

Abstract
Objective
Determine mechanisms responsible for enhanced statin efficacy in a novel statin combination we name STOX (STatin–OXysterol).
Methods
Ovarian cancer cell lines were treated with combinations of statins and oxysterols. Cell viability was determined by a modified MTT assay. Apoptosis was evaluated by immunoblotting of PARP and DAPI-mediated visualization of apoptotic nuclei. STOX effects on the expression of genes of the mevalonate pathway were assessed by real-time qPCR and immunoblotting. siRNA-mediated gene silencing was used to test the involvement of oxysterol-mediated repression of SREBP-2 in STOX synergy. The impact of statin-mediated inhibition of protein prenylation and on cholesterol homeostasis was evaluated.
Results
Oxysterols dramatically enhance cytotoxicity of statins in ovarian cancer cells through increased apoptosis. Decreased expression of SREBP-2 down-regulates the mevalonate pathway and prevents the active statin-induced sterol feedback, enhancing statin toxicity. Comparison of two ovarian cancer cell lines reveals two distinct mechanisms of statin induced toxicity, namely, dependence on protein geranylgeranylation and/ or perturbation of cellular cholesterol levels.
Conclusions
We provide evidence of statins' mechanisms of cytotoxicity in different ovarian cancer cells and discovered a new approach to significantly enhance the anti-tumor activity of statins. These observations provide a potential new path to improve statins as a treatment against ovarian cancer with obtainable dosages.
Keywords: Ovarian, cancer, Statins, SREBPs, Cholesterol
Introduction
High-grade serous epithelial ovarian cancer (EOC) is the most lethal gynecological cancer with no curative treatment and low overall survival [1–5] and new therapeutic agents are desperately needed.
Statins inhibit 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), the rate-limiting enzyme in the synthesis of mevalonate (MVA), which is a necessary precursor for synthesis of cholesterol and isoprenoids. HMGCR itself has transforming properties, suggesting that deregulation of mevalonate production is critical for some types of cancer cells [6,7]. p53 mutant cancer cells, which include 96% of EOC, may be particularly dependent on up-regulation of the MVA pathway [7,8]. Preclinical models demonstrate anti-cancer efficacy of statins [9–11] and epidemiological studies associate statin usage with reduced cancer risk and improved outcomes [12–17]. However, clinical trials revealed no clear treatment benefit [18–20]. Therefore, there is a need to improve our understanding of the regulation of mevalonate metabolism and the anti-cancer properties of statins, to identify strategies that improve statin efficacy in cancer treatments.
A major obstacle in blocking cholesterol and mevalonate synthesis in cancer cells is the multiple feedback loops compensating for any single blockage [7,11,21] (Fig. 1A). In multiple myeloma, statin-resistant cells activate a sterol feedback response when exposed to statins leading to increased expression of HMGCR (Fig. 1A), while sensitive cells do not [22]. This feedback response depends on the sterol regulatory element-binding proteins (SREBPs) [23]. Three isoforms of SREBPs are expressed from two genes. SREBP-1s regulate fatty acid synthesis and SREBP-2 regulates cholesterol metabolism [23]. High concentrations of cholesterol and its oxygenated derivatives, oxysterols, inhibit SREBPs' activation by preventing protein processing and nuclear transport [1, 3,5]. Oxysterols are also ligands for LXR, a nuclear receptor that regulates cholesterol metabolism and efflux [9]. The balance of LXR and SREBPs activity may be crucial for growth and survival of cancer cells (Fig. 1A). Synthetic LXR agonists have shown promise in some pre-clinical cancer models [12,14,15] but in ovarian cancer cells they promote survival [6].
Fig. 1.
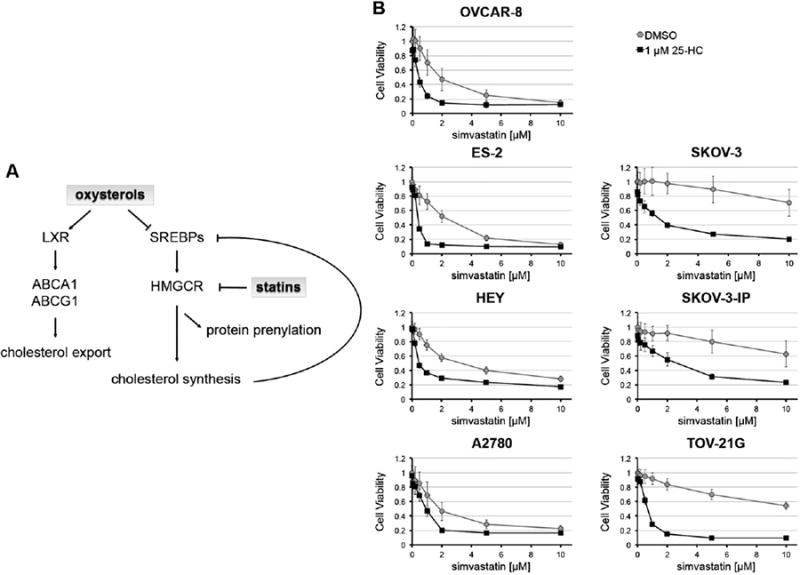
25-hydroxycholesterol synergizes with simvastatin to enhance toxicity in ovarian cancer cells. A) Schematic of the balance between LXR and SREBPs to regulate cholesterol levels. SREBPs stimulate transcription of mevalonate synthesis enzymes. Higher levels of cholesterol and related metabolites, such as oxysterols, inducea negative feedback response by inhibiting SREBPs expression and induce LXR activation. Statins' inhibition of HMGCR leads to increased SREBP-2 activation of mevalonate synthesis pathway. B) Cell viability was determined 72 h after treatment. STOX showed synergy in all tested ovarian cancer cell lines.
