

Abstract
Neurofibromatosis (NF) encompasses a group of distinct genetic disorders in which affected children and adults are prone to the development of benign and malignant tumors of the nervous system. The purpose of this review is to discuss the spectrum of CNS tumors arising in individuals with NF type 1 (NF1) and NF type 2 (NF2), their pathogenic etiologies, and the rational treatment options for people with these neoplasms. This article is a review of preclinical and clinical data focused on the treatment of the most common CNS tumors encountered in children and adults with NF1 and NF2. Although children with NF1 are at risk for developing low-grade gliomas of the optic pathway and brainstem, individuals with NF2 typically manifest low-grade tumors affecting the cranial nerves (vestibular schwannomas), meninges (meningiomas), and spinal cord (ependymomas). With the identification of the NF1 and NF2 genes, molecularly targeted therapies are beginning to emerge, as a result of a deeper understanding of the mechanisms underlying NF1 and NF2 protein function. As we enter into an era of precision oncology, a more comprehensive awareness of the factors that increase the risk of developing CNS cancers in affected individuals, coupled with a greater appreciation of the cellular and molecular determinants that maintain tumor growth, will undoubtedly yield more effective therapies for these cancer predisposition syndromes.
INTRODUCTION
Neurofibromatosis (NF) encompasses at least three distinct medical disorders, including NF type 1 (NF1), NF type 2 (NF2), and schwannomatosis.1 Each of these conditions has a different genetic etiology.2 Although all three disorders are characterized by peripheral nerve sheath tumors (neurofibromas or schwannomas), the diagnostic criteria used to render an accurate clinical diagnosis for each are quite specific and readily distinguish one disorder from another.
NF1 is the most common of the three conditions, affecting 1 in 3,000 individuals worldwide.3 As a tumor predisposition syndrome, individuals with NF1 are prone to the development of a diverse spectrum of benign and malignant cancers. Within the nervous system, the pathognomonic feature of this disorder is the formation of peripheral nerve sheath tumors (neurofibromas); however, children with NF1 manifest brain tumors (optic pathway gliomas [OPGs] and brainstem gliomas [BSGs]). Accurate estimates of the frequency of malignant tumors in NF1 have not been established; however, 5% of individuals will develop a malignant peripheral nerve sheath tumor,4 with a lifetime risk between 9% and 13%,5 whereas the prevalence of high-grade gliomas is 10 to 50 times higher than it is in the general population.6
NF2 affects 1 in 30,000 individuals worldwide and is predominantly a tumor predisposition disorder.7 Children and adults with NF2 harbor a spectrum of nervous system tumors, including cranial and peripheral nerve schwannomas, as well as meningiomas and spinal ependymomas.8 In addition, 60% to 80% of individuals with NF2 may develop early cataracts9 and retinal abnormalities (hamartomas and epiretinal membranes10) that can impair vision.11 Schwannomatosis is less common than NF2, and adults typically present with spinal and peripheral schwannomas.12 In this review, we will restrict our discussion to CNS tumors arising in children and adults with NF1 and NF2. Treatises on peripheral nervous system involvement (neurofibromas, malignant peripheral nerve sheath tumors, and schwannomas) in the neurofibromatoses can be found elsewhere.7,13-17
NEUROFIBROMATOSIS TYPE 1
Optic Gliomas
The most common brain tumor affecting individuals with NF1 is the OPG, seen in 15% to 20% of people with this condition.18 These neoplasms are classified as pilocytic astrocytomas, which do not progress to high-grade malignancies. OPGs can arise anywhere along the optic pathway from the retro-orbital optic nerve to the postchiasmal optic radiations (Figs 1A-1D); however, the majority of these gliomas are restricted to the optic nerves and chiasm. Importantly, OPGs are tumors of childhood, most frequently arising in children younger than 7 years of age (mean age, 4.5 years). Although the majority of these tumors appear in young children, late-onset OPGs have been reported.19
Fig 1.

CNS tumors in neurofibromatosis type 1. (A) Right optic nerve glioma with contrast enhancement. (B) Right optic glioma in the coronal plane, revealing asymmetry in the sizes of the optic nerves. (C) Bilateral optic chiasm and tract glioma. (D) Bilateral optic radiation glioma. (E) Left pontine glioma extending into the medulla. (F) Bilateral midbrain glioma with associated obstructive hydrocephalus (note the enlarged posterior horns of the lateral ventricles). Asterisks denote the location of the tumors in all panels.
