

Abstract
Background
Research comparing the survival of children with familial dilated cardiomyopathy (FDCM) to that of children with idiopathic dilated cardiomyopathy (IDCM) has produced conflicting results.
Methods and Results
We analyzed data from children with FDCM or IDCM using the National Heart, Lung, and Blood Institute-funded Pediatric Cardiomyopathy Registry. Compared to children with IDCM (n=647), children with FDCM (n=223) were older (mean 6.2 vs. 4.5 years, P <0.001), less often had heart failure (64% vs. 78%, P <0.001), had less-depressed mean left ventricular (LV) fractional shortening z-scores (−7.85 ± 3.98 vs. −9.06 ± 3.89, P <0.001) and lower end-diastolic dimension z-scores (4.12 ± 2.61 vs. 4.91 ± 2.57, P <0.001) at diagnosis. The cumulative incidence of death was lower for patients with FDCM compared with IDCM (P=0.04; hazard ratio 0.64, P=0.06) but no difference in risk of transplant or the combined death or transplant outcome. There was no difference in the proportion of children with echocardiographic normalization at three years of follow-up (FDCM, 30% vs. IDCM, 26%; P=0.33). Multivariable analysis showed no difference in outcomes between FDCM and IDCM but for both groups older age, congestive heart failure (CHF) and increased LV end-systolic dimension z-score at diagnosis were independently associated with an increased risk of death or heart transplantation (all Ps <0.001).
Conclusions
There was no survival difference between FDCM and IDCM after adjustment for other factors. Older age, CHF, and greater LV dilation at diagnosis were independently associated with increased risk of the combined endpoint of death or transplantation.
Clinical Trial Registration
Keywords: dilated cardiomyopathy, genetics, family history, familial cardiomyopathy, idiopathic
Subject Terms: Cardiomyopathy, Pediatrics
Reports from the National Heart, Lung, and Blood Institute-funded Pediatric Cardiomyopathy Registry (PCMR) have shown that dilated cardiomyopathy (DCM) is diagnosed in 0.57 of every 100,000 children per year in the US and has a 5-year risk of death or heart transplantation of 46%.1,2 These results are consistent with those from pediatric cohort studies in Australia and Finland.3,4
In adults, DCM occurs mainly secondary to coronary artery disease.5 In children, DCM has a wide spectrum of causes: myocarditis, neuromuscular disease, inborn errors of metabolism, malformation syndromes, FDCM and secondary forms caused by environmental or therapeutic exposures, endocrine disease, although nearly 70% of cases are categorized as idiopathic.1
Over the past 30 years, outcomes of children with cardiomyopathy have improved little and only by the introduction of heart transplantation.6 In a previous study, Towbin et al. found that the 5-year survival of children with DCM varied depending on cause,1 with the best clinical outcomes observed in children with FDCM (94%), and the worst in children with neuromuscular disorders (57%). Survival of children with idiopathic disease and malformation syndromes was intermediate (76% for both).1 Unfortunately, the same study found that a cause of DCM was identified in only 34% of children, although comprehensive etiologic testing was not always conducted.1,7 Another report from the PCMR, using competing risks analysis, showed that children who had FDCM at the time of diagnosis had a lower death rate but a similar transplant rate at 5 years after diagnosis when compared to IDCM.8
Since previous studies have suggested that outcomes in children with DCM are related to the etiology of DCM, improving the ability to identify the underlying etiology of the disease could improve risk estimates and treatment strategies. Knowing the underlying etiology of DCM is also a prerequisite for risk assessment for family members. Therefore, we undertook a new PCMR analysis comparing children with FDCM to children with IDCM with a documented negative family history.
We hypothesized that transplant-free survival (absence of death and/or heart transplantation) at time of last follow-up would be better in children with FDCM than in those with IDCM. Using data from the PCMR, we also identified and compared the predictors of the combined endpoint of death or transplant of children with FDCM to those with IDCM.
Methods
The PCMR is a cooperative effort of nearly 100 centers in the United States and Canada that enrolled children with primary cardiomyopathy between January 1990 and February 2009. The PCMR design and implementation are described in detail elsewhere.2 The study was approved by an institutional review committee and the subjects gave informed consent.
Briefly, children less than 18 years old, newly diagnosed with cardiomyopathy at participating centers, were eligible for inclusion. Children were ineligible if they had a specific secondary cause of DCM, such as pulmonary parenchymal or vascular disease, endocrine disease, rheumatic disease, immunologic disease, cardiotoxic exposures, or a congenital cardiovascular malformation that occurred independently of a malformation syndrome. All participating centers obtained Institutional Review Board approval to enroll patients in the registry.
Demographics, family history, vital and transplant status, and clinical data relevant to cardiomyopathy, including echocardiographic measurements, were collected at diagnosis and annually thereafter.
For the present analysis, the diagnosis of DCM (defined as a DCM phenotype without a mixed phenotype such as DCM with hypertrophic or restrictive features or coexisting left ventricular non-compaction) was determined using strict echocardiographic criteria, including evidence of left ventricular (LV) dilation and systolic dysfunction; semi-quantitative echocardiographic patterns of DCM; a confirmed DCM diagnosis by autopsy or endomyocardial biopsy; or other compelling clinical evidence of DCM.1 Familial DCM was defined as a patient with a clinical diagnosis of FDCM documented in the medical record by the treating cardiologist at the last known follow-up and/or at least one or more affected family members (regardless of age) noted in the medical record. This report is based on analysis of the results from the PCMR database as of January 14, 2013.