We show that the statin–oxysterol co-treatment significantly enhances statin cytotoxicity in ovarian cancer cells, likely because of the suppression of the sterol feedback induced by statins. We name this statin–oxysterol combination STOX. We identify two mechanisms that trigger statin cytotoxicity in two ovarian cancer cell lines, which are based on differential cell reliance on geranylgeranylation and ability to efficiently sustain cholesterol homeostasis. Our observations provide a new path to develop statin-based combinatorial therapy and lay the foundation to identify patients that could benefit the most from statin therapy.
Materials and methods
Reagents
Simvastatin, lovastatin, fluvastatin, 25-hydroxycholesterol, 22(S)-hydroxycholesterol, 22(R)-hydroxycholesterol, GW3965, farnesyl pyrophosphate and geranylgeranyl pyrophosphate were purchased from Sigma-Aldrich. Primary antibodies: anti-β-actin (Sigma-Aldrich, A2228), anti-SREBP-2 (Abcam, ab30682), anti-SREBP-1 (Santa Cruz Biotechnology, sc-8984), anti-HMGCR (Millipore, 07-572), and anti-PARP (Cell Signaling, 46D11). Secondary antibodies were purchased from Li-Cor Biosciences. Low-density lipoprotein (LDL) and lipoprotein-deficient serum (LPDS) were purchased from Biomedical Technologies, Inc.
Cell culture
SKOV-3 and ES-2 were purchased from the American Type Culture Collection. OVCAR-8 was purchased through the National Cancer Institute Developmental Therapeutics Program's tumor repository program. HEY and A2780 were generously provided by Dr. Barbara Vanderhyden (University of Ottawa, Ottawa, Ontario) and SKOV-3-IPcells by Mien-Chie Hung (MD Anderson Cancer Center, Houston, TX). TOV-21G was provided by Charles River Laboratories (Morrisville, NC). All cell lines used in this study except TOV-21G were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. TOV-21G was cultured in RPMI supplemented with 15% FBS, 25 mg/mL gentamicin, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere. SKOV-3, OVCAR-8, and TOV-21G cell lines were authenticated by the ATCC Cell Line Authentication Service.
RNA expression analysis
24 h after addition of drugs, total RNA was isolated using miRNeasy Kit (Qiagen) and reverse-transcribed with SuperScript III RT following manufacturers' instructions. cDNA was quantified with Quant-iT PicoGreen (Life Technologies) and real-time qPCR was performed with SYBR Green Master Mix (Applied Biosystems) on a 7900HT Fast Real-Time PCR System. All reactions were performed in triplicate using 2–5 ng cDNA. mRNA expression levels were calculated using the ΔΔCt method relative to GAPDH. Data are represented as mean ± SD of 3 biological replicates.
Protein extraction and immunoblots
Immunoblots were performed on cells treated as indicated in figure legends. Total cell lysates were obtained with lysis in RIPA buffer (Millipore). For detection of HMGCR, the cells were processed as previously described [7]. The cells were lysed in buffer A (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 10 mM NaF, 1 mM Na3VO4) supplemented with 1× EDTA-free protease inhibitor cocktail (Roche) and clarified lysates were not boiled before electrophoresis to avoid HMGCR aggregation. For mSREBP-1 and mSREBP-2 detection, cell lysates were enriched for nuclear proteins. Briefly, the cells were resuspended in hypotonic Buffer B (10 mM Hepes-KOH pH 7.4, 10 mM KCl, 250 mM sucrose, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 10 mM NaF, 1 mM Na3VO4) supplemented with 1× EDTA-free protease inhibitor cocktail (Roche) and mixed every 10 min while incubating at 4 °C for 30 min. NP-40 was added to 0.1% final concentration and incubated for an additional 10 min at 4 °C before centrifugation for 10 min at 4 °C. Pellets were resuspended in Buffer B, incubated 15 min at 4 °C before adding NP-40 to a 0.1% final concentration and centrifugation for 10 min at 4 °C. The nuclei enriched samples were lysed in RIPA buffer. Protein concentration of cleared lysates was determined by BCA assay (Pierce) and 30–50 μg of protein subject to 8% or 10% SDS/PAGE analysis. Immunoblots were imaged using the ODYSSEY CLx Infrared Imaging scanner (Li-Cor Biosciences).
Cell viability assays
After 72 h of treatment, the WST-1 assay (Clontech) was performed according to the manufacturer's instructions. Absorbance at 450 nm was measured with a SpectraMax M5 microplate reader (Molecular Devices). Absorbance at 650 nm was used as baseline background signal. Measurements were performed in triplicate and data are mean ± SD of at least two biological replicates.
Apoptosis
The cells were treated for 72h before being washed twice in PBS and fixed in 3% paraformaldehyde for 30 min at 4 °C, followed by three PBS washes. The cells were permeabilized in 0.2% Triton X-100 in PBS for 10 min and washed three times in PBS. The cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min at room temperature, followed by a wash in PBS. Nuclear staining was visualized using a Zeiss Axiovert 200M fluorescence microscope. Percentage of apoptosis was calculated as the mean ± SD of at least 3 biological replicates. For each biological replicate at least 160 cells were counted for STOX treated cells and at least 500 cells for the other conditions.
