

Abstract
Only a fraction of all smokers develop chronic obstructive pulmonary disease (COPD), suggesting a large role for genetic susceptibility. The leptin receptor (LEPR) is present in human lung tissue and may play a role in COPD pathogenesis. The present study examined the association between genetic variants in the LEPR gene and lung function decline in COPD.
In total, 429 European Americans were randomly selected from the National Heart Lung and Blood Institute Lung Health Study. 36 single nucleotide polymorphisms (SNPs) in LEPR were genotyped using the Illumina™ GoldenGate platform (Broad Institute, Cambridge, MA, USA). Mean annual decline in forced expiratory volume in 1 s % predicted over the 5-yr period was calculated using linear regression. Linear regression models were also used to adjust for potential confounders. In addition, in vivo expression of the receptor gene was assessed with immunohistochemistry on lungs from smoke-exposed inbred mice.
We identified significant associations (p<0.05) between lung function decline and 21 SNPs. Haplotype analyses confirmed several of these associations seen with individual markers. Immunohistochemistry results in inbred mice strains support a potential role of LEPR in COPD pathogenesis.
We identified genetic variants in the LEPR gene significantly associated with lung function decline in a population of smokers with COPD. Our results support a role for LEPR as a novel candidate gene for COPD.
Keywords: Chronic obstructive pulmonary disease, leptin receptor, lung function decline, polymorphisms
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death in the USA and the fifth leading cause of death worldwide, and its prevalence is expected to increase in coming decades [1, 2]. The overwhelming majority of COPD is caused by environmental exposures. In the USA, this exposure is primarily cigarette smoke (CS); however, only 15% of all smokers develop COPD. This suggests a large role for genetic susceptibility.
In addition to its role in obesity (appetite suppression), leptin has been shown to have multiple other functions, including increasing sympathetic nerve activity, maintaining reproductive function, immunity, angiogenesis, preserving normal respiratory function in the presence of obesity and cell proliferation of tracheal epithelial cells and lung growth [3]. After adjustment for obesity, the leptin pathway has been associated with inflammatory markers as well as multiple inflammatory conditions, including cardiovascular disease [4, 5]. Its potential role in the systemic inflammatory response in patients with COPD is evident from the correlation of leptin with other inflammatory markers [6–8]. Leptin has been shown to directly stimulate phagocytic activity of macrophages and enhance endotoxin-induced production of tumour necrosis factor-α, interleukin (IL)-6, and IL-12, cytokines typically involved in COPD pathogenesis [9–12]. Conversely, deficiency in leptin or its receptor may predispose sufferers to both immunodeficiency and infection [10]. Serum leptin levels are elevated in exacerbations of COPD and have also been associated with bacterial pulmonary infections, which are associated with COPD exacerbations and decline in lung function [7, 13–15]. Leptin exerts multiple effects through its leptin receptor (LEPR), located on human chromosome 1p31. The LEPR is produced in several alternatively spliced forms that share extracellular and trans-membrane domains but have varying cytoplasmic residues [16], and has a wide tissue distribution, including lung tissue [17].
We tested for association between genetic variants in the LEPR gene and lung function decline in a subset of the multicentre National Heart, Lung and Blood Institute (NHLBI)-supported Lung Health Study (LHS) cohort. In addition, the potential role of LEPR in COPD pathogenesis was further evaluated in smoke-exposed AKR/J mice, which display marked airspace enlargement by histological and morphometric criteria [18]. To determine whether the LEPR is differentially localised to resident lung cells under conditions that simulate human COPD, we performed immunohistochemistry on murine lung sections obtained from AKR/J mice exposed to 4 months of CS versus room air controls.
Identifying pathways and novel molecular targets that modify the clinical course of disease is fundamental to developing preventive strategies and novel therapies.
Methods
Subjects
Subjects participating in the current study included 429 European Americans (EA) randomly selected from a group of 4,287 participants of the multi-centre NHLBI-supported LHS for whom DNA was available. Lung function was measured annually over 5 yrs and conducted according to American Thoracic Society guidelines using identical spirometers, software, procedures and reading centre personnel [19, 20]. The quality of the spirometry testing conducted by the technicians was monitored centrally throughout the testing and comparison of baseline spirometry measures showed good reproducibility with very small mean short-term intra-individual variations in forced expiratory volume in 1 s (FEV1) [21]. Lung function data from annual visit 1 to annual visit 5 was used for the current analyses and has been shown to have a good linear fit in previous LHS analyses [19, 22]. Subjects with less than three annual lung function measurements were excluded from the analysis (n=15).