Study Variables
Demographic variables included age, sex, and race/ethnicity. Congestive heart failure (CHF) at diagnosis, as indicated in the medical record and a family history of sudden death were also included in analyses. Echocardiographic measurements, including LV end-diastolic dimension (LVEDD) and end-systolic dimension (LVESD), LV end-diastolic posterior wall thickness, end-diastolic septal thickness, and LV mass were expressed as z-scores relative to body surface area in normal children;9 LV fractional shortening (LVFS) and LV ejection fraction (LVEF) were expressed as age-adjusted z-scores.10 Data on the presence of atrial enlargement (left, right, or both), and LVEF were available for less than half of children so were not analyzed further.
Statistical Methods
A Fisher exact test was used to compare the frequencies of categorical variables; Student’s t test (for variables with normal distributions) and the Wilcoxon rank sum test (for variables with other continuous distributions) were also used to evaluate differences by the presence or absence of FDCM.
The Kaplan-Meier method was used to estimate the incidence of the composite endpoint of the earliest occurring of pre-transplant death and cardiac transplant. The two components of the composite, pre-transplant mortality and transplant, were analyzed using cumulative incidence functions (CIFs), as death is a competing risk to the occurrence of transplant, with use of Gray’s test11 for hypothesis testing. Regression modeling to compare the incidence of pre-transplant death and the incidence of transplant between the FDCM vs. IDCM groups was conducted using the subdistribution hazard model methodology of Fine and Gray.12 Similarly, the echocardiographic outcomes were analyzed in a competing risks framework with the methodology described above13. The component outcomes were echocardiographic normalization, pre-transplant death/transplant composite and persistently abnormal echocardiogram. Echocardiographic normalization was defined as having both LVEDD <2 SD above normal for body surface area (LVEDD z-score < 2) and LVFS or LVEF <2 SD below normal for age (LVFS or LVEF z-score > −2).
Cox proportional hazards regression modeling was used to assess FDCM vs. IDCM differences in the composite outcome of death/transplant, and to identify the factors that were univariately associated with the composite endpoint. To determine whether predictors of death/transplant differed for FDCM and IDCM, each covariate was tested for interaction. Multivariable modeling was used to identify independent predictors of death/transplant. The multivariable model selection procedure included all variables with a univariate P-value less than 0.20, with the exception of medications at diagnosis, left or right atrial enlargement and ejection fraction because of the large number of missing values for these variables. The procedure to construct appropriate covariate-adjusted models included all variables with a FDCM vs. IDCM univariate P-value less than 0.10. However, LVEDD rather than LV ESD was utilized for final models due to missing data in the latter.
Alpha was set at 0.05, and all tests were two-tailed. Data were analyzed with the Statistical Analysis System statistical software program, version 9.3 (SAS Institute, Cary, North Carolina), the free software package R “cmprsk” and S-PLUS version 6.1 (Insightful Corporation, Seattle, Washington).
Results
Demographic and Clinical Characteristics
As of January 2013, 1834 children with DCM were enrolled in the PCMR. Of these, 223 were identified as having FDCM and 647 as having IDCM as of their last follow-up visit. We excluded 964 patients: 357 whose family history was unknown; 153 with a diagnosis of IDCM who were found to have another etiology (myocarditis, inborn errors of metabolism, malformation syndromes, neuromuscular disorders) at follow-up; and 454 children in whom a specific etiology (other than FDCM) was identified at clinical presentation. The median follow-up time of patients who did not reach an endpoint of death or transplantation (non-transplanted survivors) was 3.7 years (interquartile range [IQR]: 1.0 to 6.9 years) for the FDCM group and 2.1 years (IQR: 0.6 to 5.1) for the IDCM group. The median follow-up time for the entire cohort across both groups was 2.4years (IQR: 0.8 to 5.6)
Children with FDCM were significantly older at presentation than children with IDCM (median, 3.3 years [IQR: 0.3 to 12.1] vs. 1.0 year [IQR: 0.3 to 9.2]), less likely to have CHF at diagnosis (64.1% vs. 78.2%, P < 0.001), and more likely to have a family history of sudden death (39.0% vs. 2.5%, P < 0.001; Table 1). Children with FDCM also had significantly lower mean z-scores for LVEDD (P = 0.006), and significantly higher mean z-scores for end-diastolic septal thickness (P = 0.034), LVFS (P < 0.001), and LVEF (P = 0.008; Table 1). Compared to children with FDCM, children with IDCM were significantly more likely to receive any cardiac medication at diagnosis (95.8% vs. 91.4%, P = 0.03) and antiarrhythmic therapy (23.5% vs. 13.7%, P = 0.02).
Table 1.