Cellular cholesterol measurement
48 h after the indicated treatment, half of the cells were lysed in RIPA buffer supplemented with 1× EDTA-free protease inhibitor cocktail (Roche) and total protein concentration determined with BCA assay (Pierce). The second half of the cells was homogenized in 1:2 (v/v) methanol:choloroform solution to extract cholesterol and lipids [18], insoluble material was pelleted and the organic phase was vacuum dried. Cellular cholesterol and lipids were measured by the Amplex Red Cholesterol Assay (Life Technologies) following manufacturers' instructions. Cholesterol content in each sample was normalized to total protein. Data are represented as mean ± SD from 6 biological replicates.
SREBP-2 suppression by siRNA
Knock-down of SREBP-2 was achieved with three individual siRNAs (Thermo Scientific Dharmacon) and control experiments were performed with non-targeting control siRNA (Thermo Scientific Dharmacon). The cells were transfected with 25 nM siRNA. SKOV-3 and OVCAR-8 were transfected using DharmaFECT 3 (Thermo Scientific Dharmacon) at 0.4% (v/v) and DharmaFECT 1 (Thermo Scientific Dharmacon) at 0.2% (v/v), respectively. After 24 h, medium was replaced with complete medium containing simvastatin. After 72 h, cell viability was measured by WST-1. SREBP-2 RNA and protein levels were assessed at 48 h after transfection.
Results
Oxysterols synergize with statins to kill ovarian cancer cells
Seven ovarian cancer cell lines were tested to explore the effect of oxysterols, which are known SREBP repressors [1,5,24], in combination with statins (STOX). Ovarian cancer cells were treated with simvastatin, 25-HC, or co-treated with simvastatin and 25-HC and cell viability was assessed at 72 h (Fig. 1B). 1 μM 25-HC does not significantly reduce cell viability as a single agent, but synergizes with simvastatin in all the tested ovarian cancer cell lines (Fig. 1 and Fig. S1). Thus, 1 μM 25-HC effectively lowers simvastatin IC50s both in statin-sensitive cell lines (OVCAR-8, ES-2, HEY, and A2780) and even more dramatically in statin-resistant cell lines (SKOV-3, SKOV-3-IP, and TOV-21G).
To test if the synergy between simvastatin and 25-HC is general and observed for other statin–oxysterol combinations, we treated ovarian cancer cells with simvastatin in combination with either 22(R)HC or 22(S)HC (Fig. S2). Cell viability measured in multiple cell lines clearly demonstrated that both oxysterols synergized with simvastatin lowering the IC50s by 3.4 to 25-fold compared to the simvastatin-alone treatment. Because lipophilic statins are more effective inhibitors than more hydrophilic statins in cancer cell lines and in vivo models [2,4,11,25], we additionally tested 25-HC in combination with fluvastatin and lovastat-in (Fig. S3). In OVCAR-8 fluvastatin alone showed high potency which was only slightly improved by the co-treatment with 25-HC, but in statin-resistant SKOV-3 cells and for lovastatin in both cell lines 25-HC significantly potentiated anti-cancer activity of statins (Fig. S3). These data demonstrate the versatility of STOX combinations on multiple ovarian cancer cell lines.
25-HC potentiates simvastatin-induced cytotoxicity by increasing apoptosis-mediated cell deathinSKOV-3 and OVCAR-8 (Fig. 2). Cleaved PARP significantly increased upon co-treatment of 25-HC and simva-statin (Fig. 2A). Moreover, DAPI apoptotic assay revealed that 25-HC increased simvastatin-induced apoptosis by 9-fold in OVCAR-8 (Fig. 2B) and by 15-fold in SKOV-3 (Fig. 2C).
Fig. 2.
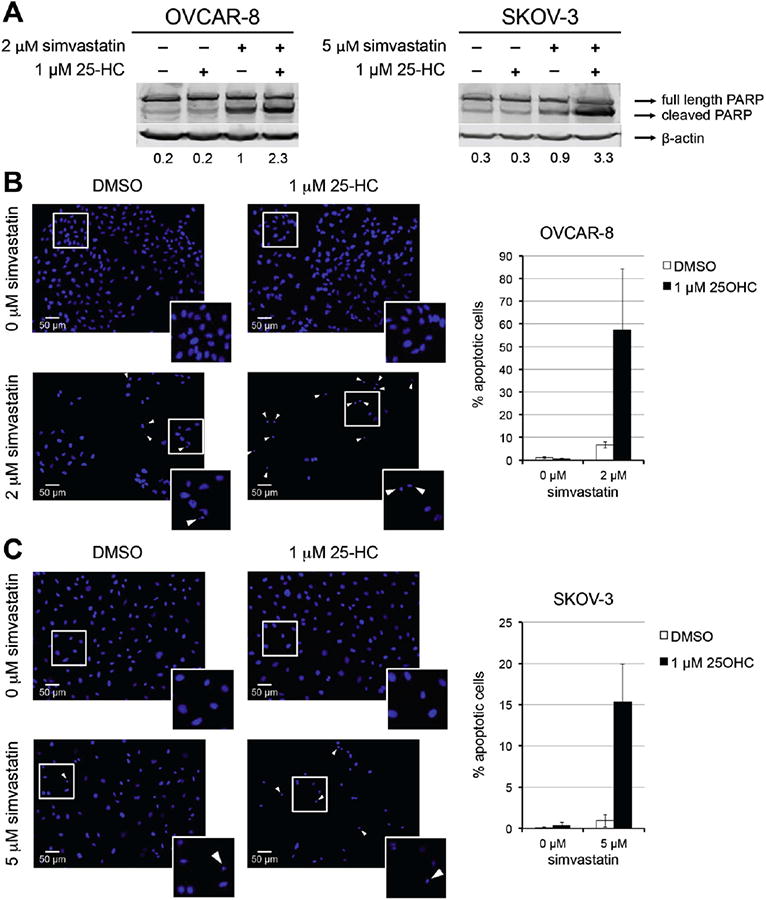
25-hydroxycholesterol combined with simvastatin increases apoptosis in ovarian cancer cell lines. A) Immunoblots of PARP after 48 h of treatment with indicated concentrations of statins and 25-HC. Band quantification represents the fraction of cleaved PARP after normalization to β-actin. B and C) DAPI stained cells after 72 h of treatment as indicated. The cells were scored as either apoptotic or non-apoptotic based on nuclear morphology. Representative images of OVCAR-8. (B) and SKOV-3 (C) are shown. Arrows indicate apoptotic cells and magnification of cells in the white squares are shown in the lower right corner. Percentages of apoptotic cells relative to vehicle are graphed.