OPGs are usually identified on magnetic resonance imaging (MRI), where they typically appear as swollen optic nerves, frequently with robust gadolinium enhancement. Some practitioners use baseline neuroimaging in early childhood; however, a normal brain MRI does not preclude the later development of an OPG.20 Moreover, initially symptomatic children are more likely to require treatment than are those with incidentally identified tumors.21 In most centers, children undergo annual age-appropriate visual assessments, beginning in the first year of life, to detect changes in visual acuity,22,23 with neuroimaging reserved for symptomatic children to track the growth of a known OPG.24
Unlike their sporadic counterparts, NF1-associated OPGs do not always exhibit progressive enlargement on neuroimaging studies, and only 30% to 50% of children with NF1-associated OPGs will experience a decline in visual acuity (two-line decrement). Fewer than 50% of initially symptomatic children will have continued visual impairment requiring treatment. The decision to treat should be based on a progressive decline in visual acuity, using age-appropriate vision tests (Teller, HOTV, and Snellen acuity cards).22
Risk factors for vision loss related to NF1-associated OPGs include young age, optic tract/radiation involvement, and female sex. In studies, children younger than 2 years of age, as well as those harboring optic tract/radiation gliomas, were more likely to experience declines in visual acuity and require treatment.25 Although the frequency of OPGs is similar in males and females with NF1, two studies found that girls with NF1-associated OPG required treatment more often than their male counterparts did,26 especially for OPGs restricted to the optic nerve.27,28
A small number of young children with NF1-associated OPGs will present with signs of precocious puberty. Most of these children harbor chiasmal tumors, requiring evaluation of the hypothalamic-pituitary-gonadal endocrine axis.29 The finding of early secondary sexual characteristics in a child with NF1 should warrant further evaluation for a chiasmal/hypothalamic glioma.
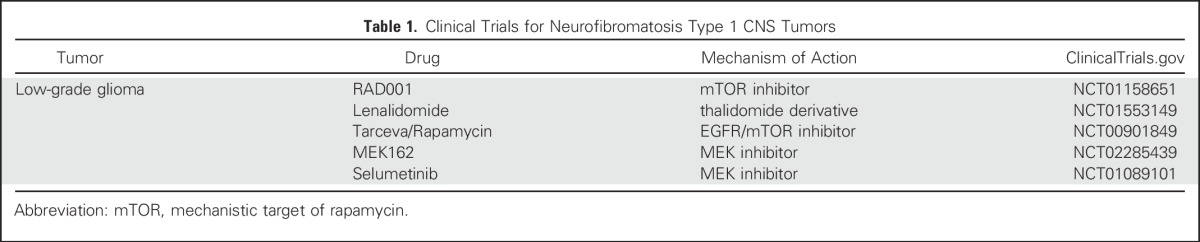
First-line treatment of symptomatic children with NF1-associated OPG is carboplatin/vincristine therapy,30 resulting in tumor stabilization in 50% to 60% of treated individuals. However, it is not clear whether this therapy results in recovery of vision, which remains a source of lifelong morbidity for children and adults with NF1.31 For this reason, therapies aimed at inhibiting the growth control pathways hyperactivated as a result of NF1 loss are currently being pursued in clinical trials (Table 1).
Table 1.
Clinical Trials for Neurofibromatosis Type 1 CNS Tumors
Brainstem Gliomas
The second most frequently encountered brain tumor in individuals with NF1 is the BSG.32 NF1-associated BSGs have been comparatively understudied relative to NF1-associated OPGs, with a small number of reports describing the clinical features in 20 to 30 children with these tumors.33 In general, NF1-associated BSGs are indolent neoplasms, often discovered as incidental findings on neuroimaging studies used to monitor the growth of a known OPG or to discover the etiologic cause of headache (Fig 1E). Similar to NF1-associated OPGs, these tumors are usually pilocytic astrocytomas, arising anywhere within the brainstem (midbrain, pons, and medulla). In contrast to OPGs, children with NF1-associated BSGs tend to be slightly older (average age, 7 to 8 years).34
When children with NF1-associated BSGs are symptomatic, they may come to medical attention because of headache with associated nausea and vomiting. These children typically harbor a midbrain tumor that results in obstructive hydrocephalus (Fig 1F) and requires ventriculoperitoneal shunt placement or an endoscopic ventriculostomy. In other cases, children may have less localizing signs or symptoms, such as cranial nerve palsies, ataxia, or hypotonia. Current medical treatment is also carboplatin/vincristine chemotherapy35; however, molecularly targeted drug therapies are also being evaluated in clinical trials.
Gliomas in Adults
Most low-grade gliomas are observed in children with NF1, but these tumors can arise, albeit less commonly, in adults with NF1.36,37 Although NF1-associated OPGs and BSGs do not progress to malignancy, young adults with NF1 are prone to the development of malignant gliomas. These tumors are uncommon, affecting fewer than 1% of all individuals with NF1; however, it should be appreciated that young adults with NF1 harbor a 10-fold to 50-fold increased risk of developing these deadly cancers.6 For this reason, adults with NF1 who present with signs of increased intracranial pressure, new-onset seizures, or neurologic deficits should be promptly evaluated with neuroimaging studies. Unfortunately, the prognosis for NF1-associated malignant glioma is dismal, and treatments are similar to those administered to adults with malignant gliomas in the general population.
NEUROFIBROMATOSIS TYPE 2
Vestibular Schwannomas
The most common brain tumor affecting individuals with NF2 is the vestibular schwannoma (VS), observed in 90% to 95% of people with NF2.9,38 Bilateral VSs are pathognomonic for this tumor predisposition condition. VSs are contrast enhancing and are best evaluated by high-resolution, contrast-enhanced T1-weighted MRI with fine continuous cuts through the internal auditory canal (Fig 2A). Given their anatomic location, people with VSs usually present with hearing loss, tinnitus, or imbalance, or a combination of these three symptoms.39
Fig 2.