Characteristics of Children with Familial Dilated Cardiomyopathy or Idiopathic Dilated Cardiomyopathy.
| Characteristic | Overall | FDCM | IDCM | P Value* |
|---|---|---|---|---|
| N† | 870 | 223 | 647 | |
| Age at diagnosis | ||||
| Mean (SD), years | 4.9 (5.9) | 6.2 (6.4) | 4.5 (5.8) | 0.001 |
| Median (IQR), years | 1.2 (0.3–10.3) | 3.3 (0.3–12.1) | 1.0 (0.3–9.2) | 0.04 |
| Age < 1 year at diagnosis, % | 47.4 | 42.2 | 49.2 | 0.07 |
| Male, % | 50.6 | 54.7 | 49.1 | 0.16 |
| Race/ethnicity, % | 0.06 | |||
| White | 54.2 | 60.1 | 52.2 | |
| Black | 21.2 | 16.1 | 23.0 | |
| Hispanic | 17.3 | 15.1 | 18.0 | |
| Other | 7.3 | 8.7 | 6.8 | |
| Congestive heart failure at diagnosis, % | 74.6 | 64.1 | 78.2 | < 0.001 |
| Family history of sudden death, % (total n) | 10.5 (727) | 39.0 (159) | 2.5 (568) | < 0.001 |
| Left ventricular echocardiographic z-scores, mean (SD) | ||||
| End-diastolic dimension (total n = 699, 175, 524) | 4.71 (2.60) | 4.12 (2.61) | 4.91 (2.57) | < 0.001 |
| End-systolic dimension (total n = 614, 160, 454) | 6.35 (3.00) | 5.79 (2.89) | 6.55 (3.01) | 0.006 |
| Fractional shortening (total n = 713, 183, 530) | −8.74 (3.95) | −7.85 (3.98) | −9.06 (3.89) | < 0.001 |
| End-diastolic posterior wall thickness (total n = 553, 145, 408) | −0.60 (2.58) | −0.63 (2.66) | −0.59 (2.56) | 0.86 |
| End-diastolic septal thickness (total n = 515, 126, 389) | −0.98 (1.92) | −0.67 (1.99) | −1.09 (1.88) | 0.03 |
| Mass (total n = 545, 143, 402) | 2.49 (2.78) | 2.32 (3.08) | 2.55 (2.67) | 0.43 |
| Medications at diagnosis (n) | ||||
| Any cardiac medication | 94.7% (786) | 91.4% (198) | 95.8% (588) | 0.03 |
| Anticongestive therapy¶ | 86% (843) | 82.2% (213) | 87.3% (630) | 0.07 |
| Antiarrhythmic therapy | 20.6% (462) | 13.7% (139) | 23.5% (323) | 0.02 |
| ACE inhibitor | 62.7% (461) | 60.9% (138) | 63.5% (323) | 0.60 |
| Beta blocker | 13.2% (461) | 15.1% (139) | 12.4% (322) | 0.46 |
| Carnitine | 1.5% (461) | 0.7% (139) | 1.9% (322) | 0.68 |
FDCM, familial dilated cardiomyopathy; IDCM, idiopathic dilated cardiomyopathy; IQR, interquartile range.
P values compare the differences between FDCM and IDCM.
Sample size is smaller than the total stated for selected variables (family history; LV mass).
Anticongestive therapy is defined as digoxin and/or diuretics
Outcomes
We observed 99 events in the 223 patients with FDCM (22 deaths, 77 transplants) and 296 events in the 647 patients with IDCM (90 deaths, 206 transplants). There was a significant difference in the cumulative incidence of pre-transplant mortality; children with FDCM fared better than those with IDCM (Gray’s test P = 0.04; Fine-Gray hazard ratio for pre-transplant death 0.64; Figure 1a). The cumulative incidence of pre-transplant death at 3 years post-cardiomyopathy was 8.8% (95 confidence interval [CI]: 5.4% to 13.2%) and 14.6% (95% CI: 11.8% to 17.8%) in the FDCM and IDCM groups, respectively. However, the hazard ratio for pre-transplant death in the FDCM vs. IDCM groups after adjustment for the presence of CHF at diagnosis did not differ from one (hazard ratio [HR]: 0.71, 95% CI 0.44 to 1.13, P = 0.14; Table 2).
Figure 1.