25-HC suppresses statin induced sterol feedback through inhibition of SREBP-2
SREBPs are repressed by high cholesterol or oxysterols concentrations by sequestering the precursor protein at the endoplasmic reticulum before its proteolysis activation [26]. We assessed the expression of SREBPs and their target genes in response to STOX treatment in OVCAR-8 and SKOV-3. Simvastatin induced a 3.5–4 fold increase of HMGCR and a 2-fold increase in mature SREBP-2 (mSREBP-2) proteinlevels (Fig. 3), suggesting an active compensatory mechanism for statin inhibition of HMGCR. 25-HC strongly repressed the expression of HMGCR and mSREBP-2, but did not significantly affect mSREBP-1 expression (Fig. 3). Notably, even when combined with simvastatin 25-HC prevents the statin-induced feedback response in both cell lines strongly repressing HMGCR and mSREBP-2 protein levels.
Fig. 3.
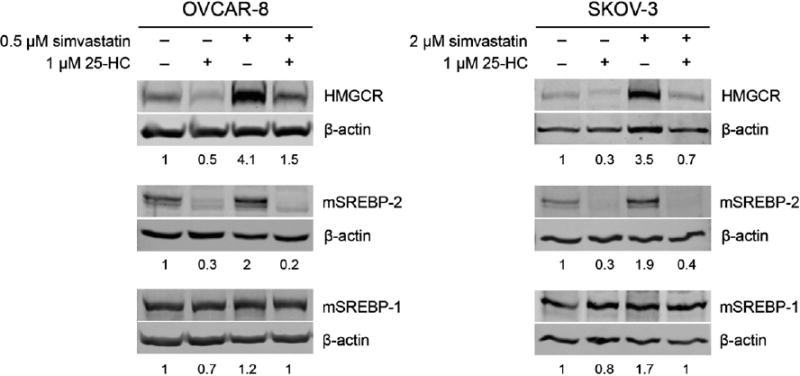
25-hydroxycholesterol inhibits the active sterol response feedback in ovarian cancer cells. The cells were exposed to treatments for 24 h before preparation of lysates for immuneblot analysis. Immunoblots were probed with anti-SREBP-1, anti-SREBP-2, or anti-HMGCR antibodies and anti-β-actin as a loading control. Band quantifications represent the ratio with β-actin and values are normalized to vehicle. The simvastatin concentration used for each cell line was chosen because it is approximately the IC50 when used in combination with 1 μM 25-HC.
SREBP-2 directly regulates cholesterol metabolism enzymes including HMGCR [10,11,27,28]. Real-time qPCR analysis revealed significant oxysterol-mediated decreases in mRNA levels in genes involved inmevalonate synthesis and cholesterol synthesis and transport (Fig. 4). 25-HC induced a strong down-regulation of SREBP-2 target genes in both OVCAR-8 and SKOV-3, which include HMGCR (Figs. 4A and D),SREBP-2 itself, the low-density lipoprotein receptor (LDLR), lanosterol synthase (LSS), and 7-dehydrocholesterol reductase (DHCR7) (Figs. 4C and F). 25-HC-mediated repression of mRNA levels was not affected by the concurrent treatment with simvastatin, similar to observations at the protein level. Interestingly, down-regulation of genes involved in fatty acid synthesis was also observed. FASN is repressed in both OVCAR-8 and SKOV-3, while SREBP-1 is repressed only in SKOV-3 (Figs. 4B, E). Overall, these data suggest a role for oxysterols in enhancing statin efficacy by potentiating the interference with the functionality of the MVA pathway.
Fig. 4.
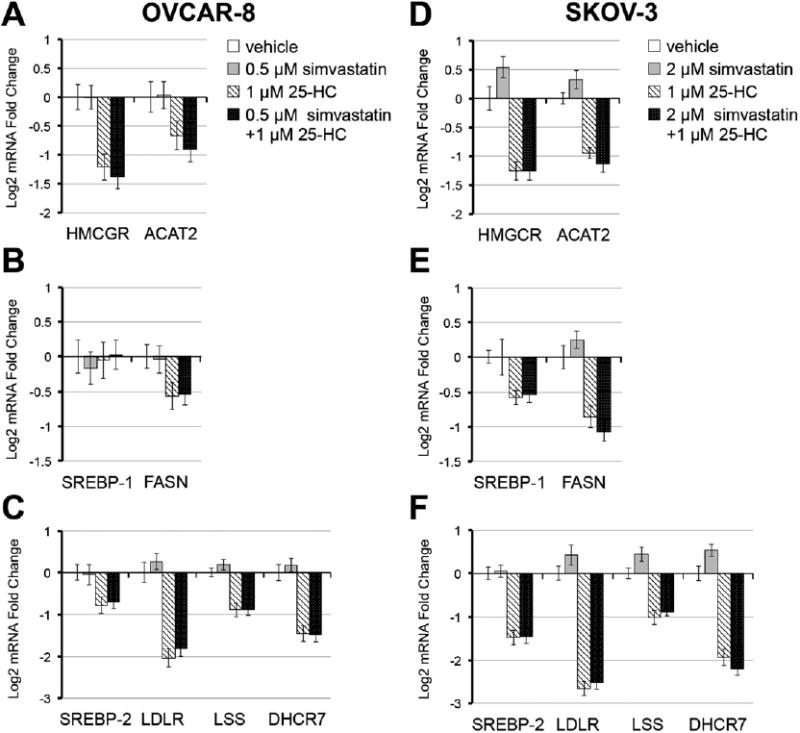
Simvastatin induces a sterol response feedback that is suppressed by 25-hydroxycholesterol. After 24 h of the indicated treatment, cells were collected for RNA analysis by real-time qPCR. A and D) Mevalonate synthesis enzymes; B and E) SREBP-1 and the SREBP-1 target, FASN; C and F) SREBP-2 and cholesterol metabolism enzymes.
