

Abstract
[Ru(bpy)2(BNIQ)]2+ (BNIQ = Benzo[c][1,7]naphthyridine-1-isoquinoline), which incorporates the sterically expansive BNIQ ligand, is a highly selective luminescent probe for DNA mismatches and abasic sites, possessing a 500-fold higher binding affinity toward these destabilized regions relative to well-matched base pairs. As a result of this higher binding affinity, the complex exhibits an enhanced steady-state emission in the presence of DNA duplexes containing a single base mismatch or abasic site compared to fully well-matched DNA. Luminescence quenching experiments with Cu(phen)22+ and [Fe(CN)6]3– implicate binding of the complex to a mismatch from the minor groove via metalloinsertion. The emission response of the complex to different single base mismatches, binding preferentially to the more destabilized mismatches, is also consistent with binding by metalloinsertion. This work shows that high selectivity toward destabilized regions in duplex DNA can be achieved through the rational design of a complex with a sterically expansive aromatic ligand.
Short abstract
The luminescent complex [Ru(bpy)2(BNIQ)]2+ selectivity targets mismatched and abasic sites in duplex DNA and exhibits an enhanced emission intensity in the presence of these defect sites relative to well-matched base pairs.
Introduction
The design of small molecules that specifically target DNA base mismatches is a promising route in the development of therapeutic and diagnostic agents directed toward mismatch repair (MMR)-deficient cancers.1 We have found that octahedral rhodium complexes with sterically expansive aromatic ligands bind to DNA mismatches with high affinity and selectivity via metalloinsertion.2 Furthermore, metalloinsertors such as [Rh(chrysi)(phen)(PPO)]2+ (Figure 1; chrysi = 5,6-chrysenequinone diimine, PPO = 2-(pyridine-2-yl)propan-2-ol) exhibit preferential cytotoxicity toward MMR-deficient compared to MMR-proficient cells.3
Figure 1.

Chemical structures of [Rh(chrysi)(phen)(PPO)]2+ (left), [Ru(Me4phen)2(dppz)]2+ (center), and [Ru(bpy)2(BNIQ)]2+ (right).
There is also interest in the design of mismatch-specific luminescent small molecules to serve as diagnostic probes for early detection of MMR-deficient cancers. While rhodium metalloinsertors are nonemissive, many other transition metal complexes, in particular those of ruthenium, iridium, rhenium, and platinum, are well-suited to serve as luminescent probes of biomolecules owing to high photostability, large Stokes shifts, long-lived excited states, and synthetic accessibility.4 The Barton group has reported [Ru(Me4phen)2(dppz)]2+ (Figure 1; Me4phen = 3,4,7,8-tetramethyl-1,10-phenanthroline, dppz = dipyrido[3,2-a:2′,3′-c]phenazine), a “light switch” complex that is highly selective toward mismatched DNA.5 This compound, a derivative of the classic DNA light switch complex [Ru(bpy)2(dppz)]2+,6 possesses bulky methyl-substituted phen ligands that discourage intercalation of the dppz ligand into well-matched sites of DNA but still allow for metalloinsertion at a mismatched site. A series of highly mismatch-specific luminescent Pt(II) complexes bearing functionalized N-heterocyclic carbene and diphosphine ligands have also been reported.7
Another logical approach in the design of a mismatch-specific luminescent transition metal complex is to modify the intercalating or inserting ligand. A hallmark of rhodium metalloinsertors is the planar, asymmetric, and sterically expansive chrysi ligand, which imparts mismatch specificity because it is too large to intercalate into well-matched duplex DNA, but inserts instead into destabilized DNA sites. While there is evidence that [Ru(bpy)2(L)]2+ complexes with sterically demanding ligands (L) bind preferentially to mismatched DNA,8 this is not necessarily accompanied by luminescence discrimination between mismatched and well-matched DNA. Ruthenium complexes bearing chrysi and related ligands are not luminescent at ambient temperature,8a and those with tpqp and tactp ligands (tpqp = 7,8,13,14-tetrahydro-6-phenylquino[8,7-k][1,8]phenanthroline; tactp = 4,5,9,18-tetraazachryseno[9,10-b]-triphenylene) do not show an increase in emission in the presence of a DNA mismatch.8c In this work, we designed and synthesized a new sterically expansive ligand BNIQ (BNIQ = benzo[c][1,7]naphthyridine-1-isoquinoline) (Scheme 1). The luminescent properties of [Ru(bpy)2(BNIQ)]2+ (Figure 1), both free and in the presence of well-matched and mismatched DNA duplexes, were explored using steady-state and excited-state lifetime measurements.
Scheme 1. Synthesis of the BNIQ Ligand.