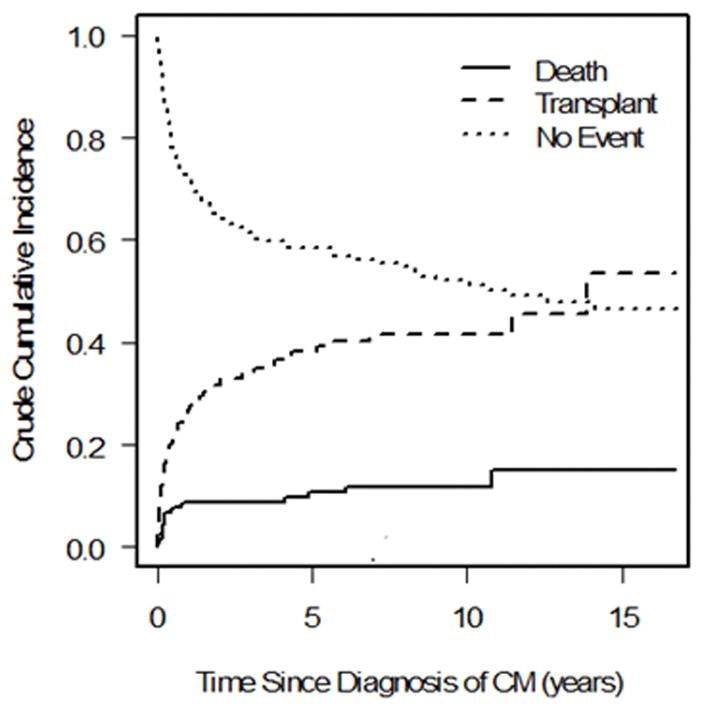
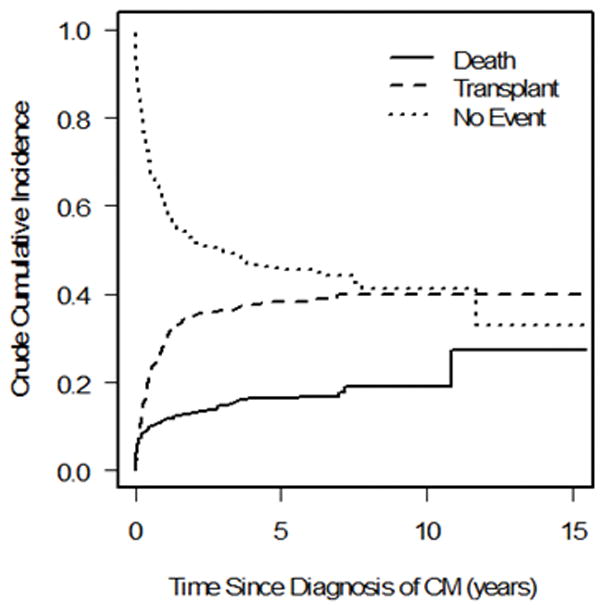
Competing outcomes analysis showing estimated proportion of patients in three mutually exclusive groups at all times after diagnosis (alive, transplanted, and died without transplant) in a) familial dilated cardiomyopathy and b) idiopathic dilated cardiomyopathy. Summation of proportions equals 1 at all time points. CM = cardiomyopathy.
Table 2.
Unadjusted and Covariate-adjusted Models for Clinical Outcome in Children with Familial DCM or Idiopathic DCM
| Outcome | N | Predictor | Hazard Ratio (95% CI) | P Value* |
|---|---|---|---|---|
| Unadjusted Models | ||||
| Death | 870 | Familial DCM vs. Idiopathic DCM | 0.64 (0.40–1.02) | 0.04/0.06 |
| Transplant | 870 | Familial DCM vs. Idiopathic DCM | 1.02 (0.78–1.32) | 0.72/0.91 |
| Death/Transplant | 870 | Familial DCM vs. Idiopathic DCM | 0.85 (0.6–1.1) | 0.15 |
| Covariate-Adjusted Models | ||||
| Death | 869 | Familial DCM vs. Idiopathic DCM | 0.71 (0.44–1.13) | 0.14 |
| Presence of CHF at diagnosis | 2.36 (1.34–4.15) | 0.003 | ||
| Transplant | 698 | Familial DCM vs. Idiopathic DCM | 1.05 (0.76–1.44) | 0.77 |
| Age at diagnosis, yr | 1.08 (1.06–1.11) | < 0.001 | ||
| Presence of CHF at diagnosis | 2.88 (1.88–4.43) | < 0.001 | ||
| LVEDD z-score at diagnosis | 1.20 (1.13–1.27) | < 0.001 | ||
| Death/Transplant | 698 | Familial DCM vs. Idiopathic DCM | 0.97 (0.74–1.28) | 0.85 |
| Age at diagnosis, yr | 1.06 (1.04–1.08) | < 0.001 | ||
| Presence of CHF at diagnosis | 3.45 (2.37–5.02) | < 0.001 | ||
| LVEDD dimension z-score at diagnosis | 1.13 (1.08–1.19) | < 0.001 |
CHF, congestive heart failure; CI, confidence interval; DCM, dilated cardiomyopathy; LVEDD, left ventricular end-diastolic dimension
Where 2 p-values are shown, the first is based on cause-specific hazard function and the second is based on the cumulative incidence functions for FDCM vs. IDCM via Fine-Gray subdistribution function. For death alone and transplant alone in adjusted modeling, the p-value is based the Fine-Gray regression. For death/transplant composite, the p-value is derived from the Wald test of the log hazard ratio estimated by Cox proportional hazards regression modeling.
There was no significant difference in the cumulative incidence of transplant (P = 0.72, Figure 1b), nor for the distributions for time to the composite outcome of death/transplant (P = 0.15, Figure 2). The cumulative incidence of transplant at 3 years with pre-transplant death as competing risk were 34.2% (95 CI: 27.5% to 41.0%) and 36.1% (95% CI: 32.1% to 40.3%) in the FDCM and IDCM groups, respectively (P = 0.997). In covariate-adjusted models for the separate outcomes of pre-transplant death and transplantation, there remained no difference between the FDCM and IDCM groups (Table 2).
Figure 2.