To test if oxysterol stimulation of LXRs contributes to the observed STOX synergy, the cells were co-treated with simvastatin and the synthetic LXR agonist GW3965 (Fig. S4). No effect or mild antagonism was observed, consistent with our previous study suggesting that LXR agonists promote ovarian cancer cell survival [6]. Therefore, these data suggest that LXR activation is not significantly contributing to STOX toxicity.
The combination of simvastatin and 25-HC effectively down-regulates SREBP-2 and suppresses the sterol feedback triggered by statins. We tested if SREBP-2 is a major contributor to the oxysterol-induced synergy with statins by siRNA-mediated silencing (Fig. 5). All three siRNAs efficiently knocked-down the SREBP-2 transcript by >80% and mSREBP-2 protein levels were reduced by 50–65% independent of statin treatment (Figs. 5A and B). Protein processing and/ or alternate splice forms could be contributing to the observed difference between the RNA and protein levels. Recapitulating the experimental conditions used to assess the STOX effect, SREBP-2 knockdown sensitized both OVCAR-8 and SKOV-3 to simvastatin (Fig. 5C). The degree of synergy is not as high as with oxysterols, likely because the siRNAs do not knock-down mSREBP-2 protein with the same efficiency. These observations support the hypothesis that SREBP-2 protein levels are critical for statin efficacy and provide a key element in the synergy between statins and oxysterols.
Fig. 5.
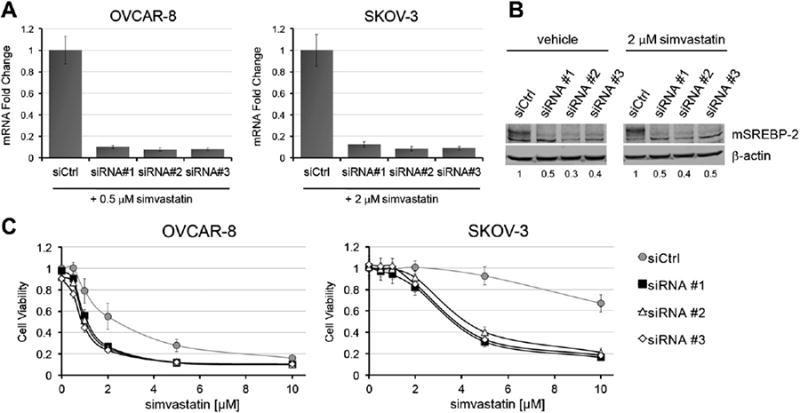
SREBP-2 knock-down sensitizes ovarian cancer cells to simvastatin. A) RNA analysis by real-time qPCR shows >80% reduction in SREBP-2 mRNA levels for each independent siRNA 48 h after transfection. Data refer to cell treated with 0.5 μM (OVCAR-8) or 2 μM (SKOV-3) simvastatin for 24 h. B) 50–70% knock-down at the protein level was obtained for each siRNA by immunoblot in SKOV-3 cells. At the time of sample collection, the cells were transfected with siRNA for 48 h while treated as indicated for 24 h. Numbers indicate the mSREBP-2/β-actin ratios. C) The cells were treated with vehicle or simvastatin 24 h after transfection and cell viability was determined after 72 h of treatment. Knock-down of SREBP-2 increases simvastatin toxicity in both SKOV-3 and OVCAR-8.
Differences in regulation of cholesterol homeostasis defines two mechanisms of statin cytotoxicity
We aimed to determine the mechanisms leading to statin and STOX-induced toxicity in ovarian cancer cells. Mevalonate is a precursor for geranylgeranyl pyrophosphate (GGPP), farnesyl pyrophosphate (FPP), and cholesterol. Geranylgeranyl and farnesyl groups are required for lipid post-translational modification of numerous proteins and are essential for proper function of many Raps superfamily proteins [16,17, 29]. Statin-induced apoptosis in cancer cells is often associated with reduced protein geranylgeranylation [7,8,10,11,25]. To assess the impact of statin-mediated inhibition of protein prenylation on viability of ovarian cancer cells, either GGPP or FPP was added to cells treated with simvastatin alone or in combination with 25-HC (Fig. 6). GGPP treatment rescued SKOV-3 cells treated with either statin or co-treated with simvastatin and 25-HC. However, GGPP only modestly inhibited simvastatin-in or STOX toxicity in OVCAR-8 cells and this partial rescue does not depend on farnesylation inhibition, since addition of FPP had no effect (Figs. 6A and B). In SKOV-3, FPP inhibited simvastatin toxicity, but only modestly counteracted the co-treatment of simvastatin and 25-HC (Fig. 6B).
Fig. 6.