CNS tumors in neurofibromatosis type 2. (A) Bilateral vestibular schwannomas with gadolinium contrast enhancement (asterisk). (B) A large meningioma in the right anterior and middle cranial fossa in the axial plane (asterisk). (C) Multiple spinal cord ependymomas (string of pearls) in the sagittal T1-weighted contrast-enhanced magnetic resonance image of the cervical spine (asterisks).
The decision to treat with either surgery and/or radiosurgery requires a consideration of the patient’s age and his or her general medical condition, hearing status, neurologic symptoms, and the size of the tumor. Many VSs can be safely monitored with serial imaging studies, coupled with assessments of hearing and other neurologic functions.40 When safely possible, the goal of surgery is complete resection of the tumor; however, when the VS is in close proximity to other cranial nerves or the brainstem, surgery may involve only a partial resection. Some patients, particularly those with larger VSs, may develop hydrocephalus, requiring a diversionary ventriculoperitoneal shunt.
Stereotactic radiosurgery is a well-tolerated option for individuals harboring smaller tumors. Although far less invasive, this procedure carries some risk of delayed hearing loss and cranial nerve dysfunction. Relative long-term hearing preservation has been reported in 33% to 53% of patients with NF2 treated with stereotactic radiosurgery.41,42 Subtotal tumor resection followed by stereotactic radiation may be used in some situations, especially for people with large tumors, to preserve the function of the facial nerve and other CNS structures.42
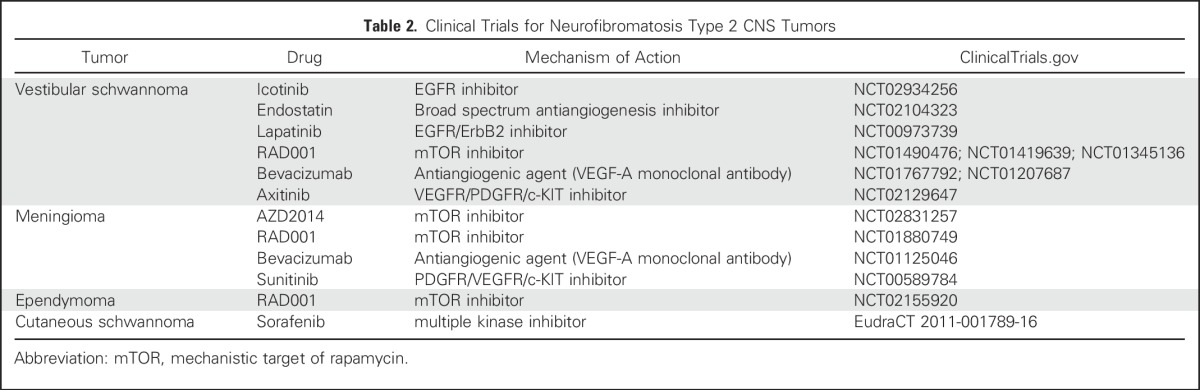
Until recently, there were no promising medical therapies for VSs, limiting the management of these tumors to surgery or radiosurgery. Over the past several years, numerous molecularly targeted treatments have emerged (Table 2). Bevacizumab is a humanized immunoglobulin monoclonal antibody specific for vascular endothelial growth factor, expressed by nearly 100% of VSs.43 Recent studies have shown that bevacizumab treatment resulted in hearing improvement and tumor shrinkage in some, but not all, patients with NF2.44
Table 2.
Clinical Trials for Neurofibromatosis Type 2 CNS Tumors
Meningiomas
Meningiomas are the second most common tumor encountered in individuals with NF2. Intracranial meningiomas are observed in 45% to 58% of people with NF2, whereas spinal meningiomas are found in approximately 20% of affected individuals. Intracranial meningiomas tend to be multiple in number and often develop at a younger age than do their sporadic counterparts.9 Meningiomas homogeneously enhance after contrast administration, and are best evaluated with contrast-enhanced T1-weighted MRI (Fig 2B).
Clinical symptoms from meningiomas are usually related to their size and anatomic location. Although most meningiomas can be safely and fully resected, surgical resection of meningiomas involving the optic nerve sheath and skull base is associated with significant neurologic morbidity.45 In situations where there is residual tumor from a partial resection, stereotactic radiosurgery has been used for local control.46 However, to date, there are no established medical treatments for NF2-associated meningiomas, and clinical studies using molecularly targeted drug therapies are currently being investigated.
Ependymomas
Ependymomas are present in 33% to 53% of individuals with NF2, with the cervical cord or cervicomedullary junction being the most common anatomic sites of involvement.47 On MRI, these tumors appear as hyperintense masses on T2-weighted sequences, which are hypointense to isointense on T1-weighted sequences. The majority of NF2-associated ependymomas are contrast-enhancing tumors (Fig 2C).
