

Abstract
Extracellular vesicles (EVs) are nanoscale membrane-formed compartments naturally secreted from cells, which are intercellular mediators regulating physiology and pathogenesis, therefore they could be a novel therapeutic carrier for targeted delivery. However, the translation of EVs is hindered by the heterogeneous composition, low yield, inefficient drug loading and unlikely scalability. Here we report a strategy to generate EVs using nitrogen cavitation (NC-EVs) that instantly disrupts neutrophils to form nanosized membrane vesicles. NC-EVs are similar to naturally secreted EVs (NS-EVs), but contain less subcellular organelles and nuclear acids. The production of NC-EVs was increased by 16 folds and is easy to scale up for clinical use compared to NS-EVs. To examine the usefulness of NC-EVs as a drug delivery platform, piceatannol (an anti-inflammation drug) was remotely loaded in NC-EVs via the pH gradient. We found that piceatannol-loaded NC-EVs dramatically alleviated acute lung inflammation/injury and sepsis induced by lipopolysaccharide (LPS). Our studies reveal that nitrogen cavitation is a novel approach to efficiently generate EVs from any cell type and could be exploited for personalized nanomedicine
1. Introduction
Synthetic nanomaterials, such as liposomes [1, 2] and nanoparticles (NPs) made from polymers [3, 4], proteins [5–8] and metals [9, 10], have been intensively studied as drug carriers and imaging agents for therapy and diagnostics, particularly in cancer [11]. To increase a targeting capacity, NPs are usually bio-functionalized via conjugating targeted antibodies or peptides to the surface of NPs [12, 13]. However, bio-functionalization of NPs still remain insufficient to replicate complex intercellular interactions present in the physiological environment in humans, thus impossibly avoiding the exposure of exogenous characteristics of synthetic NPs to the immune system [13–16]. For example, two types of well-studied nanoparticle carriers, PLGA (poly(lactic-co-glycolic acid) NPs[17] and liposomes[18]. They show no targeting properties and a short blood circulation[13]. To resolve the problems, nanoparticle surfaces are conjugated with PEG (polyethylene glycol) to increase circulation times and with antibodies or peptides for targeting. Recent studies have shown a rapid clearance of PEGylated carriers after repeated administration as a result of systemic immunogenicity [19]. In addition, the presence of targeting ligands can exert a negative influence on nanoparticle delivery by diminishing circulation times due to the enhanced immune-elimination[20]. Furthermore, the complex pathology of disease conditions cannot guarantee that a unique receptor will be substantially expressed solely on the cell population of interest, even though a highly specific targeted nanoparticle can be achieved [13]. Despite the recent innovations in nanoparticle synthesis and bioengineering [21–23], the synthetic nature of nanoparticle carriers still holds the inevitable immune response in vivo.
Intercellular communication is critical to maintain the homeostasis in a biological system [24]. The communication occurs via the direct cell-cell binding, such as via adhesion molecules or via soluble factors, and hormones secreted by cells. Studies have found that an additional mechanism regulates the intercellular interaction over both a short and long distance by releasing and taking up membrane nanosized vesicles, called extracellular vesicles (EVs) [25]. EVs are spherical and proteolipid membrane vesicles that are enriched with various proteins, nucleic acids and lipids [26]. A wide range of cellular adhesion molecules are highly expressed on EVs, therefore EVs could be a novel targeted drug delivery system [27]. EVs could overcome the limitations of synthetic nanomaterials because EVs are endogenous cellular compartments. They can be designed in personalized medicine if EVs are generated from individual patients.
Despite their potential in targeted drug delivery, the translation of EVs is encumbered by the heterogeneity in composition and size, low production yield, inefficient drug loading and unlikely scalability. Recently we have developed nitrogen cavitation (a physical force) used to disrupt cells to form membrane-formed nanovesicles [28]. Here we address whether EVs can be made using nitrogen cavitation and whether our technology is scalable for clinical use. We directly compared EVs made using nitrogen cavitation (called NC-EVs) with naturally secreted EVs (called NS-EVs) in the structure and composition, and examined the therapy of NC-EVs in treating inflammatory diseases.
Uncontrolled vascular inflammation is the underlying pathogenesis of most diseases, for example acute lung inflammation/injury (ALI) [29, 30]. Trauma and infection-induced sepsis cause ALI that quickly precipitates acute respiratory distress syndrome (ARDS). The annual ALI/ARDS incidence is 200,000 adult patients in the United States alone and aging patients are increasing [29]. This is a severe drain in the healthcare budget. Despite the medical advances in lung-protection ventilation and fluid-conservative management, the mortality is still unacceptably high at 40% [29]. There is no formally recommended pharmacological therapy in place for ALI/ARDS patients, in part because drugs cannot efficiently target inflamed lungs [31, 32].
Upregulated inflammation and increased permeability of alveolar endothelial and epithelial barriers are the hallmark of ALI/ARDS pathogenesis [29, 31, 33]. In the initial phase of ALI/ARDS, vascular inflammation promotes neutrophil (a type of white blood cell) transmigration into the lungs via several steps including neutrophil binding and adhesion to activated lung endothelial cells. The binding and adhesion require the interaction between integrin β2 on neutrophils and intercellular adhesion molecule-1 (ICAM-1) on endothelial cells [34, 35]. Inspired by this unique intercellular interaction, we propose to generate neutrophil-derived extracellular vesicles that can target inflammatory lung endothelium. The endothelial activation is mainly regulated by the NF-κB signaling pathway during the lung inflammation [36, 37]. Thus, targeting the NF-κB pathway in inflamed lung or inflamed sites using EVs would be a means to reverse the progression of ALI and infection-induced sepsis.
Here, we show that nitrogen cavitation can efficiently generate EVs-like membrane vesicles from neutrophils (called NC-EVs). It is shown that NC-EVs possess similar structures and compositions to naturally secreted EVs (called NS-EVs) after we characterized the membrane markers, vesicular size and surface charges using proteomics and cryo-TEM, but NC-EVs contained less sub-cellular organelles and genetic materials. Importantly, the production of NC-EVs was increased by 16 times higher than that of NS-EVs. We remotely loaded piceatannol in NC-EVs (Pic-NC-EVs) and showed that Pic-NC-EVs inhibited the NF-κB pathway in endothelium. Furthermore, Pic-NC-EVs significantly attenuated the inflammation response in acute lung inflammation/injury and sepsis induced by lipopolysaccharide (LPS), thus alleviating acute lung injury and increasing the mouse survival in sepsis. Our results illustrate that nitrogen cavitation is a novel strategy to efficiently generate EVs for therapies and this approach could be applied to a wide range of cell types for personalized nanomedicine.
2. Materials and Methods
2.1 Materials
Concanavalin A (Con A), LPS (Escherichia coli 0111:B4), α-methyl-D-mannoside, formaldehyde solution, hexadecyltrimethylammonium bromide, o-dianisidine hydrochloride, dimethylsulfoxide (DMSO, purity >99.5%), Histopaque 1077 and Histopaque 11191 were purchased from Sigma-Aldrich (St. Louis, MO). Rabbit Anti-Nup98 and anti-calnexin polyclonal antibodies were obtained from Bethyl Laboratories. Mouse anti-COX IV, anti-LAMP-1 monoclonal antibody, recombinant human and mouse TNF-α (carrier-free, purity 98%), Alexa Fluor@647 anti-mouse CD31 antibody, and ELISA kits for TNF-α, IL-1β and IL-6 were bought from Biolegend Inc. (San Diego, CA). Mouse anti-golgin-97 monoclonal antibody was obtained from Affymetrix. A pH measuring reagent of SNARF-1-AM, lipid staining dye DIO(3-octadecyl-2-[3-(3-octadecyl-2(3H)-benzoxazolylidene)-1-propenyl]-, perchlorate) [Ex(484 nm)\Em(501 nm)], DID (3H-Indolium, 2-(5-(1,3-dihydro-3,3-dimethyl-1-octadecyl-2H-indol-2-ylidene)-1,3-pentadienyl)-3,3-d imethyl-1-octadecyl-, perchlorate) [Ex(644 nm)\Em(655 nm)], Penicillin-streptomycin (pen-strep) and glutamine (100×) were purchased from Life Technologies(Grand Island, NY). A HL60 cell line was bought from ATCC (Manassas, VA). Anti-ICAM-1, anti-integrin β2, anti-TLR4, anti-PECAM-1, anti-PSGL-1, anti-integrin α4, anti-GAPDH antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Pierce™BCA protein assay kit and ECL western blotting substrate were obtained from Thermo Fisher Scientific (Rockford, IL). Diff-Quick dye was purchased from Polysciences Inc. (Warrington, PA). Human vascular endothelia cells (HUVECs), cell culture medium EBM supplemented with EGM Singlequot (For HUVECs) and RPMI-1640 were purchased from Lonza (Walkersville, MD). Trypsin 0.25% solution and HBSS buffer (without Ca2+, Mg2+ and phenol red) was obtained from Corning (Corning, NY). Annexin V-FITC was purchased from BD Bioscience (San Jose, CA).