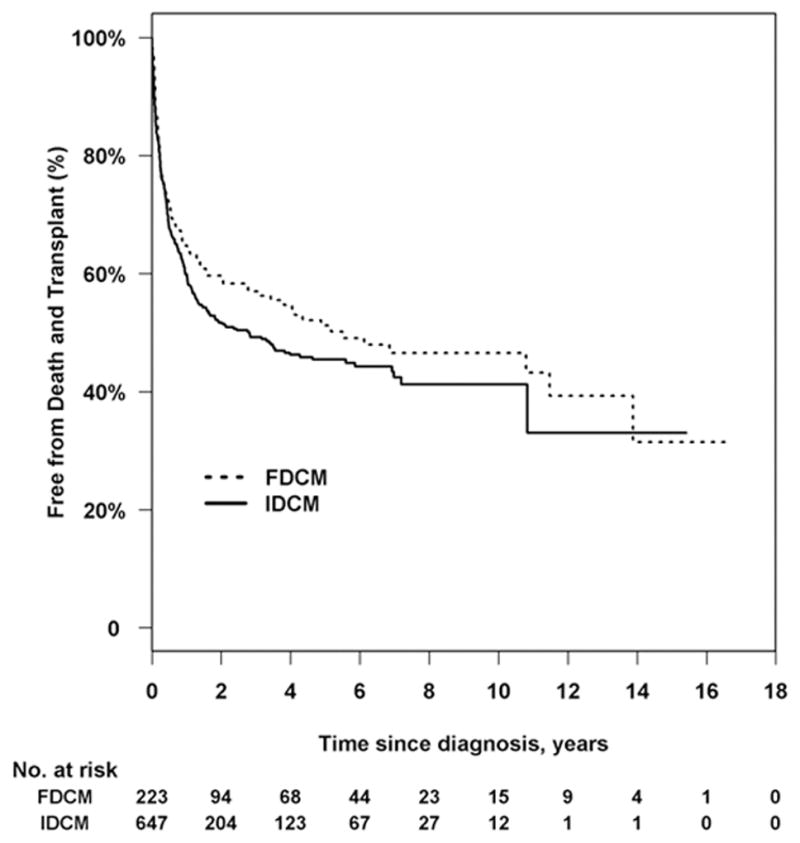
Kaplan-Meier curve comparing time to death and transplantation between children with familial dilated cardiomyopathy (FDCM) and idiopathic cardiomyopathy without a family history of cardiomyopathy (IDCM). Log-rank P = 0.15; 395 events.
At three years after diagnosis of cardiomyopathy, there was no difference in the proportion of children with echocardiographic normalization between patients with FDCM and those with IDCM (P = 0.33). The rates were 30% vs. 26%, respectively, at 3 years. We found no differences in medical therapy between the children with echo normalization and those without for both the IDCM and FDCM group except that children with FDCM who did not have echo normalization were significantly more likely to receive anticongestive therapies (P < 0.001).
Risk Factors for Death/Transplant
Since there were no differences in time to death or transplant between FDCM and IDCM groups, we combined the groups and identified univariate risk factors for the composite outcome of time to death or transplantation since diagnosis (Table 3). The risk of death or transplant was associated with older age at diagnosis (HR: 1.03, 95% CI: 1.01 to 1.05). Diagnosis after 1 year of age significantly increased the risk of death or transplant by nearly 40% (HR: 1.39, 95% CI: 1.14 to 1.70). The hazard ratio for death or transplant was 3.18 times as large for children with CHF at diagnosis as it was for those without (Table 3). For the subset of children with medication class data, antiarrhythmic use was associated with death/transplant (HR: 1.73, 95% CI: 1.22 to 2.46). We identified an interaction (P = 0.025) between DCM type (FDCM vs. IDCM) and only one clinical factor: LVEDD z-score. More abnormal dilation was a significant risk factor for both groups, but the effect was stronger for children with FDCM (HR: 1.24, 95% CI: 1.14 to 1.35 in FDCM vs. HR: 1.10, 95% CI: 1.05 to 1.16 in IDCM). However, for the entire cohort the only independent predictors of death/transplant were those shown in the multivariable model in Table 2 (LVEDD z-score without an interaction; CHF; and older age at diagnosis).
Table 3.
Univariate Analysis of Predictors of the Composite Outcome (Death or Heart Transplantation) in Children with Familial Dilated Cardiomyopathy (FDCM) or Idiopathic DCM (IDCM).
| Characteristics | n/% | Hazard Ratio (95% CI) | P Value |
|---|---|---|---|
| Type of dilated cardiomyopathy | 870 | 0.15 | |
| Familial dilated cardiomyopathy | 223 | Reference | |
| Idiopathic dilated cardiomyopathy | 647 | 1.18 (0.94–1.49) | |
| Age at diagnosis, years | 870 | 1.03 (1.01–1.05) | < 0.001 |
| Age ≥ 1 year at diagnosis | 52.6% (458/870) | 1.39 (1.14–1.70) | 0.001 |
| Male | 50.6% (440/870) | 1.12 (0.92–1.36) | 0.28 |
| Race/ethnicity | 852 | 0.03 | |
| White | 462 | Reference | |
| Black | 181 | 1.15 (0.90–1.46) | |
| Hispanic | 147 | 0.71 (0.52–0.95) | |
| Other | 62 | 0.82 (0.53–1.25) | |
| CHF present at diagnosis | 74.6% (648/869) | 3.18 (2.36–4.29) | < 0.001 |
| Family history of sudden death | −10.5% (76/727) | 1.01 (0.72–1.40) | 0.98 |
| Echocardiographic z-scores | |||
| LV end-diastolic dimension | 699 | 1.14 (1.09–1.19) | < 0.001 |
| LV end-systolic dimension | 614 | 1.16 (1.10–1.21) | < 0.001 |
| LV fractional shortening | 713 | 0.91 (0.87–0.94) | < 0.001 |
| LV end-diastolic posterior wall thickness | 553 | 1.04 (0.99–1.09) | 0.15 |
| End-diastolic septal thickness | 515 | 1.02 (0.95–1.09) | 0.67 |
| LV mass | 545 | 1.09 (1.05–1.13) | < 0.001 |
CHF, congestive heart failure; CI, confidence interval; LV, left ventricular.