Clinical symptoms of spinal cord ependymomas are variable and depend on the size and anatomic location of the tumor. In contrast to sporadic tumors, the majority of NF2-associated spinal tumors are asymptomatic. As such, fewer than 20% of patients with these tumors are symptomatic, and those with intramedullary spinal cord tumors most commonly present with back pain, weakness, or sensory disturbances.48
The management of NF2-associated ependymomas has not been firmly established. Although observation is often used to follow asymptomatic tumors, surgical resections are frequently effective and curative in patients with NF2-associated symptomatic spinal cord ependymomas. The timing of the resection is best determined by detailed neurologic surveillance to assess for early onset of symptoms.49 Because most NF2-associated spinal cord ependymomas are grade II tumors, gross total resection is often the mainstay of treatment, with radiation therapy reserved for recurrent or residual tumors. WHO grade I myxopapillary ependymoma has also been reported in patients with NF2, and these tumors are usually treated with surgery.50 Given that NF2 is a tumor predisposition syndrome, there are concerns about the additional risks of radiation therapy in this patient population. For this reason, chemotherapeutic options for the treatment of recurrent and unresectable tumors are desirable. The evaluation of molecularly targeted therapies for these tumors is currently ongoing. Bevacizumab treatment improved symptoms related to NF2-associated ependymomas.51
FUTURE THERAPIES
Molecular Etiologies
NF1 is caused by a germline mutation in the NF1 gene located on chromosome 17.52 Although this mutation alone is sufficient for some NF1-associated clinical features (eg, autism symptoms), tumor formation requires that a somatic (acquired) mutation in the remaining normal NF1 allele occur.53 In this manner, both copies of the NF1 gene are inactivated in NF1-associated cancers, resulting in loss of NF1 protein (neurofibromin) expression. Neurofibromin is a large protein (220-250 kilodaltons), which functions as a GTPase-activating protein to accelerate the conversion of active, GTP-bound RAS to its inactive, GDP-bound form.54 In this manner, neurofibromin normally suppresses RAS-mediated growth,55 such that its loss in tumor cells results in elevated RAS activity and increased cell proliferation and survival (Fig 3A). Active RAS transmits its growth-promoting signal through cascades of small proteins whose phosphorylation by kinases (eg, RAF kinase, phosphoinositide-3-kinase) leads to successive activation of RAS downstream effectors (eg, MEK, AKT). In neuroglial cells, neurofibromin loss leads to both MEK and AKT activation, which each can converge on the mechanistic target of rapamycin (mTOR).56,57 With the elucidation of the key components of the RAS signaling pathway, a number of molecularly targeted therapies have entered clinical trials, including MEK and mTOR inhibitors (Table 1).
Fig 3.

NF1 and NF2 genes/proteins. (A) The NF1 gene encodes a 2818 amino acid protein with a central RAS-GTPase-activating protein domain. Neurofibromin functions to accelerate the conversion of active GTP-bound RAS to its inactive GDP-bound conformation. Active RAS transmits its growth-promoting signal by activating MEK, AKT, and mechanistic target of rapamycin (mTOR). (B) The NF2 gene encodes a 595 amino acid protein with striking sequence similarity to molecules of the protein 4.1 (ezrin, radixin, and moesin) superfamily. Numerous mechanisms have been proposed for merlin growth regulation, including suppression of RAF/MEK, YAP, mechanistic target of rapamycin (mTOR), ErbB2, Src, FAK, and Rac1 activation. Molecularly targeted therapies that inhibit specific signaling intermediates and receptor tyrosine kinases have been evaluated in preclinical and clinical trials.
The NF2 tumor suppressor gene is located on chromosome 22q, and its protein (schwannomin or merlin) bears striking sequence similarity to a family of structural linker proteins, termed Protein 4.1 molecules.58,59 Among these Protein 4.1 family members, the subclass of molecules containing ezrin, radixin, and moesin (ERM proteins) are the most similar to merlin (Fig 3B). Unlike NF1-associated gliomas, tumor development in NF2 may not require coupling of a germline NF2 mutation with somatic NF2 loss. While NF2-associated tumors lack merlin expression, the mechanism of merlin growth suppression has not been completely elucidated. In this regard, numerous reports in different tissue types have demonstrated that merlin can control cell growth using a plethora of distinct, nonintersecting, signaling pathways, including MEK, YAP, mTOR, ErbB2, SRC, FAK, and RAC1 effectors.60-65 Some of these effectors have emerged as viable targets for therapeutic intervention, such as ErbB2 and mTOR, and agents that inhibit these signaling molecules are now in human clinical trials (Table 2).
Preclinical Models for Drug Discovery and Evaluation
Currently, there are two major barriers that limit progress in the field of NF developmental therapeutics. First, some NF-associated brain tumors (eg, optic pathway and BSGs, spinal ependymomas) are rarely biopsied or surgically removed. To circumvent this particular issue for NF1 brain tumors, the Children’s Tumor Foundation has initiated a multi-institutional international project to sequence low-grade gliomas from children with NF1. Other genomic landscaping efforts focused on brain tumors in NF2 are also in progress.
Second, unlike high-grade malignancies, it has been challenging to maintain NF-associated tumors as patient-derived xenografts (PDXs). To date, successful orthotopic transplant models have only been developed for NF2-associated meningioma.66 In the case of NF1-associated OPG, the low clonogenicity and frequent senescence of glioma tumor stem cells has precluded PDX modeling. In the absence of tractable preclinical PDX models, preclinical researchers have turned to the use of genetically engineered mouse strains.