Single nucleotide polymorphism selection and genotyping
Single nucleotide polymorphisms (SNPs) representing the LEPR gene were selected from Goldenpath (genome.ucsc.edu) and/or National Center for Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov; Bethesda, MD, USA). Criteria included: 1) SNPs approximating inter-SNP distance as close to 5 kb as were available at the time of the dbSNP Build 124 (NCBI); 2) representation of SNPs in the promoter, coding and 3′-untranslated region; and 3) SNPs with acceptable design scores according to the Illumina Assay Design Tool for genotyping on the Illumina™ GoldenGate platform (Broad Institute, Cambridge, MA, USA). Priority for selecting SNPs included: 1) regulatory and coding SNPs; 2) highly polymorphic SNPs, preferably ≥10% minor allele frequency; 3) validated SNPs; and 4) SNPs at intron/exon boundaries. A total of 36 LEPR SNPs spanning 228,294 bp on human chromosome 1p31 with an average inter-SNP distance of 6.53 kb (range 1.9–14 kb) are summarised in table 1.
Table 1. Location, minor allele frequency, and type of selected LEPR single nucleotide polymorphisms (SNPs).
| ID | SNP | Position | Region | Minor allele | MAF |
|---|---|---|---|---|---|
| 1 | rs7531867 | 65819567 | Downstream | A | 0.39 |
| 2 | rs1805096 | 65814278 | Coding exon (1019 P/P) | T | 0.39 |
| 3 | rs1892535 | 65809202 | Intron | T | 0.18 |
| 4 | rs6588153 | 65804038 | Intron | A | 0.38 |
| 5 | rs1938484 | 65793303 | Intron | A | 0.18 |
| 6 | rs8179183 | 65787973 | Coding exon (656 K/N) | C | 0.18 |
| 7 | rs3790419 | 65779130 | Coding exon (343 S/S) | G | 0.22 |
| 8 | rs3828034 | 65774346 | Intron (boundary) | C | 0.19 |
| 9 | rs12564626 | 65768563 | Intron | A | 0.45 |
| 10 | rs10443259 | 65763371 | Intron | A | 0.28 |
| 11 | rs6691346 | 65758418 | Intron | A | 0.28 |
| 12 | rs4655680 | 65753490 | Intron | T | 0.29 |
| 13 | rs1137100 | 65748462 | Coding exon (109 K/R) | G | 0.26 |
| 14 | rs10889562 | 65742466 | Intron | A | 0.22 |
| 15 | rs6702028 | 65731912 | Intron | C | 0.28 |
| 16 | rs1782763 | 65719921 | Intron | C | 0.33 |
| 17 | rs1171265 | 65715273 | Intron | A | 0.36 |
| 18 | rs1171271 | 65710811 | Intron | C | 0.27 |
| 19 | rs1782754 | 65705369 | Intron | G | 0.27 |
| 20 | rs1171279 | 65700514 | Intron | T | 0.27 |
| 21 | rs1171274 | 65692859 | Intron | C | 0.27 |
| 22 | rs10889558 | 65688987 | Intron | A | 0.27 |
| 23 | rs6694528 | 65675037 | Intron | T | 0.13 |
| 24 | rs1327121 | 65669358 | Intron | C | 0.34 |
| 25 | rs17412682 | 65664314 | Intron | C | 0.46 |
| 26 | rs2025804 | 65658142 | Intron | C | 0.34 |
| 27 | rs17127652 | 65647164 | Intron | G | 0.02 |
| 28 | rs4655811 | 65635178 | Intron | C | 0.34 |
| 29 | rs6657868 | 65625728 | Intron | A | 0.39 |
| 30 | rs9436746 | 65620494 | Intron | A | 0.4 |
| 31 | rs970468 | 65618511 | Downstream | G | 0.39 |
| 32 | rs17097182 | 65615466 | Downstream | T | 0.05 |
| 33 | rs1045895 | 65610002 | 3′ UTR | A | 0.38 |
| 34 | rs9436299 | 65604909 | Intron | C | 0.33 |
| 35 | rs1327118 | 65597590 | Promoter | C | 0.46 |
| 36 | rs10493377 | 65651840 | Promoter | G | 0.47 |
MAF: minor allele frequency; UTR: untranslated region.
Statistical analysis
Mean annual decline in lung function (post-bronchodilator FEV1 % predicted) was calculated as a linear regression slope over the 5-yr study period. Linear regression models were used to adjust for potential confounders, including baseline characteristics, smoking history (pack-yrs), age, sex, FEV1 % pred and airway reactivity (AR). AR was calculated as a quantitative measure, using the two-point slope [22]. In addition to adjusting for baseline characteristics, 1) change in body mass index (BMI), calculated as a linear regression slope and 2) smoking status at year 5, defined as “continuous smoker”, “intermittent smoker”, and “sustained quitter”, were also analysed.
Forward and backward selection was used to develop a parsimonious model. Residuals from the regression were included in a genetic additive model and the most common homozygote genotype for each SNP served as the reference category. The interaction term for smoking and genetic effect were tested using PLINK (Boston, MA, USA) [23], while testing for quantitative interaction. Each SNP locus was evaluated for Hardy–Weinberg equilibrium. All analyses were performed with StataSE, version 8.0 (Stata Corp, College Station, TX, USA) and PLINK [23].