2.2 Cell culture
Cell line HL60 was cultured in RPMI1640 medium, supplemented with 10% fetus bovine sera and antibiotics. HUVECs were cultured in EBM supplemented with SingleQuots™, containing 2% FBS, growth factors, cytokines and other supplements. Cells were digested with 0.25% Trypsin and passaged when cells reached 80% confluency. All cells were cultured in a humidified atmosphere containing 5% CO2.
2.3 NS-EV preparation and characterization
We followed a general protocol in generation of extracellular vesicles (EVs). HL60 cells were differentiated to neutrophil-like cells in RPMI1640 medium supplemented with DMSO at 1.3% (v./v.) for 4 days. NS-EVs were secreted in the culture medium of differentiated HL60 cells. The cell culture medium was collected after the centrifugation at 300 g and following the centrifugation at 2 000 g to remove cell debris. The supernatant was centrifuged at 100 000 g for 1 h and the pellet was collected, called NS-EVs. The NS-EVs were washed twice with HBSS and their size and zeta potential were measured using qNano (iZON science, MA) or dynamic light scattering. The protein and DNA contents were quantified using the BCA method (Pierce Biotechnolgy, Rockford, IL) and a double strand DNA quantification kit (Promega, Madison, WI), respectively.
2.4 Preparation of human neutrophil and HL-60 cell-derived NC-EVs
Human PMNs from healthy adult donors (The protocol of human subject has been approved by the Institutional Review Board, WSU and the execution of the experiment was fully explained to the participants. The informed consent was obtained from all participants) and activated HL60 cells were used for NC-EV preparation. Human PMNs were isolated according to the commercial guideline using Histopaque 1077 and Histopaque 1119 reagents. HL60 cells were differentiated to neutrophil-like cells in a RPMI1640 medium supplemented with 1.3% (v./v.) DMSO for 4 days. The cells were suspended in HBSS or 0.3 M Tris-HCl at 0.5–5×107/ml, and was added with MgCl2 at 2.5 mM. The cell suspension was filled in a nitrogen cavitation chamber at a pressure of 400–500 psi for 20 min at 0 °C, and subsequently the cavitation was operated to disrupt cells. The nitrogen cavitation was operated twice. The suspension was added with EDTA at 2.5 mM and was centrifuged at 2 000 g for 30 min. The resulted supernatant was centrifuged at 100 000 g and the pellet was collected, called NC-EVs. The pellet was washed with HBSS and the suspension was sonicated to make uniform size of NC-EVs. NC-EVs were further purified by ConA.
2.5 pH measurement in cells and NC-EVs
To measure cell or vesicle inner pH values, SNARF-1-AM (at 10mM) was used. The ratio of emission at 640 nm and 580 nm of SNARF-1 in a buffer with different pH was measured and transformed pH values. Using this method, we quantified the pH values in a cell or NC-EVs.
2.6 Remote drug loading in NC-EVs
The pH gradient driven loading of piceatannol inside NC-EVs. NC-EVs (10 mg) whose inner compartment is at pH=9 were mixed with 1.5 mg of piceatannol in 2ml HBSS, and the suspension was adjusted to pH=4.5. The pH gradient between the inner and outside of EVs was formed. Without the pH gradient drug loading, the preparation was conducted in the same HBSS at pH=7.4. The mixture was incubated at 37°C for 30 min. The suspension was centrifuged at 100,000 g to obtain drug-loaded vesicles. After the samples were lyophilized, the drug was extracted using ethanol and was quantified using HPLC (Waters 2690 separation module, coordinating with a Waters 486 tunable absorbance detector). The piceatannol concentration in EVs was analyzed.
2.7 Proteomics of NC-EVs and NS-EVs
NC-EVs and NS-EVs were suspended in HBSS. The proteins were extracted using Tris-buffered phenol. Protein concentrations were measured using the bicinchoninic acid (BCA) assay. The proteins of EVs were digested using the method of filter-aided sample preparation to obtain peptides for mass spectroscopy using a Q Exactive system (Thermo Scientific Inc.). The localization of proteins of NC-EVs, NS-EVs and parent cells was analyzed using Gene Ontology.
2.8 Piceatannol inhibits ICAM-1 expression via NF-κB Pathway
HUVECs were seeded in a 12-well plate at 160 000 cell per well 24 h prior treatment with TNF-α and/or piceatannol. The ICAM-1 expression was quantified by Western blot. IκBα and P65 of HUVECs treated with TNF-α or piceatannol were analyzed using Western blot after cytoplasm and nuclei components were isolated. Briefly, cells were pelleted by centrifugation and re-suspended in the cell lysis buffer [10 mM HEPES; pH 7.5, 10 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol (DTT)] and stayed on ice for 15–20 min. The supernatant was collected and the pellet of nuclei content was washed three times with a cell lysis buffer and re-suspended in the nuclear extraction buffer (containing 20 mM HEPES (pH 7.5), 400 mM NaCl, 1 mM EDTA, 1 mM DTT), and incubated on ice for 30 min. The nuclear extract is collected by centrifugation at 12,000 g for 15 min at 4°C. The protein was analyzed with WB using anti-IκBα and p65 antibodies.
2.9 Intravital microscopy of live mouse cremaster venules
To visualize the adherence of NC-EVs to vasculature in vivo, intravital microscopy was used. A CD1 mouse was firstly intrascrotally given TNF-α (500 ng in 250 μl saline). 3 h later, the vascular inflammation was expected to appear the high expression of ICAM-1. The mouse was anesthetized with i.p. administration of a mixture of ketamine and xylazine, and maintained at 37°C on a thermo-controlled rodent blanket. To conveniently infuse NC-EVs and antibodies, a tracheal tube was inserted and a right jugular vein was cannulated. The scrotum was then incised, and the testicle and surrounding cremaster muscle were exteriorized onto an intravital microscopy tray. The cremaster preparation was superfused with thermo-controlled (37°C) and aerated (95% N2, 5% CO2) bicarbonate-buffered saline throughout the experiment. Finally, Alex Fluor-647 anti-mouse CD31 antibody (2.5 μg per mouse) and NC-EVs labeled with DiO (0.1 mg/mouse) were simultaneously infused into the mouse. Images were recorded using a Nikon A1R+ laser scanning confocal microscope with a resonant scanner.
2.10 Western blot and cytokine measurement
The protein expression of NC-EVs and NS-EVs was quantified using Western blot (Bio-Rad Molecular imager (ChemiDoc XRS+, Bio-Rad, USA). The antibodies of integrin β2, LAMP-1, Calnexin, COX IV, Golgi-97, Nup98, and GAPDH were used to locate the cellular organelles. Cytokine levels in BALF and sera were determined using commercial ELISA kits for TNF-α, IL-1β and IL- 6 (Biolegend, San Diego, CA) according to the manufacturer’s instructions.
2.11 Therapy of acute lung inflammation
The Washington State University Institutional Animal Care and Use Committee approved all animal care and experimental protocols used in the studies. All experiments were made under anesthesia using intraperitoneal (i.p.) injection of a mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg) in saline.
Adult CD1 mice (18–25 g) were purchased from Harlan Labs (Madison, WI). LPS was intratracheally administrated to mice. 2 hours later, mice were randomly grouped by 3 mice per group. The mice were intravenously administrated with HBSS, piceatannol, and piceatannol-loaded NC-EVs, respectively. The dose of piceatannol is at 2mg/kg.
2.12 Lung leukocyte infiltration in bronchoalveolar lavage fluid (BALF)
At 12 h post-LPS administration, mice were anesthetized with the i.p. injection of ketamine and xylazine mixture. The trachea was cannulated, and 1 ml HBSS was infused intratracheally to collect BALF. The lavage fluid was centrifuged at 420 g for 4 min, and a cell pellet was suspended in 0.7 ml red blood cell lysis buffer (Qiagen, Valencia, CA). After 30 min, the cells were pelleted by centrifugation at 420 g for 4 min, and suspended in 0.5 ml HBSS. The total cell number was determined with a hemocytometer. Cell suspension was diluted to a final concentration at 1×105 cells/ml and a 200-μl of the suspension was spun onto a slide at 700 rpm for 5 min using a cytocentrifuge (Shandon, Southern Sewickley, PA). The slides were stained with a Diff-Quick dye, and examined at a magnification of 400 by a microscope. The percentage of neutrophils was randomly determined.
2.13 Lung edema quantified by protein concentration in BALF
The lung permeability (lung edema) was evaluated by the lavage protein content. The total protein concentrations were determined using a BCA protein assay kit (Thermo scientific, Rockford, IL).
2.14 Survival in the sepsis model
CD1 mice were randomly grouped by 9 mice per group and i.p. administrated with LPS at 22 mg/kg. 2 hours after LPS administration, the mice were intravenously received with HBSS, piceatannol (3 mg/kg) and piceatannol-loaded vesicles (piceatannol at 3 mg/kg). The mice were monitored every 6–10 h to analyze the death.
2.15 Therapy in a sepsis model
CD1 mice (18–25 g) were randomly grouped by 3 mice per group and i.p. administrated with LPS at 22 mg/kg (mouse body weight). 2 hours after LPS administration, the mice were intravenously injected with HBSS, piceatannol (3 mg/kg) and piceatannol-loaded vesicles (piceatannol at 3 mg/kg). 4 and 8 hours post-LPS administration, the blood samples and tissues were collected to analyze cytokines and MPO concentrations.