Although there are no preclinical models of NF1-associated BSGs, NF1-associated OPGs and high-grade gliomas have been generated in mice. Several high-grade glioma models have been established by coupling complete Nf1 gene inactivation with loss of other tumor suppressor genes (eg, p53, PTEN). Each of these high-grade glioma models is distinct with respect to mouse engineering and entails a variety of genetic targeting strategies, such as CRISPR/Cas9,67 standard and conditional knockout mice,68,69 and viral gene silencing.70
Similar to children with NF1, Nf1 optic gliomagenesis in mice requires a combination of a germline inactivating Nf1 gene mutation and somatic Nf1 loss in neuroglial progenitor cells.71 These preclinical models have been instructive in revealing new insights into disease pathogenesis, risk assessment, and tumor treatment. First, optic gliomagenesis requires a productive interaction between neoplastic tumor cells and non-neoplastic immune system–like cells (microglia) in the glioma microenvironment.72 These monocytes provide growth-promoting signals (eg, chemokines) for NF1-deficient neoplastic cells,73 such that silencing microglia function or chemokine signaling greatly attenuated optic glioma growth in mice.74 Second, mice with Nf1 optic gliomas exhibit reduced visual acuity, which reflects retinal ganglion cell loss.75 Using these mice, the molecular basis for the sexual dimorphism observed in female children with NF1 (reduced visual acuity) was identified.76 In addition, defining the temporal course of events in mice has revealed therapeutic windows in which treatments at the time of early retinal ganglion cell death may limit additional vision loss.77 Third, Nf1 optic glioma mice have allowed early preclinical evaluations of promising therapies that inhibit RAS downstream signaling effectors, leading to clinical trials with MEK and mTOR inhibitors.78,79
Although no models of NF2-associated ependymoma have been generated to date, Nf2 genetically engineered mouse strains with meningioma and schwannoma have been developed. Somatic Nf2 loss after subarachnoid or subdural viral injection of Cre recombinase into newborn Nf2flox/flox conditional knockout mice results in meningioma in 20% to 30% of mice by 11 months of age.80 More aggressive tumors develop when Nf2 loss is coupled with loss of other tumor suppressor genes (eg, Ink4a).81 In addition, these mice have been instructive in identifying a common cell of origin for these tumors82 and the value of HSP90 inhibitors as potential treatments for NF2-associated nervous system tumors.83 Similarly, mice with biallelic Nf2 inactivation in periostin-expressing cells develop schwannomas histologically identical to their human counterparts,84 as well as functional impairments in hearing and balance.
With the availability of an efficient Neurofibromatosis Clinical Trials Consortium85 and numerous authenticated preclinical models of NF-associated brain tumors, it now becomes possible to identify molecular targets and evaluate their efficacy in mice prior to human clinical trials. As we enter an era of precision medicine, we are poised to make outstanding progress for NF1 and NF2 CNS tumors. However, it is important to deploy these preclinical platforms in a way that most closely resembles human clinical trials, both in terms of dosing and outcome measures.86 This is particularly relevant to NF1-associated OPGs and NF2-associated VSs, where vision and hearing loss, respectively, need to be considered as success metrics in rodent drug trials. Moreover, individuals with NF-associated brain tumors represent a heterogeneous population of individuals in terms of sex,28 germline NF gene mutation,87,88 and coexisting genetic changes.89 The use of a diverse collection of mice that fully capture this heterogeneity will be critical to identify populations of persons most likely to respond to any given therapy. Third, we need to better calibrate our expectations with respect to celebrating preclinical outcomes. Setting a higher bar for tumor responses in rodent drug studies may result in greater efficacy in human clinical trials. In summary, we have come a long way since the discovery of the NF1 and NF2 genes in the early 1990s and now are uniquely positioned to find effective medical therapies for the CNS tumors arising in this population of at-risk individuals.
Footnotes
Supported by a Research Program Award from the National Institute of Neurologic Disorders and Stroke (No. 1-R35-NS097211-01; D.H.G.).