Individual haplotypes were analysed using PLINK. Pairwise linkage disequilibrium (LD) based on the D' statistic was measured using Haploview (Broad Institute) [24]. LD blocks were defined using their default algorithm [25]. Sliding windows of two to four adjacent SNPs were used to test for association. Haplotype estimates were computed using PHASE (Seattle, WA, USA) assuming no recombination [26].
Murine model
10-week-old AKR/J mice were exposed to CS 5 h·day-1, 5 days·week-1, for 6 months. The exposure was conducted by burning 2R4F reference cigarettes (University of Kentucky, Tobacco Research Institute, Lexington, KY, USA), using a TE-10 smoking machine (Teague Enterprises, Woodland, CA, USA). Each cigarette was puffed for 2 s, once every minute for a total of eight puffs, at a flow rate of 1.05 L·min-1, to provide a standard puff of 35 cm3. The smoke machine was adjusted to produce a mixture of sidestream smoke (89%) and mainstream smoke (11%). The smoke chamber was monitored daily for total suspended particles and carbon monoxide, with concentrations of 90 mg·m-3 and 350 ppm, respectively. Air-exposed control mice were housed in a filtered air environment.
Immunohistochemistry
5-μm paraformaldehyde-fixed, paraffin-embedded tissue sections from smoke-exposed and room air control AKR/J mice were deparaffinised and rehydrated in an ethanol series. Sections were blocked for nonspecific binding with 3% normal serum from chicken and incubated with the primary antibodies for 1 h at room temperature. Following incubation with the primary antibody overnight at 4°C, slides were washed with PBS Tween-20, incubated for 30 min at room temperature with an appropriate biotinylated secondary antibody and developed by using 3,3′-diaminobenzidine substrate and chromagen from Dako (Stockport, UK). Antigen retrieval was performed using citrate buffer for 30 min. Antibodies were used at the following concentrations: LEPR, ObR (goat polyclonal, I-17, 1:100; Santa Cruz Biotechnology, Santa Cruz, AZ, USA), donkey anti-goat secondary antibody (sc2020, 1:400, Santa Cruz Biotechnology).
Results
Clinical characteristics of the subjects are presented in table 2. There were no statistical differences in baseline characteristics between those included in the final analyses (n=414) compared with those excluded because more than two data points in lung function were missing (n=15, data not shown). Distributions of baseline characteristics and lung function measurements were also similar to those in the full LHS cohort from which this subset was selected (n=5,887, data not shown). Age, baseline lung function, AR, smoking status at year 5, and change in BMI were independently associated with lung function decline, and were included in the final regression analyses. There was no significant interaction between smoking status at year 5 and LEPR polymorphisms.
Table 2. Patient characteristics.
| Subjects n | 414 |
| Baseline characteristics | |
| Mean age yrs | 48.6±7.0 |
| Male % | 65.0 |
| BMI kg·m-2 | 25.4±3.6 |
| Smoking pack-yrs | 40.5±19.2 |
| Pre-BD FEV1 % | 76.3±6.4 |
| Post-BD FEV1 % | 79.2±6.2 |
| Longitudinal characteristics at 5 yrs | |
| Smoking history % | |
| Continuous | 50 |
| Intermittent now smoking | 12 |
| Intermittent now quit | 19 |
| Sustained quitter | 19 |
| Post-BD ΔFEV1 %·yr-1 | -0.74±1.63 |
Data are presented as mean±sd, unless otherwise stated. BMI: body mass index; BD: bronchodilator; FEV1: forced expiratory volume in 1 s.
Single-marker analyses
All 36 LEPR SNPs were in the Hardy–Weinberg equilibrium. All results of the two-point tests for association between LEPR markers and FEV1 % pred decline are presented in table 3. We found evidence for significant associations between 21 SNPs in the LEPR gene and FEV1 % pred decline, spanning the length of the gene from intron 2 to the 5′ end. The minor alleles of most significant SNPs were associated with attenuation in lung function decline. Of particular interest, each G allele at the functional marker rs1137100, in exon 4, which creates a lysine to arginine amino acid change, was associated with a 0.33%·yr-1 attenuation in annual loss of FEV1 % pred.
Table 3. Association of LEPR polymorphisms with lung function decline in a subset of the Lung Health Study European American cohort#.