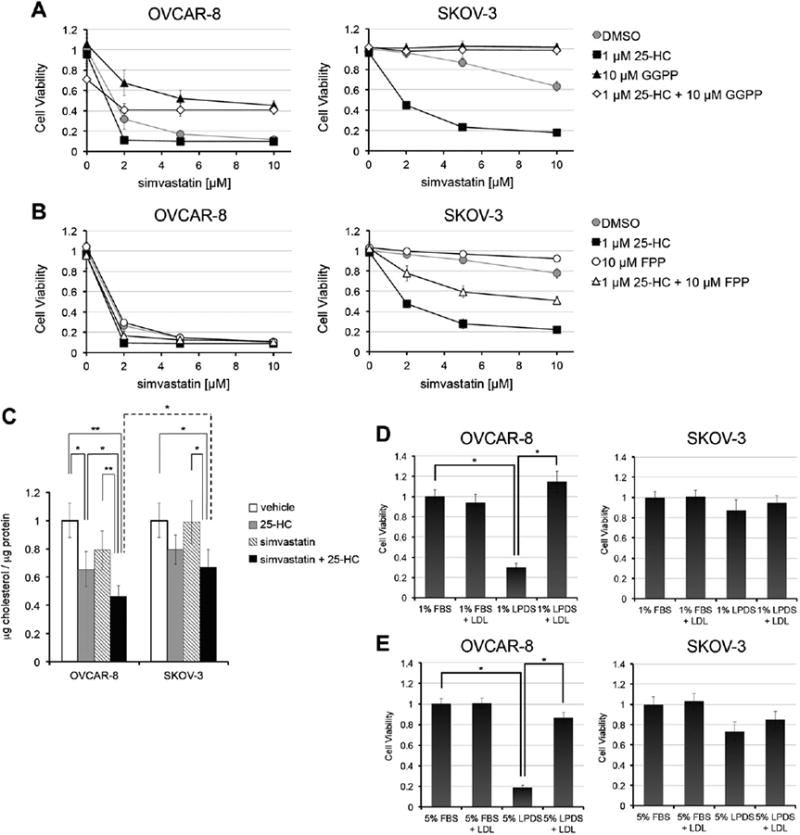
OVCAR-8, but not SKOV-3, is sensitive to statin-mediated inhibition of cholesterol synthesis. A and B) Role of protein prenylation in OVCAR-8 and SKOV-3 viability. Cell viability was assessed at 72 h. C) Cholesterol homeostasis is more affected by STOX in OVCAR-8 than in SKOV-3 cells. Both cell lines were treated with 1 μM 25-HC, while simvastatin, alone or in combination with 25-HC, was used at 2 μM for SKOV-3 and at 0.5 μM for OVCAR-8. The different simvastatin concentrations reflect the different IC50s for the combined treatment for the two cell lines at 72 h. Cellular cholesterol was measured at 48 h and values were normalized to total protein content. t-test: *: p < 0.05; **: p < 0.01. D and E) Reduced serum cholesterol concentrations adversely affect OVCAR-8 but not SKOV-3 cells. SKOV-3 and OVCAR-8 cells were culturedin 1% or 5% of FBS or LPDS medium for 72h in the presence or absence of 5 μg/mL LDL. Cell viability was determined at 72 h. t-test *: p < 0.005.
Mevalonate is also a necessary precursor for cholesterol biosynthesis. Because simvastatin and STOX-induced toxicity in OVCAR-8 is not completely due to reduced protein prenylation, we hypothesized that OVCAR-8 cells are more sensitive to changes in cholesterol levels. Both 25-HC and simvastatin alone reduced cholesterol more effectively in OVCAR-8 than in SKOV-3 cells (Fig. 6C). Notably, in both cell lines co-treatment with simvastatin and 25-HC reduced cholesterol levels more significantly than simvastatin alone. STOX lowered cholesterol levels significantly more in OVCAR-8 cells than in SKOV-3, consistent with the hypothesis that in OVCAR-8 STOX is toxic because of a combined inhibition of both protein prenylation and cholesterol synthesis.
To test the efficiency of cholesterol homeostasis in OVCAR-8 and SKOV-3 and to assess if low cholesterol levels are functionally important in OVCAR-8, the cells were grown in lipoprotein-deficient serum (LPDS) in the presence or absence of LDL for 72h (Figs. 6D and E). OVCAR-8 cell viability was reduced by >70% in LPDS and was fully restored with the addition of LDL, while SKOV-3 cells were only modestly affected by the absence of exogenous cholesterol (Figs. 6D and E). Because LPDS induces elevated OVCAR-8 cell death, further testing of statin-mediated toxicity in low cholesterol conditions was not possible. Together, these observations suggest that these two ovarian cancer cell lines are sensitive to statin because of distinct toxicity pathways.
Discussion
We have discovered a new approach to enhance statin efficacy by preventing the sterol feedback response through the oxysterol mediated suppression of SREBP-2 expression. We identified an SREBP-2 dependent survival pathway critical for statin sensitivity in ovarian cancer cells. When the sterol feedback response is repressed by oxysterols and combined with HMGCR inhibition by statins, significant enhancement of cell death is achieved. Our data suggest that suppression of the ability of ovarian cancer cells to respond to statins through SREBP-2 leads to significant improvement in statin efficacy, especially in cancer cells that are otherwise resistant to statins (Fig. 1). These observations suggest that an effective sterol feedback response is a critical parameter for statin sensitivity in ovarian cancer cells and that STOX combinations could be used to enhance statin efficacy. Serum concentrations of 2–4 μM are achievable for lipophilic statins in high dose studies [20,30]; however, significant side effects at these high doses are possible [19,20], motivating efforts to find combinations that increase statin antitumor potency. Our approach lowers the statin concentrations necessary to achieve cancer cell toxicity into the range obtainable in patients, potentially reducing the problems associated with possible side effects. We also found that STOX is effective in other cancer cell lines including breast (data not shown), suggesting that the approach could be applied to a variety of tumors.