Discussion
The study analyzes outcome and risk factors in patients with FDCM and DCM. The major finding is that the risk factors for the composite outcome of death or transplant are CHF and older age at diagnosis, particularly age at presentation older than 1 year increased the risk of death or transplant by almost 40%; dilatation of the LV (LVEDD z-score) was also a significant risk factor for death or transplant. In the subsets of children with medications data available, we found that antiarrhythmic use was associated with an increase of death or transplant; this finding may reflect the fact that patients were sicker although the possibility of a proarrhythmic effect cannot be excluded and caution should be used when starting these drugs. Our findings show that the etiology of cardiomyopathy, FDCM or IDCM was not a factor in determining the outcome; patient clinical characteristics are the major determinant.
Although there was no difference in outcome between the FDCM and IDCM groups, we found that children with FDCM were significantly older at the time of diagnosis, less likely to present with heart failure, and more likely to have a positive family history of sudden cardiac death than were children with IDCM. In addition, they showed less evidence of pathologic LV remodeling at presentation as shown by significantly less, but still abnormal, LV dilation, less septal thinning, and better, but still abnormal, LV function (LVFS or LVEF). Children with IDCM were more likely to be on cardiac medications and antiarrhythmic at diagnosis probably reflecting the fact that IDCM children were sicker at presentation as evidence that they were more likely to present with CHF.
Of children with either FDCM or IDCM, 26% to 30% experienced echocardiographic normalization over time, a finding consistent with normalization rates in a previous PCMR analysis of all IDCM patients (regardless of family history).14
Our report represents perhaps the largest study to date in children comparing clinical outcomes in FDCM and IDCM. In previous studies of patients with IDCM, further investigation of relatives found that between 20% and 50% of index cases had FDCM.15–18 Although these studies were mostly of adults, they all included pediatric cases of DCM. In our study, of the 1832 cases of pure DCM in the PCMR database, 12% of children had a diagnosis of FDCM at their most recent follow-up. Similarly, in the National Australian Childhood Cardiomyopathy Study, the prevalence of FDCM in children less than 10 years of age was 14.7%.3
Clear clinical or echocardiographic markers that differentiate FDCM from IDCM have been difficult to identify. FDCM cannot be reliably diagnosed in any individual child based solely on clinical (other than family history) or echocardiographic data, a finding consistent with other reports.15,17,19 Even an accurate family history can not necessarily identify FDCM at the time of diagnosis. Indeed, as illustrated by the PCMR experience, it is likely that a subset of the patients categorized as IDCM will be subsequently determined to have FDCM due to the manifestation of the disease in relatives at a later time and others may carry the same causative gene mutations despite the absence of a prior family history of DCM. Presently, the diagnosis of FDCM requires assembling a complete and accurate family history, as well as screening family members with echocardiography or another imaging modality. A recent review concluded that proper screening of family members with echocardiography or other assessments of LV size and function will detect FDCM in 20% to 35% of patients with IDCM.20 Genetic evaluation of these populations using newly available DCM genetic testing panels should improve identification of children with FDCM. Since most cases of IDCM are suspected to have a genetic etiology, evaluation of these children with clinically available DCM genetic testing panels is also important.