| LD block | SNP ID | SNP | β | 95% CI | p-value |
|---|---|---|---|---|---|
| 1 | 1 | rs7531867 | 0.298 | -0.111–0.298 | 0.006 |
| 2 | rs1805096 | 0.295 | -0.082–0.339 | 0.007 | |
| 2 | 3 | rs1892535 | 0.314 | -0.008–0.435 | 0.018 |
| 4 | rs6588153 | 0.292 | -0.381–0.039 | 0.007 | |
| 5 | rs1938484 | 0.351 | -0.459–0.469 | 0.007 | |
| 6 | rs8179183 | 0.109 | -0.014–0.410 | 0.429 | |
| 3 | 7 | rs3790419 | -0.09 | 0.013–0.432 | 0.473 |
| 8 | rs3828034 | 0.026 | -0.008–0.419 | 0.848 | |
| 9 | rs12564626 | 0.25 | -0.004–0.435 | 0.015 | |
| 10 | rs10443259 | 0.334 | -1.011–0.509 | 0.004 | |
| 11 | rs6691346 | 0.334 | 0.039–0.477 | 0.004 | |
| 12 | rs4655680 | 0.312 | -0.436–0.027 | 0.007 | |
| 13 | rs1137100 | 0.33 | 0.019–0.456 | 0.005 | |
| 14 | rs10889562 | -0.044 | -0.350–0.277 | 0.73 | |
| 15 | rs6702028 | 0.32 | 0.085–0.538 | 0.006 | |
| 16 | rs1782763 | 0.318 | 0.111–0.565 | 0.003 | |
| 17 | rs1171265 | 0.321 | -0.329–0.138 | 0.004 | |
| 18 | rs1171271 | 0.32 | 0.091–0.543 | 0.006 | |
| 19 | rs1782754 | 0.317 | 0.093–0.547 | 0.006 | |
| 20 | rs1171279 | -0.095 | 0.105–0.537 | 0.425 | |
| 4 | 21 | rs1171274 | 0.338 | 0.106–0.529 | 0.004 |
| 22 | rs10889558 | 0.311 | 0.093–0.547 | 0.007 | |
| 23 | rs6694528 | -0.037 | -0.296–0.207 | 0.818 | |
| 24 | rs1327121 | 0.238 | 0.103–0.557 | 0.033 | |
| 25 | rs17412682 | -0.232 | 0.087–0.537 | 0.027 | |
| 26 | rs2025804 | 0.258 | 0.106–0.562 | 0.022 | |
| 27 | rs17127652 | -0.251 | 0.106–0.562 | 0.518 | |
| 5 | 28 | rs4655811 | 0.216 | 0.049–0.452 | 0.055 |
| 29 | rs6657868 | 0.206 | -0.240–0.292 | 0.06 | |
| 30 | rs9436746 | 0.223 | -0.334–0.155 | 0.039 | |
| 6 | 31 | rs970468 | 0.198 | -0.161–0.378 | 0.068 |
| 32 | rs17097182 | 0.005 | 0.096–0.607 | 0.983 | |
| 33 | rs1045895 | -0.171 | 0.082–0.502 | 0.111 | |
| 34 | rs9436299 | 0.214 | 0.056–0.572 | 0.059 | |
| 35 | rs1327118 | 0.129 | 0.084–0.507 | 0.231 | |
| 36 | rs10493377 | 0.093 | 0.088–0.508 | 0.373 |
Examination of the linkage disequilibrium (LD) structure across these single nucleotide polymorphisms (SNPs) revealed high LD (D′ range 0.80–1.0, with R2 range 0.65–0.96) for 31 of 35 pairs of contiguous SNPs (fig. 1). In fact, 33 of the SNPs fell into one of six blocks, and most of the 21 SNPs with association signal are noted to have similar effect sizes for the minor allele and fall into one of three LD blocks (2, 3 and 4). CI: confidence interval. Bold p-values are statistically significant.
: n=414.
Haplotype analyses
Considering the clustering of most genotyped SNPs within six LD blocks with high levels of LD also observed between SNPs across different blocks, a systematic sliding window approach was implemented, considering windows of two to four SNPs/window beginning with the first (5′) marker, and working across the gene, one marker at a time. Haplotype tests revealed multiple association signals in three specific regions that overlapped with the single-SNP results described above. 25 haplotypes showed stronger association than single SNP results (fig. 2). One of the most compelling regions was at the 5′ end of LEPR (region 1) and the haplotype GCCT (rs7531867, rs1805096, rs1892535 and rs6691346), which revealed a strong association with lung function decline (p=0.003) and includes the coding-synonymous SNP, rs1805096. Furthermore, one 3-SNP and two 4-SNP window haplotypes in region 2 spanning rs10443259 to rs10889562 (introns 2 through 5) were most strongly associated with lung function decline (p=0.002), and included the functional marker rs1137100, as described previously.
Figure 2.

Haplotypes (n=25) that show greater association than single nucleotide polymorphism results.
Validation of LEPR as a candidate gene for COPD phenotypes in inbred mice
AKR/J mice, a well-characterised inbred strain, exposed to 4 months of CS not only exhibit airspace enlargement but also develop airway thickening and inflammation that is highly reminiscent of COPD [18]. We examined whether CS exposure altered LEPR expression in the lungs of this strain. Using immunohistochemistry, we observed that LEPR expression was evident in macrophages, the airways and airspace compartments in room air-exposed AKR/J mice. However, upon smoke exposure, there was a reduction of staining in the airspace and airway wall (fig. 3). These findings are consistent with CS-induced downregulation of LEPR expression in the epithelial compartments of CS-exposed mice.
Figure 3.