2.16 MPO activity measurement
Tissues were homogenized in 1 ml of PBS (pH 5) with 0.5% hexadecyltrimethylammonium bromide and 5 mM EDTA. The homogenates were sonicated, centrifuged at 20,000 × g for 20 min. The supernatant was then collected and mixed 1/30 (v/v) with an assay buffer (0.2 mg/ml o-dianisidine hydrochloride and 0.0005% H2O2). The absorption change was measured at 460 nm in 3 min, and MPO activity was calculated by the absorption rate.
2.17 Exposure of phosphatidylserine (PS) on nanovesicles
The equal weight of NC-EVs or NS-EVs were incubated with Annexin V-FITC to allow the staining of PS on EVs. The nanovesicles were purified by the centrifugation at 100,000 g 10 min after the incubation and washed with PBS. Finally, the nanovesicles were suspended in 200 μl PBS. The fluorescence intensity was measured at 525 nm using a fluorescence spectrometer.
2.18 Determine the pro-inflammation effect of NC-EVs
HUVECs were seeded in a 12-well plate at 160 000 cells per well 24 h prior the treatments with HBSS, TNF-α (25 ng/ml), NC-EVs (1.8 mg/ml) and piceatannol-loaded NC-EVs (25 ng/ml at piceatannol), respectively. 4 h later, the cells were then washed and lysed to determine the ICAM-1 expression using western blot.
2.19 Statistical analysis
Data are expressed as means ± SD. Statistical analysis was conducted using one-way ANOVA or Student’s t-Test.
3. Results
3.1 Efficient Generation of EVs using Nitrogen Cavitation (NC-EVs)
Cells secret EVs spontaneously or upon the activation under a pathological condition as shown in Fig. 1A. Based on the biogenesis mechanism, EVs are composed of exosomes, microvesicles and apoptotic bodies [38, 39]. Despite the apparent differences in the mechanism of biogenesis, it is experimentally difficult to separate the subpopulations of EVs. However, EVs structurally possess a bilayer membrane directly derived from a source cell. Since the cell membrane is a key component to form EVs, we reasoned that EVs could be made if we physically disrupt a cell, and subsequently the membrane forms EVs. We have developed a means to instantly break a cell using nitrogen cavitation [28], and subsequently centrifuged and made the uniform size of membrane vesicles from neutrophil-like HL60 cells [40] (called NC-EVs) (Figure 1A). The cryo-TEM image (Fig. 1B) showed that NC-EVs are spherically vesicular, similar to EVs naturally secreted from the HL 60 culture medium (called NS-EVs).
Figure 1.
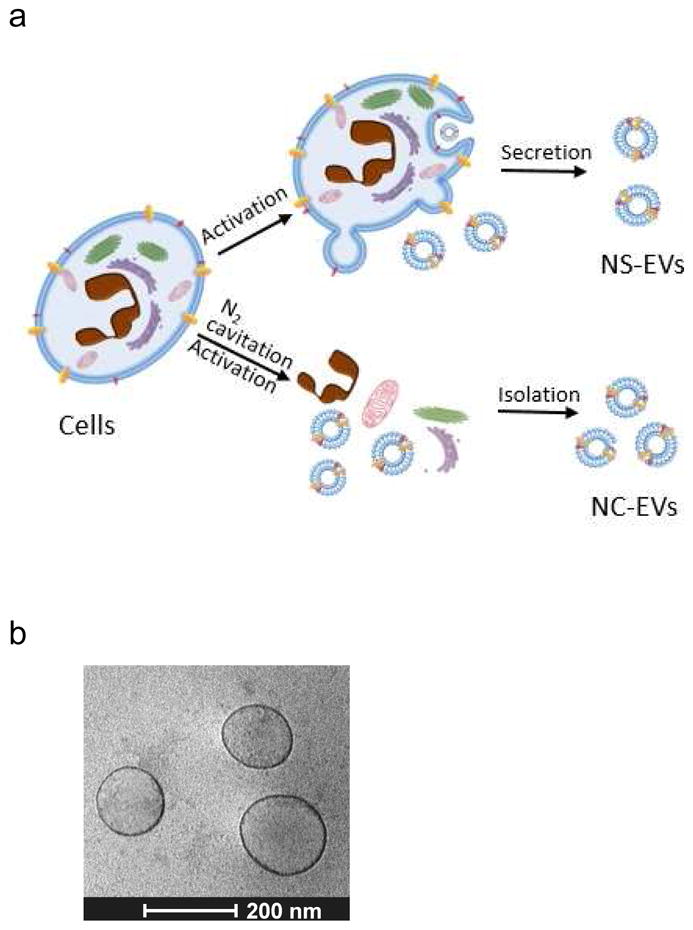


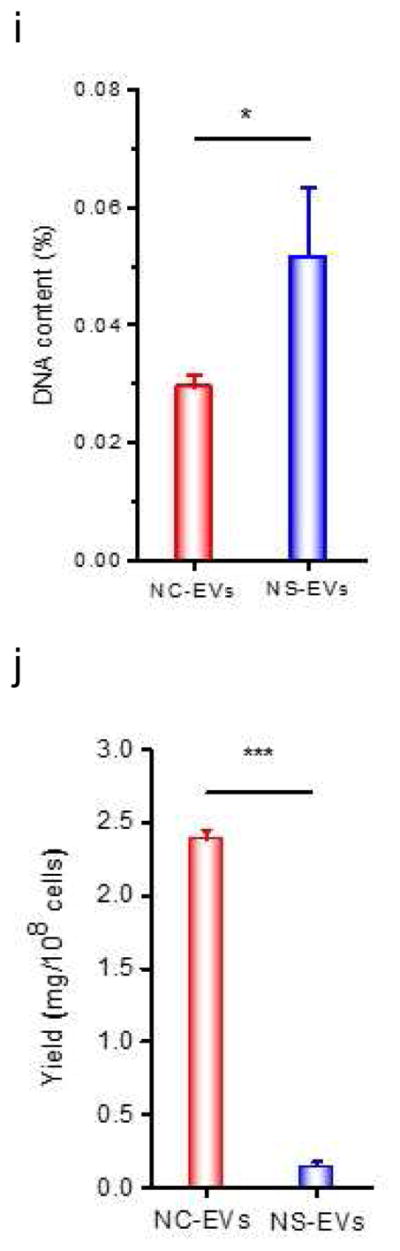
Efficient generation of extracellular vesicles (EVs) using nitrogen cavitation. (a) Schematics shows the generation of EVs from natural secretion (NS-EVs) and using nitrogen cavitation (NC-EVs). (b) Cryo-TEM image of NC-EVs. (c) Western blot of NC-EVs, NS-EVs and their parent cells. The same amount of proteins was loaded in each sample. Quantification analysis on the composition of NC-EVs (d) and NS-EVs (e) based on Western blot. Sizes (f), zeta potentials (g), proteins (h), DNA (i), and production yield (j) of NC-EVs and NS-EVs. All data expressed as means ± SD (n=3–6). * and ** represent p<0.05 and 0.01 respectively by t-Test.
We measured the proteins in NS-EVs and NC-EVs using Western blot (Fig. 1C). In contrast, NC-EVs showed the higher expression of integrin β2, a membrane marker of activated neutrophils [5]. We also examined whether NC-EVs or NS-EVs mixed with subcellular organelles since the purity of EVs is important as a drug delivery platform [41, 42]. The Western blot showed that NC-EVs are lack of ERs (Calnexin, a marker of ERs), lysosomes (lysosomal associated membrane protein 1, LAMP-1) and mitochondria (Cytochrome c Oxidase IV, COX IV) compared to NS-EVs. A quantitative analysis (Fig. 1D and E) of Western blot indicates that NC-EVs did not contain the organelles, such as mitochondria, Golgi bodies (Golgi-97) and nuclei (Nup98, nuclear membrane protein). The lysosomes and ERs in NC-EVs are extremely lower than that in NS-EVs. The targeting ligands, integrin β2, were enriched in NC-EVs by 7 times higher than that in NS-EVs. Furthermore, NC-EVs showed the similar size, surface zeta potential and proteins to NS-EVs (Fig. 1F–H).
Genetic materials contained in EVs could cause a side effect in drug delivery [41], we therefore investigated DNA contents in NC-EVs and NS-EVs (Fig. 1I). The study indicates that NC-EVs contained lower genetic molecules than NS-EVs. Further, the production yield of EVs was determined based on the same quantity of cells. The result shows that the yield of NC-EVs was increased by 16 folds compared to NS-EVs (Fig. 1J).