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
CNS Tumors in Neurofibromatosis
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Jian Campian
Honoraria: Novocure
Consulting or Advisory Role: Novocure
David H. Gutmann
Patents, Royalties, Other Intellectual Property: NF1 Gene Patent
REFERENCES
- 1.Kresak JL, Walsh M: Neurofibromatosis: A review of NF1, NF2, and schwannomatosis. J Pediatr Genet 5:98-104, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blakeley JO, Plotkin SR: Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro-oncol 18:624-638, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson JL, Gutmann DH: Neurofibromatosis type 1. Handb Clin Neurol 132:75-86, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Ingham S, Huson SM, Moran A, et al. : Malignant peripheral nerve sheath tumours in NF1: Improved survival in women and in recent years. Eur J Cancer 47:2723-2728, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Evans DG, Huson SM, Birch JM: Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res 2:17, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutmann DH, Rasmussen SA, Wolkenstein P, et al. : Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1). Neurology 59:759-761, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Karajannis MA, Ferner RE: Neurofibromatosis-related tumors: Emerging biology and therapies. Curr Opin Pediatr 27:26-33, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans DG: Neurofibromatosis type 2. Handb Clin Neurol 132:87-96, 2015 [DOI] [PubMed] [Google Scholar]
- 9.Parry DM, Eldridge R, Kaiser-Kupfer MI, et al. : Neurofibromatosis 2 (NF2): Clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet 52:450-461, 1994 [DOI] [PubMed] [Google Scholar]
- 10. doi: 10.1016/s0002-9394(14)70558-6. Meyers SM, Gutman FA, Kaye LD, et al: Retinal changes associated with neurofibromatosis 2. Trans Am Ophthalmol Soc 93:245-252, 1995; discussion 252-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sisk RA, Berrocal AM, Schefler AC, et al. : Epiretinal membranes indicate a severe phenotype of neurofibromatosis type 2. Retina 30:S51-S58, 2010. (4, suppl) [DOI] [PubMed] [Google Scholar]
- 12.Merker VL, Esparza S, Smith MJ, et al. : Clinical features of schwannomatosis: A retrospective analysis of 87 patients. Oncologist 17:1317-1322, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilton DA, Hanemann CO: Schwannomas and their pathogenesis. Brain Pathol 24:205-220, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koontz NA, Wiens AL, Agarwal A, et al. : Schwannomatosis: The overlooked neurofibromatosis? AJR Am J Roentgenol 200:W646-W653, 2013 [DOI] [PubMed] [Google Scholar]
- 15.Carroll SL: Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol 123:321-348, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu-Emerson C, Plotkin SR: The neurofibromatoses. Part 2: NF2 and schwannomatosis. Rev Neurol Dis 6:E81-E86, 2009 [PubMed] [Google Scholar]
- 17.Lin AL, Gutmann DH: Advances in the treatment of neurofibromatosis-associated tumours. Nat Rev Clin Oncol 10:616-624, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Listernick R, Charrow J, Greenwald MJ, et al. : Optic gliomas in children with neurofibromatosis type 1. J Pediatr 114:788-792, 1989 [DOI] [PubMed] [Google Scholar]
- 19.Listernick R, Ferner RE, Piersall L, et al. : Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology 63:1944-1946, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Listernick R, Charrow J, Greenwald M: Emergence of optic pathway gliomas in children with neurofibromatosis type 1 after normal neuroimaging results. J Pediatr 121:584-587, 1992 [DOI] [PubMed] [Google Scholar]
- 21.King A, Listernick R, Charrow J, et al. : Optic pathway gliomas in neurofibromatosis type 1: The effect of presenting symptoms on outcome. Am J Med Genet A 122A:95-99, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Fisher MJ, Avery RA, Allen JC, et al. : Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology 81:S15-S24, 2013. (21, suppl 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Listernick R, Ferner RE, Liu GT, et al. : Optic pathway gliomas in neurofibromatosis-1: Controversies and recommendations. Ann Neurol 61:189-198, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Listernick R, Charrow J: Knowledge without truth: Screening for complications of neurofibromatosis type 1 in childhood. Am J Med Genet A 127A:221-223, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Fisher MJ, Loguidice M, Gutmann DH, et al. : Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: A multicenter retrospective analysis. Neuro-oncol 14:790-797, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diggs-Andrews KA, Brown JA, Gianino SM, et al. : Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol 75:309-316, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher MJ, Loguidice M, Gutmann DH, et al. : Gender as a disease modifier in neurofibromatosis type 1 optic pathway glioma. Ann Neurol 75:799-800, 2014 [DOI] [PubMed] [Google Scholar]
- 28. Diggs-Andrews KA, Brown JA, Gianino SM, et al: Reply: To PMID 24375753. Ann Neurol 75:800-801, 2014. [DOI] [PubMed]
- 29.Habiby R, Silverman B, Listernick R, et al. : Precocious puberty in children with neurofibromatosis type 1. J Pediatr 126:364-367, 1995 [DOI] [PubMed] [Google Scholar]
- 30.Mahoney DH, Jr, Cohen ME, Friedman HS, et al. : Carboplatin is effective therapy for young children with progressive optic pathway tumors: A Pediatric Oncology Group phase II study. Neuro-oncol 2:213-220, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dodgshun AJ, Elder JE, Hansford JR, et al. : Long-term visual outcome after chemotherapy for optic pathway glioma in children: Site and age are strongly predictive. Cancer 121:4190-4196, 2015 [DOI] [PubMed] [Google Scholar]