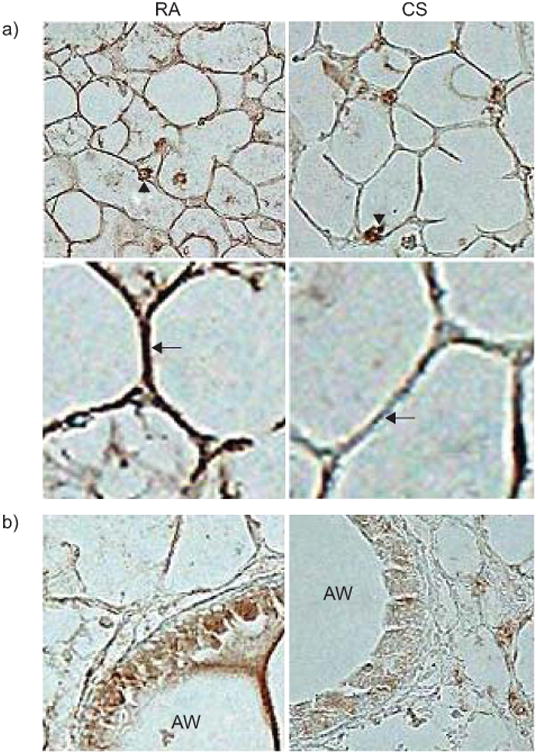
Reduced leptin receptor expression in lung parenchyma of AKR/J mice exposed to 4 months of cigarette smoke (CS) or room air. Representative images of a minimum of three mice per condition. a) Decreased immunohistochemical staining of leptin receptor (brown) in alveolar walls of smoke-exposed mice using polyclonal antibody against ObR and avidin-biotin-peroxidase complex method. Arrowheads denote preserved leptin receptor staining of alveolar macrophages after CS exposure. Arrows show reduced airspace wall staining in smoke-exposed mice. b) Reduced leptin receptor expression is also evident in airway epithelial cells of smoke-exposed mice. RA: room air exposed; AW: airway lumen.
Discussion
We identified 21 SNPs in the LEPR gene that were significantly associated with lung function decline in an EA population with COPD and our haplotype analysis supported results from SNP analysis. The association signals observed across these SNPs likely represent two or three signals in the gene. Furthermore, the potential role of the LEPR in lung architecture and COPD phenotypes is supported by an AKR/J murine model showing decreased LEPR expression in airway wall and epithelium after smoke exposure. Our results identify LEPR as a novel candidate gene for COPD.
To our knowledge, no previous studies have investigated the role of genetic polymorphisms in the LEPR gene and COPD or its associated phenotypes. The phenotype that we studied was rate of decline of lung function rather than COPD per se. The rationale for this was that rate of decline in lung function is a more precise phenotype than a single cross-sectional measurement of lung function needed to define COPD, and may serve as a more genetically homogeneous phenotype. We identified 21 SNPs in the LEPR gene that were significantly associated with lung function decline in a EA population with COPD. One of the most compelling regions was at the 5′ end of LEPR (region 1), which includes part of the intracellular domain of the receptor [16] and may result in different signalling potency. Most variants identified in LEPR were associated with an attenuation of lung function decline, and notably, the G variant at the nonsynonymous SNP rs1137100, which codes for an amino acid substitution in the extracellular domain of the LEPR [27], was associated with a 0.33%·yr-1 attenuation in loss of FEV1 % pred. Airflow obstruction that occurs in COPD is caused by a mixture of small airway disease, parenchymal destruction and increased airways responsiveness that develops over decades [28]. Thus, over time, these genetic variants may have a substantial impact on disease progression. Over a 40-yr period, a person homozygous for the G allele at rs1137100 will have a FEV1 % pred that is 26.4% higher compared with a homozygote for the major allele. Given a minor allele frequency of 0.26, as seen in our cohort, we might extrapolate 7% of the population to be homozygote for the minor allele (GG), 38% to be heterozygote (AG) and 55% to be homozygote for the major allele (AA). Even after adjusting for smoking status, this genetic variant could explain the difference between mild and moderate or moderate and very severe COPD under current Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria [29].
The dearth of well-characterised populations of COPD subjects with longitudinal lung function data comparable to the LHS adds to the uniqueness of our population, but limits the opportunity to replicate these results. However, finding multiple significant SNPs in a single gene reduces the likelihood of spurious results due to multiple testing, though it may also be due in part to the high degree of LD in this gene.
Furthermore, when using a false discovery rate of 0.2, six SNPs would remain statistically significantly associated with lung function decline (p<0.006) in the present study. Previous studies have shown genetic variants in LEPR to be associated with markers of inflammation, including C-reactive protein (CRP) and fibrinogen levels, lending support to the hypothesis that the leptin pathway has a physiological influence on inflammatory traits. Specifically, the minor allele (T) for the rs1805096 locus was associated with lower levels of fibrinogen, CRP and IL-6 levels in a previous study of healthy EAs [30]. Both lower CRP and IL-6 levels have been associated with attenuated lung function decline in the LHS cohort [31]. Interestingly, the minor allele (T) in the coding SNP rs1805096 was also associated with lower rates of lung function decline in our study (0.295 %·yr-1; p=0.007). Therefore, we hypothesise one potential mechanism of the disease-modifying effects of the LEPR gene may be mediated through inflammatory mechanisms. The notion that leptin may function as an immunomodulatory cytokine has become increasingly accepted. The association between leptin and muscle wasting and cachexia in COPD is well established [32]; and the presence of leptin in induced sputum of patients with moderate COPD and its association with other inflammatory markers [33] suggests leptin may be involved in the local inflammatory response in COPD.