3.2 Proteomics of EVs
Fig. 2A shows the SDS-PAGE of HL-60 cells and their NC-EVs, indicating that NC-EVs have the similar protein pattern to their parent cells. Furthermore, we performed proteomics on NS-EVs, NC-EVs and their source of HL 60 cells (Fig. 2B and 2C). The numbers of proteins in NS-EVs and NC-EVs dramatically reduced compared to that in a whole cell of HL-60, and some proteins were shared by EVs with their parent cell (Fig. 2B), suggesting that EVs are derived from their parent cells. We further analyzed the membrane proteins (Fig. 2C), and observed the similar trend of protein distributions as in Fig. 2B. We also found that the number of proteins shared by both NC-EVs and NS-EVs is 28 when we analyzed all proteins in the samples (Fig. 2B), and then it became 26 when we analyzed the membrane proteins (Fig. 2C). This interesting observation suggests that those proteins might be associated with cell membrane since NC-EVs and NS-EVs are made of membrane vesicles. It is noted that a portion of proteins were present neither in NC-EVs and NS-EVs. This implies that the protein composition in NC-EVs may be different from in NS-EVs, consistent with Western blot in Fig. 1C–E showing that there are some organelles in NS-EVs, but not in NC-EVs. Collectively, the proteomics qualitatively suggest that both NS-EVs and NC-EVs resemble their parent cells, and contained many membrane proteins of their parent cells, which is consistent with the studies in Fig. 1.
Figure 2.

Proteomics of EVs and their source cell. (a) Protein profile of NC-EVs and their parent cell (activated HL60 cells) by SDS-PAGE. The total proteins (b) and the membrane proteins (c) in NC-EVs, NS-EVs and their parent cell analyzed with mass spectroscopy.
3.3 pH gradient-driven loading of piceatannol in NC-EVs
It was reported that doxorubicin can be remotely loaded in preformed liposomes via a pH gradient [43, 44], so we asked whether this approach could be applied to load a therapeutic inside NC-EVs. To test this hypothesis, we chose piceatannol (an anti-inflammation drug and a weak acid) to be loaded in NC-EVs with a high pH value inside (Fig. 3A). We first developed a fluorescence sensor (SNARD-1) [45] to measure the inner pH values of HL60 cells and their NC-EVs in HBSS at pH=7.4 and confirmed that this approach can accurately measure pH values in a cell and NC-EVs (Fig. 3B). When we disrupted cells to make NC-EVs in a HBSS buffer at pH=7.4, the inner pH value of NC-EVs was same as that of used HBSS, implying that the pH value inside NC-EVs might be determined by the used buffer. To achieve a high pH value in NC-EVs, we chose the 0.3 M Tris-HCl (pH=9.0) buffer when we disrupted cells to form NC-EVs. The fluorescence sensor (SNARD-1) indicated that the pH value inside NC-EVs was 9, which was consistent with the used buffer (Fig. 3B). The result indicates that we can alter pH values inside NC-EVs to form a pH gradient for drug loading (Fig. 3B).
Figure 3.
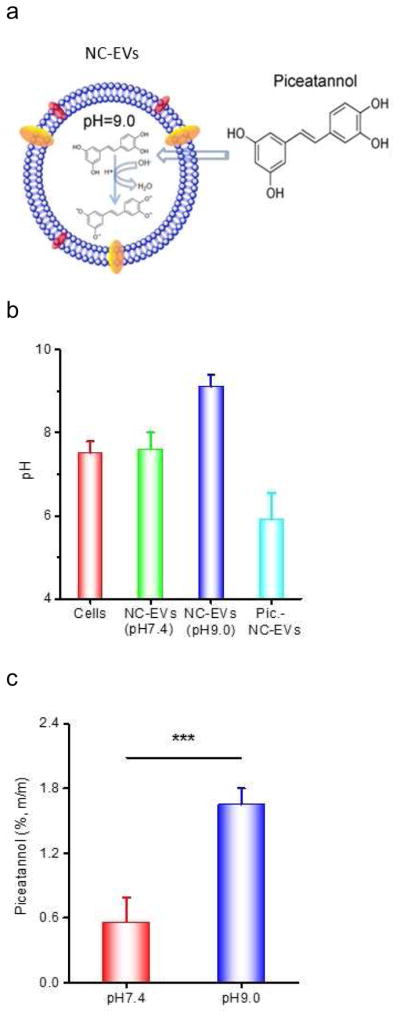
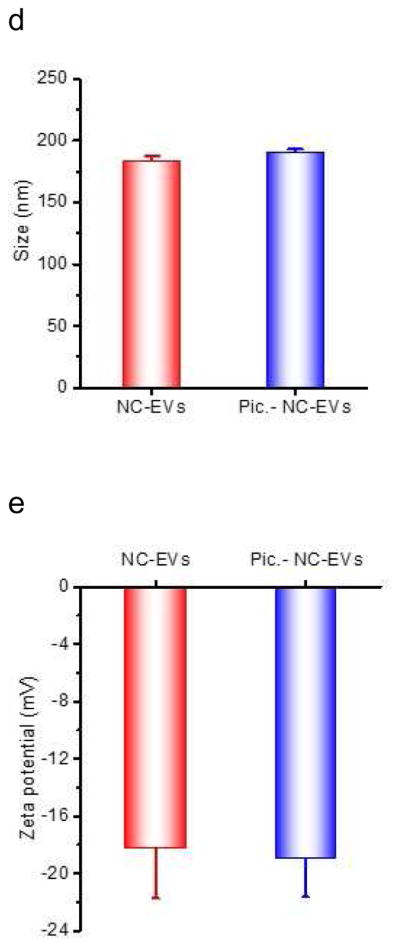
Remote loading of piceatannol inside NC-EVs driven by a pH gradient. (a) Mechanism for loading of piceatannol in NC-EVs. (b) pH values inside NC-EVs and their parent cells determined by SNARF-1 (n=3). (c) Loading efficiency of piceatannol in NC-EVs preformed at indicated pH buffers (n=3). Size (d) and surface charge (e) of NC-EVs with pre- and post-piceatannol loading (n=5). All data are expressed as means ± SD. ***P<0.001 (student’s t-test).
Piceatannol is a weak acid molecule, and increases the solubility in the basic environment [46]. When there was no pH gradient (both inside and outside pH of NC-EVs were 7.4), the piceatannol loading was 0.6% (drug(w)/vesicles(w)). However, when NC-EVs had a pH gradient where the inside pH was 9.0 and the outside pH was 7.4, the drug loading efficiency of piceatannol increased to 1.7% (w/w) indicating that the drug increase would be associated with the pH gradient (Fig. 3C). When we measured the pH values inside NC-EVs after loading piceatannol (Fig. 3B), we found that the pH value dropped to 6.5, lower than the initial value of pH=9, implying the existence of piceatannol inside EVs. Using the remote loading of piceatannol in NC-EVs, we increased the loading efficiency by 3 times (Fig. 3C). Moreover, we characterized the size and surface charges of NC-EVs, finding that loading of piceatannol inside NC-EVs does not alter their size and surface zeta potentials (Fig. 3D and E).
3.4 Piceatannol-loaded NC-EVs attenuated acute lung inflammation via NF-κB pathway
To prevent acute lung inflammation/injury caused by endothelial activation, we addressed whether piceatannol could inhibit the ICAM-1 upregulation in endothelial cells via the NF-κB signaling pathway. After human umbilical vein endothelial cells (HUVECs) were treated with TNF-α, the ICAM-1 expression dramatically increased and was in a time-dependent manner (Fig. 4A). We further examined whether the upregulation of ICAM-1 was determined by the NF-κB pathway. The proteins of IκBα kinase and phosphorylation of p65 are associated with the activation of NF-κB pathway [47]. It was observed that IκBα decreased and p65 protein increased accordingly (Fig. 4A), implying that the ICAM-1 expression was regulated by the NF-κB signaling. In contrast, the endothelial cells treated with both TNF-α and piceatannol suppressed the ICAM-1 expression via inhibition of degradation of IκBα, thus avoiding the translocation of p65 protein into a nucleus. The result indicates that piceatannol can inhibit the activation of NF-κB pathway, thus attenuating the expression of adhesion molecules, ICAM-1.
Figure 4.