- 32.Guillamo JS, Créange A, Kalifa C, et al. : Prognostic factors of CNS tumours in neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain 126:152-160, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Ullrich NJ, Raja AI, Irons MB, et al. : Brainstem lesions in neurofibromatosis type 1. Neurosurgery 61:762-766, 2007; discussion 766-767 [DOI] [PubMed] [Google Scholar]
- 34.Mahdi J, Shah AC, Sato A, et al. : A multi-institutional study of brainstem gliomas in children with neurofibromatosis type 1. Neurology 88:1584-1589, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brossier NM, Gutmann DH: Improving outcomes for neurofibromatosis 1-associated brain tumors. Expert Rev Anticancer Ther 15:415-423, 2015 [DOI] [PubMed] [Google Scholar]
- 36.Strowd RE, III, Rodriguez FJ, McLendon RE, et al. : Histologically benign, clinically aggressive: Progressive non-optic pathway pilocytic astrocytomas in adults with NF1. Am J Med Genet A 170:1455-1461, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gutmann DH, James CD, Poyhonen M, et al. : Molecular analysis of astrocytomas presenting after age 10 in individuals with NF1. Neurology 61:1397-1400, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Evans DG, Huson SM, Donnai D, et al. : A clinical study of type 2 neurofibromatosis. Q J Med 84:603-618, 1992 [PubMed] [Google Scholar]
- 39.Asthagiri AR, Parry DM, Butman JA, et al. : Neurofibromatosis type 2. Lancet 373:1974-1986, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slattery WH, III, Fisher LM, Iqbal Z, et al. : Vestibular schwannoma growth rates in neurofibromatosis type 2 natural history consortium subjects. Otol Neurotol 25:811-817, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Phi JH, Kim DG, Chung HT, et al. : Radiosurgical treatment of vestibular schwannomas in patients with neurofibromatosis type 2: Tumor control and hearing preservation. Cancer 115:390-398, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Mathieu D, Kondziolka D, Flickinger JC, et al. : Stereotactic radiosurgery for vestibular schwannomas in patients with neurofibromatosis type 2: An analysis of tumor control, complications, and hearing preservation rates. Neurosurgery 60:460-468, 2007, discussion 468-470 [DOI] [PubMed] [Google Scholar]
- 43.Dalgorf DM, Rowsell C, Bilbao JM, et al. : Immunohistochemical investigation of hormone receptors and vascular endothelial growth factor concentration in vestibular schwannoma. Skull Base 18:377-384, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plotkin SR, Stemmer-Rachamimov AO, Barker FG, II, et al. : Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med 361:358-367, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Couldwell WT, Fukushima T, Giannotta SL, et al. : Petroclival meningiomas: Surgical experience in 109 cases. J Neurosurg 84:20-28, 1996 [DOI] [PubMed] [Google Scholar]
- 46.Wentworth S, Pinn M, Bourland JD, et al. : Clinical experience with radiation therapy in the management of neurofibromatosis-associated central nervous system tumors. Int J Radiat Oncol Biol Phys 73:208-213, 2009 [DOI] [PubMed] [Google Scholar]
- 47.Mautner VF, Tatagiba M, Lindenau M, et al. : Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. AJR Am J Roentgenol 165:951-955, 1995 [DOI] [PubMed] [Google Scholar]
- 48.Chang UK, Choe WJ, Chung SK, et al. : Surgical outcome and prognostic factors of spinal intramedullary ependymomas in adults. J Neurooncol 57:133-139, 2002 [DOI] [PubMed] [Google Scholar]
- 49.Hoshimaru M, Koyama T, Hashimoto N, et al. : Results of microsurgical treatment for intramedullary spinal cord ependymomas: Analysis of 36 cases. Neurosurgery 44:264-269, 1999 [DOI] [PubMed] [Google Scholar]
- 50.Plotkin SR, O’Donnell CC, Curry WT, et al. : Spinal ependymomas in neurofibromatosis Type 2: A retrospective analysis of 55 patients. J Neurosurg Spine 14:543-547, 2011 [DOI] [PubMed] [Google Scholar]
- 51.Farschtschi S, Merker VL, Wolf D, et al. : Bevacizumab treatment for symptomatic spinal ependymomas in neurofibromatosis type 2. Acta Neurol Scand 133:475-480, 2016 [DOI] [PubMed] [Google Scholar]
- 52.Viskochil D, Buchberg AM, Xu G, et al. : Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62:187-192, 1990 [DOI] [PubMed] [Google Scholar]
- 53.Gutmann DH, McLellan MD, Hussain I, et al. : Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res 23:431-439, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu GF, O’Connell P, Viskochil D, et al. : The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62:599-608, 1990 [DOI] [PubMed] [Google Scholar]
- 55.Basu TN, Gutmann DH, Fletcher JA, et al. : Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 356:713-715, 1992 [DOI] [PubMed] [Google Scholar]
- 56.Dasgupta B, Yi Y, Chen DY, et al. : Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res 65:2755-2760, 2005 [DOI] [PubMed] [Google Scholar]
- 57.Jiang Y, Scheinberg JA, Senevirathne R, et al. : The efficacy of short and repeated high-pressure processing treatments on the reduction of non-O157:H7 Shiga-toxin producing Escherichia coli in ground beef patties. Meat Sci 102:22-26, 2015 [DOI] [PubMed] [Google Scholar]
- 58.Bianchi AB, Hara T, Ramesh V, et al. : Mutations in transcript isoforms of the neurofibromatosis 2 gene in multiple human tumour types. Nat Genet 6:185-192, 1994 [DOI] [PubMed] [Google Scholar]
- 59.Rouleau GA, Merel P, Lutchman M, et al. : Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363:515-521, 1993 [DOI] [PubMed] [Google Scholar]