Inbred mice or guinea pigs subjected to chronic CS exposure have been shown to be invaluable models of CS-induced parenchymal lung disease [34, 35]. Furthermore, the use of animal models of complex human diseases to parse candidate genes identified in broad genetic or genomic surveys is a standard approach to initial pathway validation, especially if additional populations are not available for replication and therapeutic targets are an ostensible goal [36, 37]. We noted a reduction in staining in the airspace wall and airway epithelial compartment, but retained expression in macrophages in AKR/J smoke-exposed mice. This is consistent with findings of Bruno et al. [13] who found decreased expression of leptin and its receptor in smokers and subjects with mild to severe COPD as compared with healthy nonsmoking subjects. Accordingly, reduced leptin receptor expression (acquired or genetic) may be a critical predisposing factor to CS-induced lung disease.
In summary, the current study is the first to report an association between LEPR polymorphisms and COPD. Our results support the role of the leptin pathway, and particularly the LEPR in COPD and lung function decline. Specifically, individuals with the minor allele at the selected polymorphisms were less susceptible to loss of lung function and COPD progression. The SNP and haplotype tests point to three clusters of signal highlighting two potential loci considering the linkage disequilibrium between these signals: the functional SNP rs137100 and at the synonymous coding SNP rs1805096. While these two SNPs are not in LD with each other (D′=0.45, R2=0.125), haplotypes of significance that include each SNP overlap neighbouring LD blocks, making it somewhat difficult to tease apart these two signals. It is necessary to replicate our findings in other populations; however, our results identify LEPR as a novel target in COPD and lung function loss.
Figure 1.
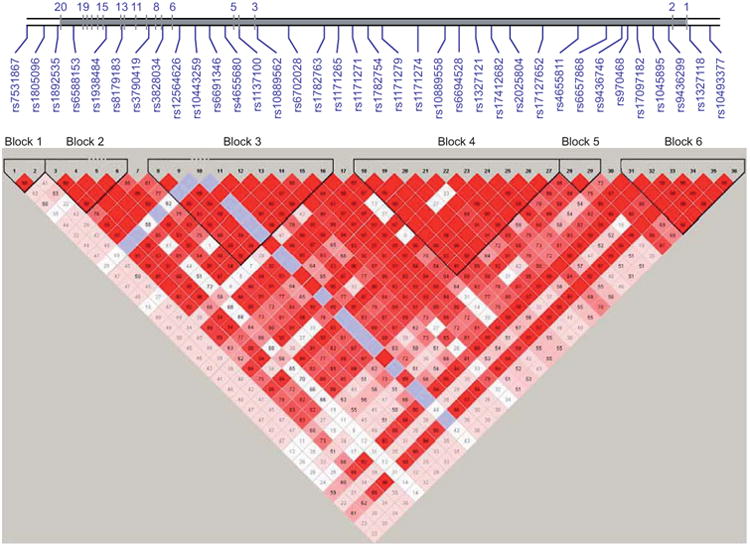
Pairwise linkage disequilibrium (LD) in subjects is represented as red squares for strong LD, blue squares for nonsignificant LD, and white squares for little or no LD. LD blocks are identified as noted.
Acknowledgments
We would like to thank: P. Chi, M. Campbell, A.L. Scott, A.E. Jedlicka and M.V. Mintz (Malaria Research Institute, Gene Array Core Facility, Johns Hopkins University, Baltimore, MD, USA); A.F. Scott, K. Doheny, R. Ashworth and C. Boehm (Center for Inherited Disease Research Institute, Johns Hopkins University); and H. Voelker and K. Farnell (LHS Data Coordinating Center, University of Minnesota, Minneapolis, MN, USA).