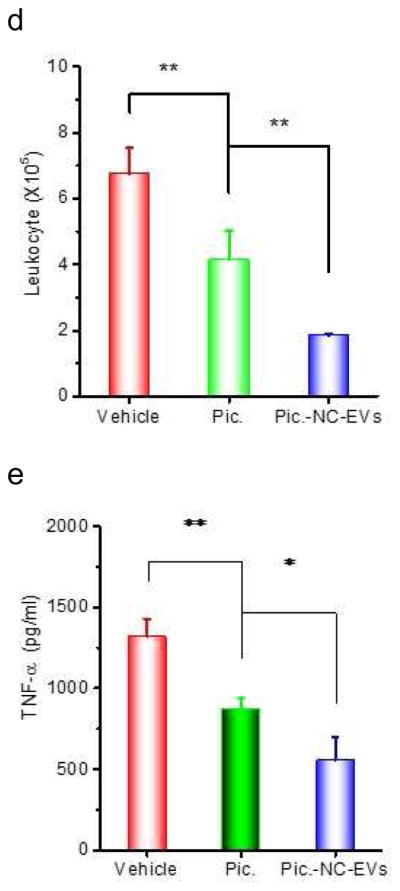
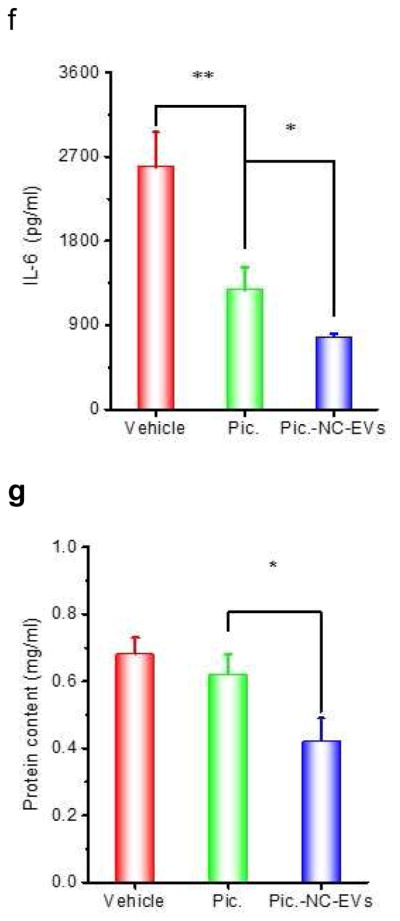
Piceatannol-loaded NC-EVs (pic-NC-EVs) attenuate mouse acute lung inflammation/injury via the NF-κB pathway. (a) Piceatannol inhibits the expression of ICAM-1 on endothelial cells via the NF-κB pathway. The transcriptional factors, including IκBα in cytoplasm and p65 in a nucleus were detected by Western blot. (b) Intravital microscopy image of cremaster venules of a live mouse intrascrotally treated with TNF-α (0.5 μg) after i.v. infusion of DiO-fluorescently-labeled NC-EVs (green) and Alex-Fluor-647-labeled anti-CD31(pink). NC-EVs adhere to the surface of a venule. The image was taken by the lasers at 488 nm and 640 nm using A1R+ resonant-scanning confocal microscope. Neutrophils (c) and total leukocytes (d) in the lung lavage fluid were analyzed after 12 h post LPS (10 mg/kg) challenge. TNF-α (e), IL-6 (f) and protein concentrations (g) in the lung lavage fluid were measured. All data are expressed as means ± SD. *P<0.05, and **P<0.01 (student’s t-test) compared to control except specially indicated.
To investigate the uptake of NC-EVs by endothelium in vivo, we established the intravital microscopy of cremaster venules in the TNF-α treated mouse model[28]. It is noted that NC-EVs were adherent to inflammatory venules induced by TNF-α (Fig. 4B). We observed that the adherent NC-EVs rarely detached from venules in the imaging period of 30–60 min, implying that it was likely that NC-EVs were internalized by activated endothelium. This uptake was confirmed when we studied the uptake of NC-EVs in endothelial cells in vitro and the bio-distribution in inflamed lungs [28].
NC-EVs are generated by breaking cell membrane to form nanovesicles, so we do not know whether the inner leaflet component is exposed out during the formation of membrane nanovesicles. For example, phosphatidylserine (PS) flips out on NC-EVs could cause pro-inflammation [48] that diminishes the application of NC-EVs as a drug carrier. Because annexin V specifically binds PS [49], we developed a fluorescence assay in which we fluorescently labeled annexin V with FITC. We incubated FITC-labeled annexin V with NC-EVs and NS-EVs, respectively, purified them by centrifugation and measured their fluorescence. The result shows that both NC-EVs and NS-EVs exhibit the similar fluorescence intensity (Fig. S1), suggesting that they have the similar amount of PS. Furthermore, we addressed whether NC-EVs can cause the inflammation in endothelial cells. We measured ICAM-1 expression levels in HUVEVs using Western blot (Fig. S2). After the treatment with NC-EVs without loading of piceatannol, we measured the expression of ICAM-1 that is comparable with that when HBSS was used as a control. When HUVEVs were treated with TNF-α, the ICAM-1 expression dramatically increased. The result indicates that NC-EVs do not promote the pro-inflammation. Combined the studies on PS externalization (Fig. S1) and pro-inflammation (Fig. S2), the result suggests that the PS level in NC-EVs might be very low because we did not observe the pro-inflammation (Fig. S2). When HUVECs were treated with piceatannol-loaded NC-EVs, the ICAM-1 expression was decreased (Fig. S2).
To test therapeutic effect of piceatannol-loaded NC-EVs in a mouse model of acute lung inflammation/injury, we intravenously administrated piceatannol or piceatannol-loaded NC-EVs to mice 2 h after the lungs were challenged by intratracheal LPS (10 mg/kg (mouse weight). 12 h later, we collected the lung lavage fluid and analyzed the infiltration of leukocytes and neutrophils (Fig. 4C and D), and cytokines (TNF-α and IL-6) (Fig. 4E and F). In contrast with HBSS or piceatannol treatment, piceatannol-loaded NC-EVs significantly reduced the lung infiltration of leukocytes and cytokine secretion (TNF-α and IL-6). The lung permeability is a hallmark of lung edema in acute lung inflammation/injury, and the permeability is characterized by the accumulation of plasma proteins in the lungs [37]. The piceatannol treatment did not improve the lung edema compared to the control (HBSS) (Fig. 4G). However, when piceatannol-loaded NC-EVs were used, the permeability of plasma proteins in lungs dramatically decreased, suggesting the reversal of lung edema (Fig. 4G).
3.5 NC-EVs prevent sepsis-induced inflammation increasing the mouse survival
Sepsis is a systemic inflammation induced by infection, and is the life-threatening disease [50]. The molecular mechanism of sepsis is strongly relevant to vascular activation and a cytokine storm [51, 52]. We examined whether piceatannol-loaded NC-EVs could alleviate sepsis induced by LPS, thus increasing the mouse survival. Mice were i.p. injected with LPS at a lethal dose (22 mg/kg), and 2 h after the LPS injection of piceatannol or piceatannol-loaded NC-EVs were intravenously administrated to the mice and the mouse death was monitored (Fig. 5A). The free piceatannol did not protect the mice from death compared to the control (HBSS), but piceatannol-loaded NC-EVs prevent 80% of the mice from death.
Figure 5.
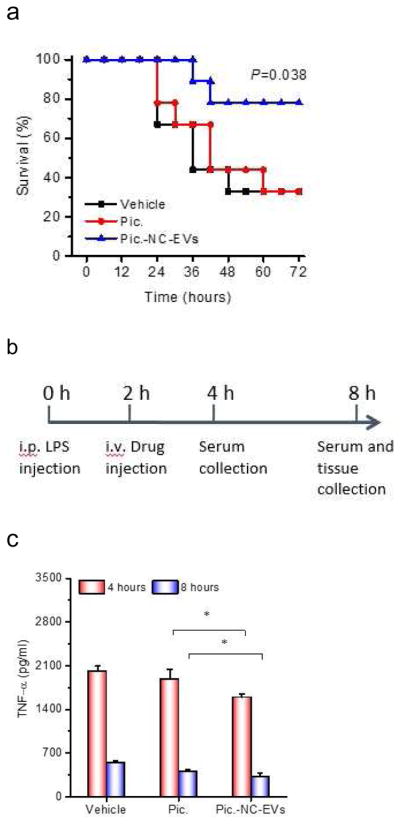
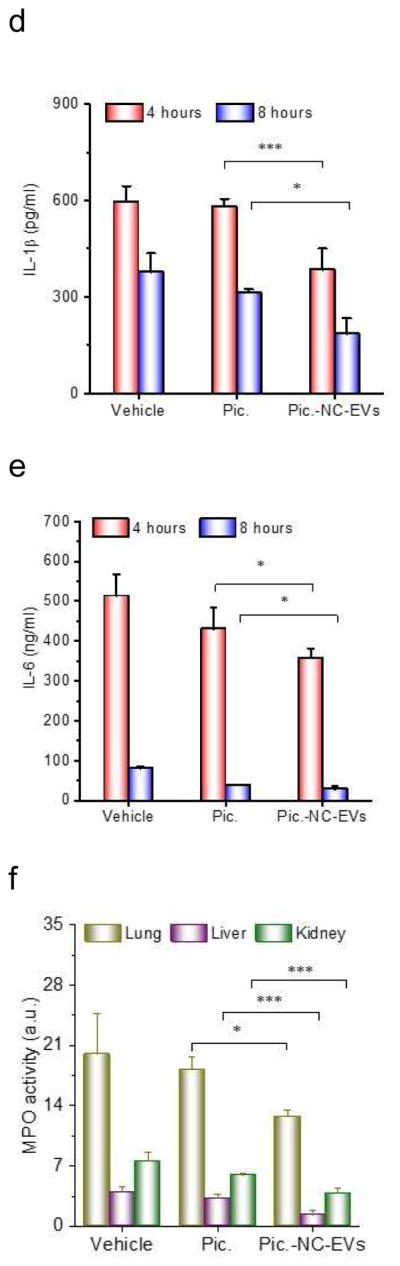
Piceatannol-loaded NC-EVs (pic-NC-EVs) protect mice from death in a sepsis model. (a) Mouse survival in sepsis induced by LPS (22 mg/kg) after the i.v administration of piceatannol and pic-NC-EVs. Statistical analysis was done using Kaplan-Meier method (n=9). (b) Schematic of experimental protocol in the sepsis study. TNF-α (c), IL-1β (d) and IL-6 (e) in the blood plasma 4 and 8 h after administration of LPS (22 mg/kg). (f) MPO in mouse lung, liver and kidney. All data are expressed as means ± SD (n=3). *P<0.05, **P<0.01 and ***P<0.001 compared to control (student’s t-test) except specially indicated.
