pyrrolysyl tRNA synthetase/tRNA pair to bacterial cells for the genetic encoding of a hydroxycoumarin lysine. The application of the hydroxycoumarin caging group,[8c] compared to the more common ortho-nitrobenzyl caging group, provides improved expression levels of the caged UvrD protein, imposes significant steric hindrance on the active site, and enables rapid light-triggered activation. In addition, we have utilized three different fluorescence-based assays with different substrates for the characterization of the enzymatic activities of the light-activated helicase. Importantly, the caged enzyme was completely inactive in all three assays and the overall activity of the optically triggered UvrD was comparable to that of wild-type UvrD, demonstrating excellent OFF to ON switching of the helicase activity. Moreover, the targeted, highly conserved lysine site in the ATPase domain can also be found in other helicase superfamily I & II proteins, such as PcrA, UvrB, Rep, and Srs2, as well as eukaryotic helicases XPB and XPD, further expanding the general applicability of this approach. Since XPD deletion is inviable,[12] this approach could help resolve the precise roles of XPB and XPD ATPase and helicase activities in TFIIH. This seven-subunit core complex, has a dual role in transcription promoter opening as well as damage verification and strand opening during NER. Interestingly, XPD may be involved in other cellular functions beyond transcription and NER. Thus, having the ability to photoactivate its helicase function could provide insight into some of its additional potential functions in the cell.[13] Furthermore, the developed methodology may enable optical control of ATP-dependent DNA and RNA helicases involved in a large number of nucleic acid transactions, and maybe generally applied to ATP-dependent enzymes in complex cellular milieus. Future studies integrating this methodology into cellular experiments, as well as, single-molecule imaging techniques will provide further mechanistic insight into DNA repair pathways.
Supplementary Material
Acknowledgments
We thank the Kisker lab (University of Wurzburg) for their gift of a vector containing the uvrD gene. This work was supported through funding from the National Science Foundation (CBET-1603930 to A.D.) and the National Institutes of Health (5R01ES019566 to B.V.H.). This project used the UPCI Cancer Biomarkers Facility that is supported in part by award P30CA047904.
References
- 1.Fischer CJ, Maluf NK, Lohman TM. J Mol Biol. 2004;344:1287–1309. doi: 10.1016/j.jmb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 2.a) Lahue RS, Au KG, Modrich P. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]; b) Husain I, Van Houten B, Thomas DC, Abdel-Monem M, Sancar A. Proc Natl Acad Sci U S A. 1985;82:6774–6778. doi: 10.1073/pnas.82.20.6774. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ali JA, Lohman TM. Science. 1997;275:377–380. doi: 10.1126/science.275.5298.377. [DOI] [PubMed] [Google Scholar]
- 3.Kad NM, Van Houten B. Prog Mol Biol Transl Sci. 2012;110:1–24. doi: 10.1016/B978-0-12-387665-2.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brosh RM., Jr Nat Rev Cancer. 2013;13:542–558. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Compe E, Egly JM. Annu Rev Biochem. 2016;85:265–290. doi: 10.1146/annurev-biochem-060815-014857. [DOI] [PubMed] [Google Scholar]
- 6.a) Gardner L, Deiters A. Curr Opin Chem Biol. 2012;16:292–299. doi: 10.1016/j.cbpa.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baker AS, Deiters A. ACS Chem Biol. 2014;9:1398–1407. doi: 10.1021/cb500176x. [DOI] [PubMed] [Google Scholar]; c) Brieke C, Rohrbach F, Gottschalk A, Mayer G, Heckel A. Angew Chem Int Ed Engl. 2012;51:8446–8476. doi: 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]; d) Shell TA, Lawrence DS. Acc Chem Res. 2015;48:2866–2874. doi: 10.1021/acs.accounts.5b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Wan W, Tharp JM, Liu WR. Biochim Biophys Acta. 2014;1844:1059–1070. doi: 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chin JW. Annu Rev Biochem. 2014;83:379–408. doi: 10.1146/annurev-biochem-060713-035737. [DOI] [PubMed] [Google Scholar]; c) Young TS, Schultz PG. J Biol Chem. 2010;285:11039–11044. doi: 10.1074/jbc.R109.091306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Hemphill J, Chou CJ, Chin JW, Deiters A. J Am Chem Soc. 2013;135:13433–13439. doi: 10.1021/ja4051026. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Engelke H, Chou C, Uprety R, Jess P, Deiters A. ACS Synth Biol. 2014;3:731–736. doi: 10.1021/sb400192a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Luo J, Uprety R, Naro Y, Chou C, Nguyen DP, Chin JW, Deiters A. J Am Chem Soc. 2014;136:15551–15558. doi: 10.1021/ja5055862. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Luo J, Arbely E, Zhang J, Chou C, Uprety R, Chin JW, Deiters A. Chem Commun. 2016;52:8529–8532. doi: 10.1039/c6cc03934k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hemphill J, Borchardt EK, Brown K, Asokan A, Deiters A. J Am Chem Soc. 2015;137:5642–5645. doi: 10.1021/ja512664v. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Gautier A, Nguyen DP, Lusic H, An W, Deiters A, Chin JW. J Am Chem Soc. 2010;132:4086–4088. doi: 10.1021/ja910688s. [DOI] [PubMed] [Google Scholar]
- 9.Lee JY, Yang W. Cell. 2006;127:1349–1360. doi: 10.1016/j.cell.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epshtein V, Kamarthapu V, McGary K, Svetlov V, Ueberheide B, Proshkin S, Mironov A, Nudler E. Nature. 2014;505:372–377. doi: 10.1038/nature12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Tessmer I, Croteau DL, Erie DA, Van Houten B. Nano Lett. 2008;8:1631–1637. doi: 10.1021/nl080316l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Boer J, Donker I, de Wit J, Hoeijmakers JH, Weeda G. Cancer Res. 1998;58:89–94. [PubMed] [Google Scholar]
- 13.Houten BV, Kuper J, Kisker C. DNA Repair. 2016;44:136–142. doi: 10.1016/j.dnarep.2016.05.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.