The principal investigators and senior staff of the clinical and coordinating centres, the National Heart Lung and Blood Institute (Bethesda, MD, USA) are as follows. M.D. Altose (Principal Investigator) and C.D. Deitz (Project Coordinator): Case Western Reserve University, Cleveland, OH, USA. M.S. Eichenhorn (Principal Investigator), K.J. Braden (Project Coordinator) and R.L. Jentons (Project Coordinator): Henry Ford Hospital, Detroit, MI, USA. R.A. Wise (Principal Investigator), C.S. Rand (Co-Principal Investigator) and K.A. Schiller (Project Coordinator): Johns Hopkins University School of Medicine, Baltimore, MD, USA. P.D. Scanlon (Principal Investigator), G.M. Caron (Project Coordinator), K.S. Mieras and L.C. Walters: Mayo Clinic, Rochester, MN, USA. A.S. Buist (Principal Investigator), L.R. Johnson (LHS Pulmonary Function Coordinator) and V.J. Bortz (Project Coordinator): Oregon Health Sciences University, Portland, OR, USA. W.C. Bailey (Principal Investigator) and L.B. Gerald (Project Coordinator): University of Alabama at Birmingham, Birmingham, AL, USA. D.P. Tashkin (Principal Investigator) and I.P. Zuniga (Project Coordinator): University of California, Los Angeles, CA, USA. N.R. Anthonisen (Principal Investigator, Steering Committee Chair), J. Manfreda (Co-Principal Investigator), R.P. Murray (Co-Principal Investigator), S.C. Rempel-Rossum (Project Coordinator): University of Manitoba, Winnipeg, MB, Canada. J.E. Connett (Principal Investigator), P.L. Enright, P.G. Lindgren, P. O'Hara (LHS Intervention Coordinator), M.A. Skeans and H.T. Voelker: University of Minnesota Coordinating Center, Minneapolis, MN, USA. R.M. Rogers (Principal Investigator) and M.E. Pusateri (Project Coordinator): University of Pittsburgh, Pittsburgh, PA, USA. R.E. Kanner (Principal Investigator) and G.M. Villegas (Project Coordinator): University of Utah, Salt Lake City, UT, USA. S.S. Hurd (Former Director, Division of Lung Diseases), J.P. Kiley (Former Project Officer and Director, Division of Lung Diseases), G. Weinmann (Former Project Officer and Director, Airway Biology and Disease Program, DLD), M.C. Wu (Division of Epidemiology and Clinical Applications): National Heart, Lung, and Blood Institute staff, Bethesda, MD, USA.
Support statement: This study was supported by the National Heart Lung and Blood Institute (grants HL076322, HL066583 and HL010342; Bethesda, MD, USA) and the National Institute on Ageing (AG21057; Bethesda). This study was also supported in part by the Intramural Research Program of the National Human Genome Research Institute and National Institutes of Health (Bethesda, MD, USA). Genotyping services were provided by the Johns Hopkins University (Baltimore, MD, USA) under US Federal Government contract number N01-HV-48195 from the NHLBI. K.C. Barnes was supported in part by the Mary Beryl Patch Turnbull Scholar Program (Baltimore, MD, USA).
Footnotes
Statement of Interest: None declared.
References
- 1.Mannino DM, Homa DM, Akinbami LJ, et al. Chronic obstructive pulmonary disease surveillance – United States, 1971–2000. Respir Care. 2002;47:1184–1199. [PubMed] [Google Scholar]
- 2.Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet. 1997;349:1498–1504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 3.O'Donnell CP, Tankersley CG, Polotsky VP, et al. Leptin, obesity, and respiratory function. Respir Physiol. 2000;119:163–170. doi: 10.1016/s0034-5687(99)00111-5. [DOI] [PubMed] [Google Scholar]
- 4.Aizawa-Abe M, Ogawa Y, Masuzaki H, et al. Pathophysiological role of leptin in obesity-related hypertension. J Clin Invest. 2000;105:1243–1252. doi: 10.1172/JCI8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barouch LA, Berkowitz DE, Harrison RW, et al. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 2003;108:754–759. doi: 10.1161/01.CIR.0000083716.82622.FD. [DOI] [PubMed] [Google Scholar]
- 6.Calikoglu M, Sahin G, Unlu A, et al. Leptin and TNF-α levels in patients with chronic obstructive pulmonary disease and their relationship to nutritional parameters. Respiration. 2004;71:45–50. doi: 10.1159/000075648. [DOI] [PubMed] [Google Scholar]
- 7.Creutzberg EC, Wouters EF, Vanderhoven-Augustin IM, et al. Disturbances in leptin metabolism are related to energy imbalance during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162:1239–1245. doi: 10.1164/ajrccm.162.4.9912016. [DOI] [PubMed] [Google Scholar]
- 8.Schols AM, Creutzberg EC, Buurman WA, et al. Plasma leptin is related to proinflammatory status and dietary intake in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:1220–1226. doi: 10.1164/ajrccm.160.4.9811033. [DOI] [PubMed] [Google Scholar]
- 9.Loffreda S, Yang SQ, Lin HZ, et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998;12:57–65. [PubMed] [Google Scholar]
- 10.Madiehe AM, Mitchell TD, Harris RB. Hyperleptinemia and reduced TNF-α secretion cause resistance of db/db mice to endotoxin. Am J Physiol Regul Integr Comp Physiol. 2003;284:R763–R770. doi: 10.1152/ajpregu.00610.2002. [DOI] [PubMed] [Google Scholar]