Sepsis is strongly associated with neutrophil tissue infiltration and a cytokine storm in a short period [53], so we established a protocol (Fig. 5B) to address the molecular mechanism of the protection of mouse death when Pic-NC-EVs were used. 2 h and 6 h after i.v. injection of piceatannol or piceatannol-loaded NC-EVs, we measured the cytokine levels and found that TNF-α, IL-1β and IL-6 significantly decreased after piceatannol-loaded NC-EVs were used compared with piceatannol or HBSS (Fig. 5C–E). Furthermore, we measured MPO (a marker of neutrophils) in lungs [54, 55], liver and kidney at 6 h after i.v. injection of drugs. The result indicates that piceatannol-loaded NC-EVs decreased neutrophil tissue infiltration in lung, liver and kidney (Fig. 5F).
3.6 Human neutrophil-derived NC-EVs
To address the translational potential of our NC-EVs technology, we generated EVs from human neutrophils using the same approach described above. We measured the size and surface charge of human neutrophil NC-EVs using dynamic light scattering (Fig. 6A), consistent with NC-EVs made from HL 60 cells. The Western blot showed that NC-EVs contained many membrane adhesion proteins from their source cell (Fig. 6B), such as TLR4 (Toll-like receptor 4), PECAM-1 (Platelet endothelial cell adhesion molecule-1) and PSGL-1 (P-selectin glycoprotein ligand-1). We also quantified the proteins and DNA in human neutrophil NC-EVs (Fig. 6C). It is noted that proteins possessed 55% of the weight of NC-EVs, indicating NC-EVs are formed from the cell plasma membrane because the plasma membrane is comprised of proteins at roughly 50% by weight [28]. However, the DNA content was extremely low.
Figure 6.
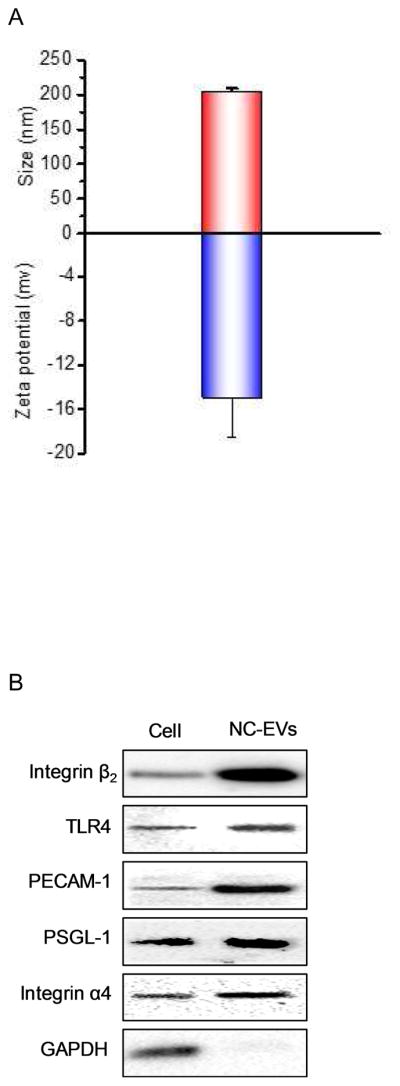
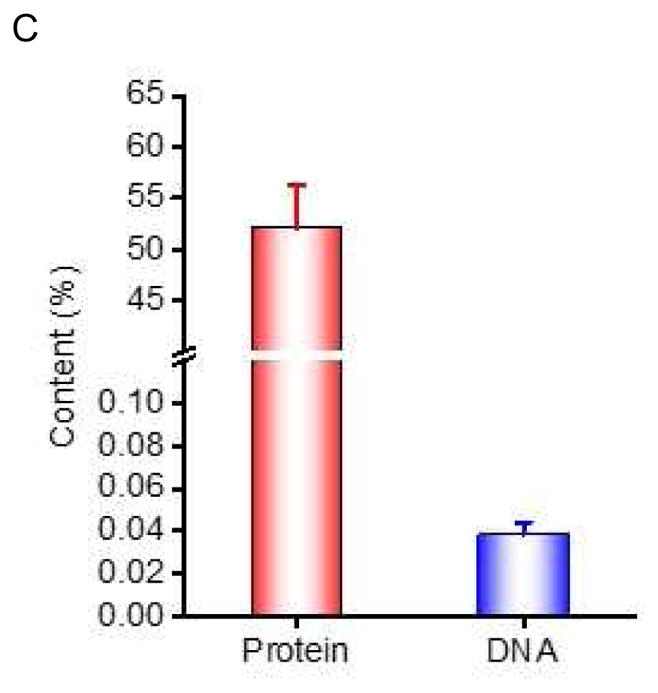
Human neutrophil-derived NC-EVs. (a) Size and zeta potential of human neutrophil-derived NC-EVs. (N=6). (b) Western blot of human neutrophils and their NC-EVs. (c) Protein and DNA contents in human neutrophil-derived NC-EVs. The protein content was measured by BCA method and the DNA content was measured by a commercial kit (n=3).
4. Discussion
Extracellular vesicles (EVs) are the natural secretion from a cell to form membrane nanoscale vesicles, and contain many adhesion molecules that specifically interact with target cells. EVs play a central role in transferring biomolecules between cells, implying that EVs could be a carrier to deliver therapeutics [39, 42]. However, there are several hurdles to apply EVs in drug delivery. To generate massive EVs, it is required to increase a culture medium volume and then to concentrate them. This process is time-consuming and is difficult to scale up for clinical use. Importantly, the reproducibility of EVs strongly depends on cell living environments (such as physiological or pathological conditions) [56], and complex subpopulations of EVs are inevitably distinguished and separated because three types of EVs (exosomes, microvesicles and apoptotic bodies) are similar in structure and size [38, 57].
EVs are spherical and cell membrane-formed vesicles. They are like synthetic liposomes in structure and size, but EVs possess many membrane proteins that could be utilized for binding of EVs to targeted cells. EVs are the endogenous compartment derived from cells so they are biocompatible and less immune responses compared to synthetic NPs. Those features enable EVs to become an excellent drug targeted delivery system [58]. The structure of EVs is formed by a cell membrane, we therefore hypothesized whether we can develop a means to instantly break a cell and release intracellular contents, and subsequently broken cell membranes reform nanosized vesicles. Here we show that nitrogen cavitation can generate nanosized vesicles (Fig. 1B). The nitrogen cavitation is a physical force that rapidly disrupts cell membrane, and instantly intracellular contents are released and disrupted plasma membrane encloses to form nanosized vesicles. Therefore, nitrogen cavitation could generate the EVs-like compartments. To examine this hypothesis, we systemically compared EVs made from nitrogen cavitation (NC-EVs) with naturally secreted EVs (NS-EVs) in protein profile, composition, size and production yield. Cryo-TEM clearly shows that NC-EVs are spherical and vesicular, and the thickness of the shell is 3–4 nm that is equivalent to that of cell plasma membrane [59]. The result indicates that NC-EVs are formed from the cell membrane. Furthermore, the studies on the protein profile, total protein amount, size and surface charges of EVs (Fig. 1C–H) indicate that NC-EVs are very similar to NS-EVs. NC-EVs however contain less subcellular organelles, such as lysosomes and ERs, and the compartments of mitochondria and Golgi are undetectable, but a membrane marker, integrin β2 in NC-EVs dramatically increase (Fig. 1C–E). When we further measured the DNA content, we found NC-EVs contained 2 times of DNA less than NS-EVs (Fig. 1I). The production yield of NC-EVs is increased by 16 times compared to that of NS-EVs (Fig. 1J). The proteomics of NC-EVs (Fig. 2A–C) shows that NS-EVs and NC-EVs are directly made from their parent cell (HL-60 cells). Collectively, the direct comparison between NC-EVs and NS-EVs clearly demonstrates that we have developed a novel approach using nitrogen cavitation to efficiently generate EVs from a neutrophil-like cell (HL-60 cell), and NC-EVs contain less the genetic content and subcellular organelles. NS-EVs are complex and heterogeneous in composition and sometimes deliver unwanted DNA/RNA or signaling proteins resulting in pathogenesis, so it is difficult to predict therapeutic outcomes if they are used as a drug carrier [58]. For instance, leukocyte-derived microvesicles play an opposite role in endothelium that is either increase of endothelial integrity or dysregulation [60]. NC-EVs made from nitrogen cavitation show the dramatic decrease of unwanted subcellular compartments and DNA, therefore we expect that NC-EVs could be an excellent candidate for drug delivery.