A second major discovery of this study is the identification of the different mechanisms through which statin achieves toxicity in two ovarian cancer cell lines, which differ in their ability to regulate cholesterol homeostasis. In OVCAR-8 cells statin toxicity is only partially dependent on inhibition of geranylgeranylation and cells are more susceptible to STOX-mediated decrease of cholesterol level and low cholesterol environments (Fig. 6). Together, these observations suggest that in OVCAR-8 cells STOX causes toxicity by a combination of reduced cholesterol availability and reduced geranylgeranylation. OVCAR-8 cells may have an inefficient cholesterol synthesis pathway and/or poor cholesterol scavenging capability leading to their higher sensitivity to extracellular cholesterol depletion. SKOV-3 cells on the other hand are not significantly affected by STOX-induced reduction of cholesterol, as suggested by the nearly complete inhibition of statin toxicity by the addition of GGPP. Nor are SKOV-3 cells affected by low cholesterol growth conditions (Figs. 6D and E). SKOV-3 cells may be able to tolerate low cholesterol levels, efficiently increase cholesterol biosynthesis, and/or may be able to scavenge cholesterol from the environment better than OVCAR-8 cells. Cancer cells can be good cholesterol scavengers to overcome decreased mevalonate synthesis [31]. These observations suggest that ovarian cancer cells are sensitive to statins due to multiple toxicity mechanisms. Understanding the balance of these mechanisms and how they are regulated to support survival is critical to understand the potential for statin therapy in ovarian cancer.
The importance of the feedback response and the central role played by SREBP-2 in modulating sensitivity to statin and mediating STOX synergy is illustrated by the increased statin cytotoxicity in SREBP-2-silenced cells. Both pharmacological (Fig. 3) and genetic manipulation (Fig. 5) of SREBP-2 increased statin sensitivity. Reduction of SREBP-2 levels, by either 25-HC or gene knock-down, did not significantly affect growth and survival of ovarian cancer cells in the absence of statins. SREBP-2 regulates the mevalonate biosynthesis pathway, including HMGCR. It is likely that repression of the whole pathway contributes to decreased mevalonate synthesis and synergy with statins. Inhibiting the cancer cells' response to statins by blocking SREBP-2 expression with oxysterols is just one approach. Other candidates include tocotrienols, which reduce viability of prostate cancer cells by repressing SREBP-2 expression [32]. Other molecules such as fatostatin inhibit both SREBP-1 and SREBP-2 and could also be combined with statins [8].
Our data suggest that in some ovarian cancer cells SREBP-1 could also support survival. SREBP-1 may be overexpressed in ovarian tumors and contribute to tumor growth and survival [21,33,34]. SREBP-1 is modestly down-regulated by STOX treatment in SKOV-3 cells, consistent with lower FASN mRNA expression (Fig. 4). An alternative possibility is that SREBP-2 is regulating fatty acid synthesis by cross-talk at some SREBP-1 targets, as has been reported [7,11,28]. Both mechanisms could lead to decreased expression of FASN as observed in both SKOV-3 and OVCAR-8. The different transcriptional response in the two cell lines suggests that deregulation of the fatty acid pathway is not the basis for the statin–oxysterol synergy exhibited in both OVCAR-8 and SKOV-3. Nonetheless, the larger decrease in FASN in SKOV3 may contribute to the greater synergy observed in this cell line compared to OVCAR-8, as fatty acid synthesis may be critical for cancer cell survival [22,34].
In summary, we find that inhibiting the sterol feedback response with agents such as oxysterols greatly enhance the efficacy of statins for cancer therapy. This new combination could form the basis of a new therapeutic approach, targeting metabolism in ovarian cancer.
Statins are being tested in combination with a variety of new and established chemotherapeutics [10,23,25,35,36]. STOX can in principle be combined with other agents that synergize with statins, as STOX acts through the same mechanisms as statins alone [10,23]. One of the outstanding challenges is to determine which types of ovarian tumors are more like OVCAR-8 and which are more like SKOV-3, in order to optimize treatments and identify patients who could benefit most from statin therapy.
Supplementary Material
Highlights.
Oxysterols synergize with statins enhancing apoptosis in ovarian cancer cells.
SREBP-2 is a key regulator of statin response in ovarian cancer cells.
Ovarian cancer cells are sensitive to statins by at least two distinct mechanisms.
Acknowledgments
We thank the Brown University Center for Genomics and Proteomics, partially supported by the NIH(NIGMS P30GM103410, NCRR P30RR031153, P20RR018728, S10RR02763), NSF (EPSCoR 0554548), and the Rhode Island Hospital. This work was supported by the Mary Kay Foundation.
Abbreviations
- 25-HC
25-hydroxycholesterol
- 22(R)HC
22(R)-hydroxycholesterol
- 22(S)HC
22(S)-hydroxycholesterol
- HMGCR
3-hydroxy-3-methylglutaryl-coenzyme A reductase
- LDL
low-density lipoprotein
- STOX
STatin–OXysterol co-treatment
Footnotes
Conflict of interest statement: The authors have no conflict of interest to declare.