- 11.Gan WQ, Man SF, Senthilselvan A, et al. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59:574–580. doi: 10.1136/thx.2003.019588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willemse BW, ten Hacken NH, Rutgers B, et al. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005;26:835–845. doi: 10.1183/09031936.05.00108904. [DOI] [PubMed] [Google Scholar]
- 13.Bruno A, Chanez P, Chiappara G, et al. Does leptin play a cytokine-like role within the airways of COPD patients? Eur Respir J. 2005;26:398–405. doi: 10.1183/09031936.05.00092404. [DOI] [PubMed] [Google Scholar]
- 14.Kanner RE, Anthonisen NR, Connett JE. Lower respiratory illnesses promote FEV1 decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. Am J Respir Crit Care Med. 2001;164:358–364. doi: 10.1164/ajrccm.164.3.2010017. [DOI] [PubMed] [Google Scholar]
- 15.Wedzicha JA. Airway infection accelerates decline of lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1757–1758. doi: 10.1164/ajrccm.164.10.2108049a. [DOI] [PubMed] [Google Scholar]
- 16.Lee GH, Proenca R, Montez JM, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 17.Tsuchiya T, Shimizu H, Horie T, et al. Expression of leptin receptor in lung: leptin as a growth factor. Eur J Pharmacol. 1999;365:273–279. doi: 10.1016/s0014-2999(98)00884-x. [DOI] [PubMed] [Google Scholar]
- 18.Guerassimov A, Hoshino Y, Takubo Y, et al. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Respir Crit Care Med. 2004;170:974–980. doi: 10.1164/rccm.200309-1270OC. [DOI] [PubMed] [Google Scholar]
- 19.Anthonisen NR, Connett JE, Kiley JP, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study JAMA. 1994;272:1497–1505. [PubMed] [Google Scholar]
- 20.Kanner RE, Connett JE, Williams DE, et al. Effects of randomized assignment to a smoking cessation intervention and changes in smoking habits on respiratory symptoms in smokers with early chronic obstructive pulmonary disease: the Lung Health Study. Am J Med. 1999;106:410–416. doi: 10.1016/s0002-9343(99)00056-x. [DOI] [PubMed] [Google Scholar]
- 21.Enright PL, Connett JE, Kanner RE, et al. Spirometry in the Lung Health Study: II. Determinants of short-term intraindividual variability Am J Respir Crit Care Med. 1995;151:406–411. doi: 10.1164/ajrccm.151.2.7842199. [DOI] [PubMed] [Google Scholar]
- 22.Tashkin DP, Altose MD, Connett JE, et al. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The Lung Health Study Research Group Am J Respir Crit Care Med. 1996;153:1802–1811. doi: 10.1164/ajrccm.153.6.8665038. [DOI] [PubMed] [Google Scholar]
- 23.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett JC, Fry B, Maller J, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 25.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 26.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson DB, Ravussin E, Bennett PH, et al. Structure and sequence variation at the human leptin receptor gene in lean and obese Pima Indians. Hum Mol Genet. 1997;6:675–679. doi: 10.1093/hmg/6.5.675. [DOI] [PubMed] [Google Scholar]
- 28.Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765–773. doi: 10.1016/S0140-6736(07)61380-4. [DOI] [PubMed] [Google Scholar]
- 29.Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555. doi: 10.1164/rccm.200703-456SO. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YY, Gottardo L, Mlynarski W, et al. Genetic variability at the leptin receptor (LEPR) locus is a determinant of plasma fibrinogen and C-reactive protein levels. Atherosclerosis. 2007;191:121–127. doi: 10.1016/j.atherosclerosis.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 31.Man SF, Connett JE, Anthonisen NR, et al. C-reactive protein and mortality in mild to moderate chronic obstructive pulmonary disease. Thorax. 2006;61:849–853. doi: 10.1136/thx.2006.059808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takabatake N, Nakamura H, Minamihaba O, et al. A novel pathophysiologic phenomenon in cachexic patients with chronic obstructive pulmonary disease: the relationship between the circadian rhythm of circulating leptin and the very low-frequency component of heart rate variability. Am J Respir Crit Care Med. 2001;163:1314–1319. doi: 10.1164/ajrccm.163.6.2004175. [DOI] [PubMed] [Google Scholar]
- 33.Broekhuizen R, Vernooy JH, Schols AM, et al. Leptin as local inflammatory marker in COPD. Respir Med. 2005;99:70–74. doi: 10.1016/j.rmed.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 34.Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol. 2008;294:L612–L631. doi: 10.1152/ajplung.00390.2007. [DOI] [PubMed] [Google Scholar]
- 35.Brusselle GG, Bracke KR, Maes T, et al. Murine models of COPD. Pulm Pharmacol Ther. 2006;19:155–165. doi: 10.1016/j.pupt.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Shapiro SD, Demeo DL, Silverman EK. Smoke and mirrors: Mouse models as a reflection of human chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:929–931. doi: 10.1164/rccm.2408007. [DOI] [PubMed] [Google Scholar]
- 37.Debouck C, Goodfellow PN. DNA microarrays in drug discovery and development. Nat Genet. 1999;21(1):48–50. doi: 10.1038/4475. [DOI] [PubMed] [Google Scholar]