- 60.Shapiro IM, Kolev VN, Vidal CM, et al. : Merlin deficiency predicts FAK inhibitor sensitivity: A synthetic lethal relationship. Sci Transl Med 6:237ra68, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garcia C, Gutmann DH: Nf2/merlin controls spinal cord neural progenitor function in a Rac1/ErbB2-dependent manner. PLoS One 9:e97320, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giovannini M, Bonne NX, Vitte J, et al. : mTORC1 inhibition delays growth of neurofibromatosis type 2 schwannoma. Neuro-oncol 16:493-504, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ammoun S, Hanemann CO: Emerging therapeutic targets in schwannomas and other merlin-deficient tumors. Nat Rev Neurol 7:392-399, 2011 [DOI] [PubMed] [Google Scholar]
- 64.Garcia-Rendueles ME, Ricarte-Filho JC, Untch BR, et al. : NF2 loss promotes oncogenic RAS-induced thyroid cancers via YAP-dependent transactivation of RAS proteins and sensitizes them to MEK inhibition. Cancer Discov 5:1178-1193, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Striedinger K, VandenBerg SR, Baia GS, et al. : The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia 10:1204-1212, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burns SS, Chang LS: Generation of noninvasive, quantifiable, orthotopic animal models for NF2-associated schwannoma and meningioma. Methods Mol Biol 1427:59-72, 2016 [DOI] [PubMed] [Google Scholar]
- 67.Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, et al. : Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun 6:7391, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kwon CH, Zhao D, Chen J, et al. : Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res 68:3286-3294, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reilly KM, Loisel DA, Bronson RT, et al. : Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet 26:109-113, 2000 [DOI] [PubMed] [Google Scholar]
- 70.Marumoto T, Tashiro A, Friedmann-Morvinski D, et al. : Development of a novel mouse glioma model using lentiviral vectors. Nat Med 15:110-116, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bajenaru ML, Hernandez MR, Perry A, et al. : Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res 63:8573-8577, 2003 [PubMed] [Google Scholar]
- 72.Pong WW, Higer SB, Gianino SM, et al. : Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol 73:303-308, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Solga AC, Pong WW, Kim KY, et al. : RNA sequencing of tumor-associated microglia reveals Ccl5 as a stromal chemokine critical for neurofibromatosis-1 glioma growth. Neoplasia 17:776-788, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daginakatte GC, Gutmann DH: Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet 16:1098-1112, 2007 [DOI] [PubMed] [Google Scholar]
- 75.Hegedus B, Hughes FW, Garbow JR, et al. : Optic nerve dysfunction in a mouse model of neurofibromatosis-1 optic glioma. J Neuropathol Exp Neurol 68:542-551, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. doi: 10.1084/jem.20160447. Toonen JA, Solga AC, Ma Y, et al: Estrogen activation of microglia underlies the sexually dimorphic differences in Nf1 optic glioma-induced retinal pathology. J Exp Med 214:17-25, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. doi: 10.1093/neuonc/now267. Toonen JA MY, Ma Y, Gutmann DH: Defining the temporal course of murine neurofibromatosis-1 optic gliomagenesis reveals a therapeutic window to attenuate retinal dysfunction. Neuro-oncol 10.1093/neuonc/now267 [epub ahead of print on December 29, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hegedus B, Banerjee D, Yeh TH, et al. : Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res 68:1520-1528, 2008 [DOI] [PubMed] [Google Scholar]
- 79.Kaul A, Toonen JA, Cimino PJ, et al. : Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro-oncol 17:843-853, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kalamarides M, Niwa-Kawakita M, Leblois H, et al. : Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev 16:1060-1065, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kalamarides M, Stemmer-Rachamimov AO, Takahashi M, et al. : Natural history of meningioma development in mice reveals: A synergy of Nf2 and p16(Ink4a) mutations. Brain Pathol 18:62-70, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalamarides M, Stemmer-Rachamimov AO, Niwa-Kawakita M, et al. : Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene 30:2333-2344, 2011 [DOI] [PubMed] [Google Scholar]
- 83.Tanaka K, Eskin A, Chareyre F, et al. : Therapeutic potential of HSP90 inhibition for neurofibromatosis type 2. Clin Cancer Res 19:3856-3870, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gehlhausen JR, Park SJ, Hickox AE, et al. : A murine model of neurofibromatosis type 2 that accurately phenocopies human schwannoma formation. Hum Mol Genet 24:1-8, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gutmann DH, Blakeley JO, Korf BR, et al. : Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin Investig Drugs 22:443-462, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gutmann DH: Eliminating barriers to personalized medicine: Learning from neurofibromatosis type 1. Neurology 83:463-471, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Toonen JA, Anastasaki C, Smithson LJ, et al. : NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum Mol Genet 25:1703-1713, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gutmann DH, Hirbe AC, Haipek CA: Functional analysis of neurofibromatosis 2 (NF2) missense mutations. Hum Mol Genet 10:1519-1529, 2001 [DOI] [PubMed] [Google Scholar]
- 89.Kaul A, Toonen JA, Gianino SM, et al. : The impact of coexisting genetic mutations on murine optic glioma biology. Neuro-oncol 17:670-677, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]