While we used 15–20 ml of the cell suspension in the current experiment, the cavitation chamber can be scalable to tens of liters for clinical use without loss of reproducibility as the fast and uniform force are applied to break cells [61]. Unlike other lysis methods, there is no heat or chemical damage to membrane proteins during nitrogen cavitation and NC-EVs are protected from oxidation by nitrogen gas. Nitrogen cavitation is fast and uniform because the same disruptive forces are applied within each cell and throughout the sample, ensuing reproducible cell-free homogenates [61]. In contrast with the production of NS-EVs, here we have developed a novel approach using nitrogen cavitation to make NC-EVs from neutrophils with the low cost, high yield, reproducibility and scalability. Most importantly, nitrogen cavitation can be applied to any cell types to generate EVs.
We have developed a remote loading of piceatannol inside NC-EVs (Fig. 3A–E) based on the pH gradient [62–64]. This novel approach allows to significantly increase drug loading in the preformed NC-EVs. In contrast, NS-EVs have limited methods to alter pH values inside EVs since the generation of EVs is usually at a neutral pH. Changing buffers in the process of nitrogen cavitation could be a novel approach to achieve required pH values to establish a pH gradient between the inner and outer NC-EVs. Using this approach, we have achieved the loading of piceatannol by 3 times compared to without the pH gradient. Due to the targeting feature and efficient drug loading of NC-EVs, our delivery system could surpass the current technologies used in the production of EVs.
While piceatannol was reported to inhibit the expression of COX-2, MMP9 and Cyclin D1 that are dependent on the NF-κB pathway [65, 66], it is not clear whether it inhibits the ICAM-1 expression in endothelium via NF-κB signaling. We investigated the downstream pathway regulating the ICAM-1 expression and found that TNF-α activates the degradation of IκBα and subsequently the p65 protein translocates into a nucleus after phosphorylation. This activates the NF-κB pathway to upregulate the ICAM-1 expression (Fig. 4A). The high expression of integrin β2 enables NC-EVs to bind the inflamed vasculature as evidenced by intravital microscopy of cremaster venules (Fig. 4B). Before we examined the usefulness of NC-EVs as a drug carrier, we studied whether NC-EVs can cause proinflammation because phosphatidylserine (PS) could flips out on the surface of NC-EVs. We measured the PS externalization and ICAM-1 levels using fluorescence assay and Western blot, respectively (Fig. 1S and 2S). We found that NC-EVs do not promote inflammation. Nitrogen cavitation rapidly breaks a cell and instantly formed closed nanovesicles (as shown in Fig. 1B), therefore PS might fail to translocate from the inner to the outer leaflet of the membrane, unlikely the externalization of PS in apoptosis [49].
To test whether piceatannol-loaded NC-EVs can prevent acute lung inflammation/injury, we systemically studied leukocyte infiltration, cytokine release and lung edema after mice were treated with NC-EVs or free piceatannol (Fig. 4C–G). The remarkable reduction of lung inflammation and lung edema represents the enhanced efficacy of piceatannol after it was loaded in NC-EVs. Most importantly, in the mouse sepsis model induced by LPS, piceatannol-loaded NC-EVs increased the mouse survival by 80%, however there was no protection of sepsis when piceatannol was used (Fig. 5A). The sepsis protection by piceatannol-loaded NC-EVs could be associated with the reduction of neutrophil infiltration and the cytokine storm in lungs, livers and kidneys (Fig. 5C–F). The enhanced therapy in two models of acute lung inflammation and sepsis after administration of piceatannol-loaded NC-EVs is associated with the targeting of NC-EVs to inflamed sites. When we studied the therapy of acute lung inflammation, we compared NC-EVs of HL-60 cells with that of red blood cells. We found that the integrin β2 on nanovesicles is required to increase the binding of endothelium [28]. Nanovesicles derived from red blood cells do not express integrin β2, like liposomes, therefore we expect that NC-EVs derived from neutrophils increase the efficacy of piceatannol in anti-inflammation therapies. Antibody-conjugated nanoparticles [67] could be compared with our delivery system in the future. This comparison might position our technology in translation and marketability.
We have also examined whether we can translate this technology, so we studied human neutrophil NC-EVs using the same approach developed here. The results are promising and we are able to generate human neutrophils EVs that contain all kinds of membrane proteins from the source of neutrophils (Fig. 6A–C), consistent with the results of HL-60 cells. Importantly, we demonstrate that our NC-EVs technology could make the translation of EVs and individualized nanomedicine possible. For example, we expect that we will draw the blood from a patient, and isolate neutrophils, and then we will use NC-EVs technology developed here to generate patient-derived nanovesicles. Finally, we will load a drug inside the nanovesicles and give back to the same patient. Because human neutrophils are more than 50% of blood leukocytes [68], we expect that we might generate enough nanovesicles from a patient for clinic use. If not, we might make nanovesicles via a blood bank. If using this approach, immunogenicity of nanovesicles would be further investigated before they are used in clinic.
5. Conclusion
In summary, we have established a novel strategy to generate EVs based on the force-induced disruption of the cell membrane using nitrogen cavitation, and the technology is simple, fast and scalable in the potential of clinical use. The physical force from nitrogen cavitation preserves the functions of cell membrane proteins of EVs. Our approach is able to make EVs from any cell types so this top-down approach can be utilized to generate personalized nanomedicine. The high production yield, scalability, reproducibility and remote drug loading make NC-EVs possible to become a novel targeted drug delivery platform. While we have demonstrated dramatic therapy of NC-EVs in acute lung injury and sepsis, this platform could be envisioned in a wide range of applications in medicine.
Supplementary Material
Acknowledgments
The work was supported by NIH grants R01GM16823 and K25HL111157 to Z. W. We thank Drs. Jing Wang and David Gang (Washington State University, USA) for performing Mass spectroscopy in the Tissue Imaging and Proteomics Laboratory, Washington State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wijagkanalan W, Kawakami S, Higuchi Y, Yamashita F, Hashida M. Intratracheally instilled mannosylated cationic liposome/NFkappaB decoy complexes for effective prevention of LPS-induced lung inflammation. J Control Release. 2011;149:42–50. doi: 10.1016/j.jconrel.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 2.Barenholz Y, Peer D. Liposomes and other assemblies as drugs and nano-drugs: from basic and translational research to the clinics. J Control Release. 2012;160:115–6. doi: 10.1016/j.jconrel.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Frede A, Neuhaus B, Klopfleisch R, Walker C, Buer J, Muller W, et al. Colonic gene silencing using siRNA-loaded calcium phosphate/PLGA nanoparticles ameliorates intestinal inflammation in vivo. J Control Release. 2016;222:86–96. doi: 10.1016/j.jconrel.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 4.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Li J, Cho J, Malik AB. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat Nanotechnol. 2014;9:204–10. doi: 10.1038/nnano.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu D, Gao J, Wang Z. Neutrophil-Mediated Delivery of Therapeutic Nanoparticles across Blood Vessel Barrier for Treatment of Inflammation and Infection. ACS Nano. 2015;9:11800–11. doi: 10.1021/acsnano.5b05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Q, Wang X, Wang C, Feng L, Li Y, Liu Z. Drug-Induced Self-Assembly of Modified Albumins as Nano-theranostics for Tumor-Targeted Combination Therapy. ACS Nano. 2015;9:5223–33. doi: 10.1021/acsnano.5b00640. [DOI] [PubMed] [Google Scholar]
- 8.Chu D, Zhao Q, Yu J, Zhang F, Zhang H, Wang Z. Nanoparticle Targeting of Neutrophils for Improved Cancer Immunotherapy. Adv Healthc Mater. 2016;5:1088–93. doi: 10.1002/adhm.201500998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song J, Yang X, Jacobson O, Lin L, Huang P, Niu G, et al. Sequential Drug Release and Enhanced Photothermal and Photoacoustic Effect of Hybrid Reduced Graphene Oxide-Loaded Ultrasmall Gold Nanorod Vesicles for Cancer Therapy. ACS Nano. 2015;9:9199–209. doi: 10.1021/acsnano.5b03804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu M, Zheng J. Clearance Pathways and Tumor Targeting of Imaging Nanoparticles. ACS Nano. 2015;9:6655–74. doi: 10.1021/acsnano.5b01320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:185–98. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 12.Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655–72. doi: 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng CJ, Tietjen GT, Saucier-Sawyer JK, Saltzman WM. A holistic approach to targeting disease with polymeric nanoparticles. Nat Rev Drug Discov. 2015;14:239–47. doi: 10.1038/nrd4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118–21. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108:10980–5. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z. Anticancer Platelet-Mimicking Nanovehicles. Adv Mater. 2015;27:7043–50. doi: 10.1002/adma.201503323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161:505–22. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 18.Ozpolat B, Sood AK, Lopez-Berestein G. Liposomal siRNA nanocarriers for cancer therapy. Adv Drug Deliv Rev. 2014;66:110–6. doi: 10.1016/j.addr.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui J, Li C, Wang C, Li Y, Zhang L, Yang H. Repeated injection of pegylated liposomal antitumour drugs induces the disappearance of the rapid distribution phase. J Pharm Pharmacol. 2008;60:1651–7. doi: 10.1211/jpp/60.12.0011. [DOI] [PubMed] [Google Scholar]