References
- 1.Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 2.Romero I, Bast RCJ. Minireview: human ovarian cancer: biology, current management, and paths to personalizing therapy. Endocrinology. 2012;153:1593–602. doi: 10.1210/en.2011-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun LP, Li L, Goldstein JL, Brown MS. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem. 2005;280:26483–90. doi: 10.1074/jbc.M504041200. [DOI] [PubMed] [Google Scholar]
- 4.Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer. 2010;10:803–8. doi: 10.1038/nrc2946. [DOI] [PubMed] [Google Scholar]
- 5.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104:6511–8. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller DH, Fischer AK, Chu KF, Burr R, Hillenmeyer S, Brard L, et al. T0901317 inhibits cisplatin-induced apoptosis in ovarian cancer cells corrected. Int J Gynecol Cancer. 2011;21:1350–6. doi: 10.1097/IGC.0b013e318228f558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clendening JW, Pandyra A, Boutros PC, Ghamrasni El S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A. 2010;107:15051–6. doi: 10.1073/pnas.0910258107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–58. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–31. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 10.Osmak M. Statins and cancer: current and future prospects. Cancer Lett. 2012;324:1–12. doi: 10.1016/j.canlet.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 11.Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene. 2012;31:4967–78. doi: 10.1038/onc.2012.6. [DOI] [PubMed] [Google Scholar]
- 12.Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1:442–56. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med. 2012;367:1792–802. doi: 10.1056/NEJMoa1201735. [DOI] [PubMed] [Google Scholar]
- 14.Jakobsson T, Treuter E, Gustafsson JA, Steffensen KR. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci. 2012;33:394–404. doi: 10.1016/j.tips.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 15.Chuu CP, Lin HP. Antiproliferative effect of LXR agonists T0901317 and 22(R)-hydroxycholesterol on multiple human cancer cell lines. Anticancer Res. 2010;30:3643–8. [PubMed] [Google Scholar]
- 16.Lavie O, Pinchev M, Rennert HS, Segev Y, Rennert G. The effect of statins on risk and survival of gynecological malignancies. Gynecol Oncol. 2013;130:615–9. doi: 10.1016/j.ygyno.2013.05.025. [DOI] [PubMed] [Google Scholar]
- 17.Elmore RG, Ioffe Y, Scoles DR, Karlan BY, Li AJ. Impact of statin therapy on survival in epithelial ovarian cancer. Gynecol Oncol. 2008;111:102–5. doi: 10.1016/j.ygyno.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Folch J, Lees M, Stanley GHS. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 19.Sondergaard TE, Pedersen PT, Andersen TL, Søe K, Lund T, Ostergaard B, et al. A phase II clinical trial does not show that high dose simvastatin has beneficial effect on markers of bone turnover in multiple myeloma. Hematol Oncol. 2009;27:17–22. doi: 10.1002/hon.869. [DOI] [PubMed] [Google Scholar]
- 20.van der Spek E, Bloem AC, van de Donk NWCJ, Bogers LH, van der Griend R, Kramer MH, et al. Dose-finding study of high-dose simvastatin combined with standard chemotherapy in patients with relapsed or refractory myeloma or lymphoma. Haematologica. 2006;91:542–5. [PubMed] [Google Scholar]
- 21.Gabitova L, Gorin A, Astsaturov I. Molecular pathways: sterols and receptor signaling in cancer. Clin Cancer Res. 2014;20:28–34. doi: 10.1158/1078-0432.CCR-13-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clendening JW, Pandyra A, Li Z, Boutros PC, Martirosyan A, Lehner R, et al. Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma. Blood. 2010;115:4787–97. doi: 10.1182/blood-2009-07-230508. [DOI] [PubMed] [Google Scholar]
- 23.Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. 2012;23:65–72. doi: 10.1016/j.tem.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong Y, Lee JN, Lee PCW, Goldstein JL, Brown MS, Ye J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006;3:15–24. doi: 10.1016/j.cmet.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Robinson E, Nandi M, Wilkinson LL, Arrow smith DM, Curtis ADM, Richardson A. Preclinical evaluation of statins as a treatment for ovarian cancer. Gynecol Oncol. 2013;129:417–24. doi: 10.1016/j.ygyno.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. GenesDev. 2009;23:2578–91. doi: 10.1101/gad.1854309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seo YK, Jeon TI, Chong HK, Biesinger J, Xie X, Osborne TF. Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. 2011;13:367–75. doi: 10.1016/j.cmet.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–69. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 30.Thibault A, Samid D, Tompkins AC, Figg WD, Cooper MR, Hohl RJ, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res. 1996;2:483–91. [PubMed] [Google Scholar]
- 31.Williams KJ, Argus JP, Zhu Y, Wilks MQ, Marbois BN, York AG, et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 2013;73:2850–62. doi: 10.1158/0008-5472.CAN-13-0382-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol. 2009;16:882–92. doi: 10.1016/j.chembiol.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Nie LY, Lu QT, Li WH, Yang N, Dongol S, Zhang X, et al. Sterol regulatory element-binding protein 1 is required for ovarian tumor growth. Oncol Rep. 2013;30:1346–54. doi: 10.3892/or.2013.2575. [DOI] [PubMed] [Google Scholar]
- 34.Currie E, Schulze A, Zechner R, Walther TC, Farese RV. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–61. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Lan T, Hou J, Zhang J, An Y, Tie L, et al. Atorvastatin sensitizes human non-small cell lung carcinomas to carboplatin via suppression of AKT activation and up regulation of TIMP-1. Int J Biochem Cell Biol. 2012;44:759–69. doi: 10.1016/j.biocel.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 36.Taylor-Harding B, Orsulic S, Karlan BY, Li AJ. Fluvastatin and cisplatin demonstrate synergistic cytotoxicity in epithelial ovarian cancer cells. Gynecol Oncol. 2010;119:549–56. doi: 10.1016/j.ygyno.2010.08.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.