For many years, genetic testing for DCM in children has been used infrequently in clinical practice because of the high cost with traditional sequencing methods and the relatively low probability of identifying a mutation in one of the many DCM genes. However, large gene panels are now available as a result of the development of next-generation sequencing approaches which can screen a larger number of genes simultaneously at lower cost and also result in a higher number of positive results.20–22 Thus, incorporating genetic testing and genetic counseling into the care of patients with DCM is recommended.23,24 In a Australian prospective study of sudden cardiac death in children and young adults the authors showed
Genetic testing performed at autopsy was able to identify a likely cause of death in 27% of the cases of unexplained sudden cardiac death.25 Although the cost-effectiveness of genetic testing in DCM has not yet been evaluated, studies on the cost-effectiveness of genetic testing in hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and long QT syndrome indicate an overall benefit over screening via cardiac surveillance.26,27
Strengths and Limitations of the Study
The PCMR is the largest longitudinal study of cardiomyopathy in children, including more than 3,500 affected children. However, the diagnosis of FDCM or evidence of affected family members in children with IDCM was identified only from the medical record. Specifically, the diagnosis of FDCM was based on a notation of FDCM in the medical record without any a priori PCMR requirement for data on detailed pedigrees or from comprehensive genetic testing (which has evolved considerably over the life of the PCMR) to establish a diagnosis of FDCM. It is important to note that family history is dynamic and needs to be revisited at each clinical encounter since children initially diagnosed as IDCM, may later be categorized as FDCM, given their updated family history or new genetic testing data. Related to this approach for identifying FDCM, it is possible that the patients in the FDCM group appear to have better survival than IDCM since longer follow-up allows more opportunity to be classified as FDCM. Furthermore, screening echocardiograms of family members were not available. Medication class was only available for a subset of patients. Almost 30% of the IDCM cases in the PCMR could not be used in the analysis as a result of inconclusive family histories. Determining whether or not a patient has familial disease is often difficult, and there is evidence that correct classification requires consistent and ongoing echocardiographic investigation of relatives, who may present with DCM at various ages.17,28 Finally, we found no statistically significant difference in the time to death/transplant (43% vs. 51% for FDCM vs. IDCM at 3 years). This difference may be considered clinically significant, but we had limited statistical power due to the shorter follow-up time in the IDCM group.
Echocardiographic screening of relatives has shown that relying on family history alone to identify FDCM may not identify asymptomatic family members with cardiomyopathy or detect whether relatives reported to have DCM may actually have had other cardiac diseases.28 Because our study relied solely on the family history found in the medical record to make the diagnosis of FDCM, some children may have been misclassified with respect to final DCM category. Changes in clinical protocols addressing the acquisition and documentation of complete pedigrees and comprehensive family screening in this population over the course of this study could also have affected the percentage of children classified as FDCM.
Future Directions
Nearly half of our FDCM cases were not identified as such at their initial clinical presentation. Our findings suggest that at least the first-degree relatives of children presenting with DCM without a clear non-familial cause should undergo echocardiographic screening. Additionally, state-of-the art clinical genetic testing of the proband, and if positive, of other family members, should be used to more accurately make the diagnosis of FDCM in the affected child. Advances in clinical genetic testing for DCM, including the development of more comprehensive panels and cost-effective whole-genomic analysis, will eventually provide a more rapid and definitive diagnosis of FDCM in children.
Conclusions
At diagnosis in both FDCM and IDCM, older age, the presence of CHF, and increased LV dilation are independently associated with poor outcomes. At the time of diagnosis, children eventually categorized as FDCM appear to be at an earlier stage of the disease with less LV remodeling, and a lower prevalence of heart failure, when compared to children with IDCM.
In summary, recognition of FDCM is important regarding the implications for screening other family members, but not for the management of the affected individual, since the outcomes are related to patient factors, not whether the child has FDCM or IDCM.
Clinical Perspective.
Our report represents perhaps the largest study to date comparing clinical outcomes in Familial Dilated Cardiomyopathy (FDCM) and Idiopathic Dilated Cardiomyopathy (IDCM) in children. We found that children with FDCM were significantly older at the time of diagnosis, less likely to present with heart failure, and more likely to have a positive family history of sudden cardiac death than were children with IDCM. We found no covariate-adjusted difference between the FDCM and IDCM groups in time to death, transplant, or the combined endpoint of death/transplantation. They also had less evidence of pathologic LV remodeling at presentation as shown by significantly less, but still highly abnormal, LV dilation, less septal thinning, and better, but still highly abnormal, LV function (LVFS or LVEF). Our findings suggest that at least the first-degree relatives of children presenting with DCM without a clear non-familial cause should undergo echocardiographic screening. Additionally, state-of-the art clinical genetic testing of both the proband, and if positive, of other family members, should be used to more accurately make the diagnosis of FDCM in the affected child. Advances in clinical genetic testing for DCM, including the development of more comprehensive genetic testing panels and cost-effective whole-genomic analysis, will eventually provide a more rapid and definitive diagnosis of FDCM in children. Since most cases of IDCM are suspected to have a genetic etiology evaluation of these children with clinical available DCM genetic testing panels is also important. In summary, recognition of FDCM is important regarding the implications for screening other family members, but not for the management of the affected individual, since the outcomes are related to patient factors, not whether the child has FDCM or IDCM.
Acknowledgments
We thank the Children’s Cardiomyopathy Foundation for its ongoing and consistent support for the work of the PCMR.
Funding Sources: The PCMR is supported by grants from the National Heart, Lung, and Blood Institute (R01 HL53392, R01 HL111459, R01 HL109090; SEL, Principal Investigator) and the Children’s Cardiomyopathy Foundation. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI.
Footnotes
Disclosures: None.