- 20.Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8:137–43. doi: 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Tiruppathi C, Minshall RD, Malik AB. Size and dynamics of caveolae studied using nanoparticles in living endothelial cells. ACS Nano. 2009;3:4110–6. doi: 10.1021/nn9012274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Tiruppathi C, Cho J, Minshall RD, Malik AB. Delivery of nanoparticle: complexed drugs across the vascular endothelial barrier via caveolae. IUBMB Life. 2011;63:659–67. doi: 10.1002/iub.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13:813–27. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vader P, Breakefield XO, Wood MJ. Extracellular vesicles: emerging targets for cancer therapy. Trends Mol Med. 2014;20:385–93. doi: 10.1016/j.molmed.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zomer A, Maynard C, Verweij FJ, Kamermans A, Schafer R, Beerling E, et al. In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell. 2015;161:1046–57. doi: 10.1016/j.cell.2015.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi DS, Kim DK, Kim YK, Gho YS. Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass spectrometry reviews. 2015;34:474–90. doi: 10.1002/mas.21420. [DOI] [PubMed] [Google Scholar]
- 27.Vader P, Mol EA, Pasterkamp G, Schiffelers RM. Extracellular vesicles for drug delivery. Adv Drug Deliv Rev. 2016;106:148–56. doi: 10.1016/j.addr.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Gao J, Chu D, Wang Z. Cell membrane-formed nanovesicles for disease-targeted delivery. J Control Release. 2016;224:208–16. doi: 10.1016/j.jconrel.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–40. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–63. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinarello CA. Anti-inflammatory Agents: Present and Future. Cell. 2010;140:935–50. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z. Imaging Nanotherapeutics in Inflamed Vasculature by Intravital Microscopy. Theranostics. 2016;6:2431–8. doi: 10.7150/thno.16307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chow CW, Herrera Abreu MT, Suzuki T, Downey GP. Oxidative stress and acute lung injury. Am J Respir Cell Mol Biol. 2003;29:427–31. doi: 10.1165/rcmb.F278. [DOI] [PubMed] [Google Scholar]
- 34.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–90. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–75. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 36.Strnad J, Burke JR. IkappaB kinase inhibitors for treating autoimmune and inflammatory disorders: potential and challenges. Trends Pharmacol Sci. 2007;28:142–8. doi: 10.1016/j.tips.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Garrean S, Gao XP, Brovkovych V, Shimizu J, Zhao YY, Vogel SM, et al. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol. 2006;177:4853–60. doi: 10.4049/jimmunol.177.7.4853. [DOI] [PubMed] [Google Scholar]
- 38.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–83. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.SELA, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–57. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 40.Jacob C, Leport M, Szilagyi C, Allen JM, Bertrand C, Lagente V. DMSO-treated HL60 cells: a model of neutrophil-like cells mainly expressing PDE4B subtype. Int Immunopharmacol. 2002;2:1647–56. doi: 10.1016/s1567-5769(02)00141-8. [DOI] [PubMed] [Google Scholar]
- 41.Schafer ST, Franken L, Adamzik M, Schumak B, Scherag A, Engler A, et al. Mitochondrial DNA: An Endogenous Trigger for Immune Paralysis. Anesthesiology. 2016;124:923–33. doi: 10.1097/ALN.0000000000001008. [DOI] [PubMed] [Google Scholar]
- 42.Kanada M, Bachmann MH, Hardy JW, Frimannson DO, Bronsart L, Wang A, et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc Natl Acad Sci U S A. 2015;112:E1433–42. doi: 10.1073/pnas.1418401112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fritze A, Hens F, Kimpfler A, Schubert R, Peschka-Suss R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Bba-Biomembranes. 2006;1758:1633–40. doi: 10.1016/j.bbamem.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 44.Alyane M, Barratt G, Lahouel M. Remote loading of doxorubicin into liposomes by transmembrane pH gradient to reduce toxicity toward H9c2 cells. Saudi Pharm J. 2016;24:165–75. doi: 10.1016/j.jsps.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wieder ED, Hang H, Fox MH. Measurement of intracellular pH using flow cytometry with carboxy-SNARF-1. Cytometry. 1993;14:916–21. doi: 10.1002/cyto.990140810. [DOI] [PubMed] [Google Scholar]
- 46.Joguparthi V, Anderson BD. Liposomal delivery of hydrophobic weak acids: enhancement of drug retention using a high intraliposomal pH. J Pharm Sci. 2008;97:433–54. doi: 10.1002/jps.21135. [DOI] [PubMed] [Google Scholar]
- 47.Gasparini C, Celeghini C, Monasta L, Zauli G. NF-kappaB pathways in hematological malignancies. Cell Mol Life Sci. 2014;71:2083–102. doi: 10.1007/s00018-013-1545-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Setty BN, Betal SG. Microvascular endothelial cells express a phosphatidylserine receptor: a functionally active receptor for phosphatidylserine-positive erythrocytes. Blood. 2008;111:905–14. doi: 10.1182/blood-2007-07-099465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–20. [PubMed] [Google Scholar]
- 50.Philips CA, Sarin SK. Sepsis in cirrhosis: emerging concepts in pathogenesis, diagnosis and management. Hepatol Int. 2016;10:871–82. doi: 10.1007/s12072-016-9753-2. [DOI] [PubMed] [Google Scholar]
- 51.Aikawa N. Cytokine storm in the pathogenesis of multiple organ dysfunction syndrome associated with surgical insults. Nihon Geka Gakkai Zasshi. 1996;97:771–7. [PubMed] [Google Scholar]
- 52.Wang HX, Liu M, Weng SY, Li JJ, Xie C, He HL, et al. Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World journal of gastroenterology. 2012;18:119–25. doi: 10.3748/wjg.v18.i2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Jong HK, van der Poll T, Wiersinga WJ. The systemic pro-inflammatory response in sepsis. J Innate Immun. 2010;2:422–30. doi: 10.1159/000316286. [DOI] [PubMed] [Google Scholar]
- 54.Schultz J, Kaminker K. Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch Biochem Biophys. 1962;96:465–7. doi: 10.1016/0003-9861(62)90321-1. [DOI] [PubMed] [Google Scholar]
- 55.Mullane KM, Kraemer R, Smith B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods. 1985;14:157–67. doi: 10.1016/0160-5402(85)90029-4. [DOI] [PubMed] [Google Scholar]
- 56.Ohno S, Ishikawa A, Kuroda M. Roles of exosomes and microvesicles in disease pathogenesis. Adv Drug Deliv Rev. 2013;65:398–401. doi: 10.1016/j.addr.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 57.Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol. 2013;113:1–11. doi: 10.1007/s11060-013-1084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ingato D, Lee JU, Sim SJ, Kwon YJ. Good things come in small packages: Overcoming challenges to harness extracellular vesicles for therapeutic delivery. J Control Release. 2016;241:174–85. doi: 10.1016/j.jconrel.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 59.Nagle JF, Tristram-Nagle S. Structure of lipid bilayers. Biochim Biophys Acta. 2000;1469:159–95. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Angelillo-Scherrer A. Leukocyte-derived microparticles in vascular homeostasis. Circ Res. 2012;110:356–69. doi: 10.1161/CIRCRESAHA.110.233403. [DOI] [PubMed] [Google Scholar]
- 61.Simpson RJ. Disruption of cultured cells by nitrogen cavitation. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.prot5513. pdb prot5513. [DOI] [PubMed] [Google Scholar]
- 62.Fritze A, Hens F, Kimpfler A, Schubert R, Peschka-Suss R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim Biophys Acta. 2006;1758:1633–40. doi: 10.1016/j.bbamem.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 63.Wang C, Wang X, Zhong T, Zhao Y, Zhang WQ, Ren W, et al. The antitumor activity of tumor-homing peptide-modified thermosensitive liposomes containing doxorubicin on MCF-7/ADR: in vitro and in vivo. Int J Nanomedicine. 2015;10:2229–48. doi: 10.2147/IJN.S79840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deng Z, Xiao Y, Pan M, Li F, Duan W, Meng L, et al. Hyperthermia-triggered drug delivery from iRGD-modified temperature-sensitive liposomes enhances the anti-tumor efficacy using high intensity focused ultrasound. J Control Release. 2016 doi: 10.1016/j.jconrel.2016.10.030. [DOI] [PubMed] [Google Scholar]
- 65.Wung BS, Hsu MC, Wu CC, Hsieh CW. Piceatannol upregulates endothelial heme oxygenase-1 expression via novel protein kinase C and tyrosine kinase pathways. Pharmacol Res. 2006;53:113–22. doi: 10.1016/j.phrs.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 66.Ashikawa K, Majumdar S, Banerjee S, Bharti AC, Shishodia S, Aggarwal BB. Piceatannol inhibits TNF-induced NF-kappaB activation and NF-kappaB-mediated gene expression through suppression of IkappaBalpha kinase and p65 phosphorylation. J Immunol. 2002;169:6490–7. doi: 10.4049/jimmunol.169.11.6490. [DOI] [PubMed] [Google Scholar]
- 67.Howard M, Zern BJ, Anselmo AC, Shuvaev VV, Mitragotri S, Muzykantov V. Vascular targeting of nanocarriers: perplexing aspects of the seemingly straightforward paradigm. ACS Nano. 2014;8:4100–32. doi: 10.1021/nn500136z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. 2014;9:181–218. doi: 10.1146/annurev-pathol-020712-164023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.