References
- 1.Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 2.Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, Colan SD. The incidence of pediatric cardiomyopathy in two regions of the united states. N Engl J Med. 2003;348:1647–1655. doi: 10.1056/NEJMoa021715. [DOI] [PubMed] [Google Scholar]
- 3.Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, Weintraub RG. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348:1639–1646. doi: 10.1056/NEJMoa021737. [DOI] [PubMed] [Google Scholar]
- 4.Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, Tikanoja T, Paavilainen T, Simell O. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am J Epidemiol. 1997;146:385–393. doi: 10.1093/oxfordjournals.aje.a009291. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association scientific statement from the Council On Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 6.Strauss A, Lock JE. Pediatric cardiomyopathy—a long way to go. N Engl J Med. 2003;348:1703–1705. doi: 10.1056/NEJMe030027. [DOI] [PubMed] [Google Scholar]
- 7.Cox GF, Sleeper LA, Lowe AM, Towbin JA, Colan SD, Orav EJ, Lurie PR, Messere JE, Wilkinson JD, Lipshultz SE. Factors associated with establishing a causal diagnosis for children with cardiomyopathy. Pediatrics. 2006;118:1519–1531. doi: 10.1542/peds.2006-0163. [DOI] [PubMed] [Google Scholar]
- 8.Alvarez JA, Orav EJ, Wilkinson JD, Fleming LE, Lee DJ, Sleeper LA, Rusconi PR, Colan SD, Hsu DT, Canter CE, Webber SA, Cox GF, Jefferies JL, Towbin JA, Lipshultz SE. Competing risks for death and cardiac transplantation in children with dilated cardiomyopathy: results from the Pediatric Cardiomyopathy Registry. Circulation. 2011;124:814–23. doi: 10.1161/CIRCULATIONAHA.110.973826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sluysmans T, Colan SD. Theoretical and empirical derivation of cardiovascular allometric relationships in children. J Appl Physiol. 2005;99:445–457. doi: 10.1152/japplphysiol.01144.2004. [DOI] [PubMed] [Google Scholar]
- 10.Colan SD, Parness IA, Spevak PJ, Sanders SP. Developmental modulation of myocardial mechanics: age- and growth-related alterations in afterload and contractility. J Am Coll Cardiol. 1992;19:619–629. doi: 10.1016/s0735-1097(10)80282-7. [DOI] [PubMed] [Google Scholar]
- 11.Gray R. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. [Google Scholar]
- 12.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Statist Assoc. 1999;94:496–509. [Google Scholar]
- 13.Astin PC, Lee DS, Fine JP. Introduction to the analysis of survival data in the presence of competing risks. Circulation. 2016;133:601–609. doi: 10.1161/CIRCULATIONAHA.115.017719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsang TS, Barnes ME, Gersh BJ, Bailey KR, Seward JB. Left atrial volume as a morphophysiologic expression of left ventricular diastolic dysfunction and relation to cardiovascular risk burden. Am J Cardiol. 2002;90:1284–1289. doi: 10.1016/s0002-9149(02)02864-3. [DOI] [PubMed] [Google Scholar]
- 15.Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, Rodenheffer RJ, Chesebro JH, Tazelaar HD. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. doi: 10.1056/NEJM199201093260201. [DOI] [PubMed] [Google Scholar]
- 16.Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2033–2040. doi: 10.1016/j.jacc.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 17.Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A, Miococ S, Vatta M, Pinamonti B, Muntoni F, Caforio AL, McKenna WJ, Falaschi A, Giacca M, Camerini F. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. J Am Coll Cardiol. 1999;34:181–190. doi: 10.1016/s0735-1097(99)00172-2. [DOI] [PubMed] [Google Scholar]
- 18.Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, Ware SM. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail. 2012;18:396–403. doi: 10.1016/j.cardfail.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moretti M, Merlo M, Barbati G, Di Lenarda A, Brun F, Pinamonti B, Gergori D, Mestroni L, Sinagra G. Prognostic impact of familial screening in dilated cardiomyopathy. Eur J Heart Fail. 2010;12:922–927. doi: 10.1093/eurjhf/hfq093. [DOI] [PubMed] [Google Scholar]
- 20.Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morales A, Hershberger RE. Genetic evaluation of dilated cardiomyopathy. Curr Cardiol Rep. 2013;15(7):375. doi: 10.1007/s11886-013-0375-1. [DOI] [PubMed] [Google Scholar]
- 22.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 23.Ackerman MJ, Priori DG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 24.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail. 2009;15(2):83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, Davis AM, Thompson T, Connell V, Wallace J, Naylor C, Crawford J, Love DR, Hallam L, White J, Lawrence C, Lynch M, Morgan N, James P, du Sart D, Puranik R, Langlois N, Vohra J, Winship I, Atherton J, McGaughran J, Skinner JR, Semsarian C. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N Engl J Med. 2016;374(25):2441–52. doi: 10.1056/NEJMoa1510687. [DOI] [PubMed] [Google Scholar]
- 26.Wordsworth S, Leal J, Blair E, Legood R, Thomson K, Seller A, Taylor J, Watkins H. DNA testing for hypertrophic cardiomyopathy: a cost effectiveness model. Eur Heart J. 2010;31:926–935. doi: 10.1093/eurheartj/ehq067. [DOI] [PubMed] [Google Scholar]
- 27.Sabter-Molina M, Garcia-Molan E, Tovar I, Ruiz-Espejo F, Gimeno JR, Valdes M. Cost effectiveness of genetic studies in inherited heart diseases. Cardiogenetics. 2013;3:28–30. [Google Scholar]
- 28.Johnson RA, Palacios I. Dilated cardiomyopathies of the adult (first of two parts) N Engl J Med. 1982;307:1051–1058. doi: 10.1056/NEJM198210213071704. [DOI] [PubMed] [Google Scholar]