Experimental Procedures
Materials and Methods
All chemicals and starting materials were purchased from commercial vendors and used as received. Ru(bpy)2Cl2 was prepared according to the literature.9 UV–visible spectra were recorded on a Beckman DU 7400 UV–visible spectrophotometer (Beckman Coulter) from 200 to 800 nm. Oligonucleotides were synthesized using standard phosphoramidite chemistry at Integrated DNA Technologies (Coralville, IA) and purified by HPLC using a C18 reverse-phase column (Varian, Inc.) on a Hewlett-Packard 1100 HPLC. The copper complex Cu(phen)22+ was generated in situ by combining CuCl2 with phen ligands in a 1:3 ratio.
Synthesis
5-Amino-2-chloropyridine (1)
2-Chloro-5-nitropyridine (5.49 g, 34.6 mmol) was added in portions to a solution of SnCl2·2H2O (39.36 g, 17.4 mmol) in 50 mL of concentrated HCl at 0 °C. The reaction was allowed to warm to room temperature and stirred for 4 h, followed by the addition of 5 M Na2CO3 (75 mL) and NH4OH (50 mL). The product was extracted from the reaction mixture with CH2Cl2 (5 × 100 mL). The combined organic layers were dried over MgSO4 and filtered, and the solvent was evaporated to give 1 (3.9 g, 88%). 1H NMR (500 MHz, CDCl3) δ 7.88 (dd, J = 3.0, 0.6 Hz, 1H), 7.11 (dd, J = 8.5, 0.6 Hz, 1H), 7.01–6.98 (m, 1H), 3.62 (s, 2H). ESI(+)MS (m/z): [M + H]+ calcd 129.0, found 129.3.
tert-Butyl (6-Chloro-3-pyridinyl) carbamate (2)
Synthesized using an adapted procedure.10 A solution of 1 (1.83 g, 14.2 mmol) and Boc2O (4.74 g, 21.8 mmol) in dry dioxane (15 mL) was heated under reflux under an Ar atmosphere for 45 h. During the course of the reaction, an additional 0.81 g (3.7 mmol) of Boc2O was added to the reaction mixture. The reaction mixture was poured into water (15 mL), and the product was extracted with EtOAc (2 × 25 mL). The combined organic layers were washed with brine (15 mL), dried over Mg2SO4, and filtered, and the solvent was evaporated. The residue was run through a plug of silica to give 2 (3.1 g, 96%). 1H NMR (500 MHz, CDCl3) δ 8.29 (dd, J = 2.9, 0.6 Hz, 1H), 7.99 (s, 1H), 7.28 (dt, J = 8.7, 0.6 Hz, 1H), 6.86 (s, 1H), 1.55 (s, 9H). ESI(+)MS (m/z): [M + H]+ calcd 229.1, found 229.1.
tert-Butyl (6-Chloro-4-iodo-3-pyridinyl) Carbamate (3)
To a solution of TMEDA (3.8 mL, 25.3 mmol) in dry THF (15 mL) under Ar was added nBuLi (10 mL, 2.5 M in hexanes) dropwise at −78 °C. The solution was stirred for 30 min, and subsequently a solution of 2 (1.37 g, 6 mmol) in dry THF (5 mL) under Ar was added dropwise. The solution was stirred at −78 °C for 1 h, and then a solution of I2 (3.23 g, 12.7 mmol) in 5 mL of dry THF under Ar was added dropwise. The reaction was allowed to warm to room temperature and stirred overnight. The reaction was cooled to −78 °C, and 20 mL of saturated NH4Cl was added. The aqueous and organic layers were separated, and the product was further extracted from the aqueous phase with EtOAc (3 × 30 mL). The organic layers were combined, dried over MgSO4, and filtered, and the solvent was evaporated. The crude material was purified by silica gel column chromatography (9:1 hexanes/EtOAc) to give 3 (0.60 g, 28%). 1H NMR (500 MHz, CDCl3) δ 8.98 (s, 1H), 7.76 (s, 1H), 6.69 (s, 1H), 1.59 (s, 9H). ESI(+)MS (m/z): [M + H]+ calcd 355.0, found 354.8.
tert-Butyl (6-Chloro-4-(2-formylphenyl)-3-pyridinyl) Carbamate (4)
A solution of 3 (0.604 g, 1.71 mmol), 2-formylphenylboronic acid (0.384 g, 2.56 mmol), Pd(PPh3)4 (0.0986 g, 5 mol %), 2 M Na2CO3 (3 mL), EtOH (2 mL), and toluene (12 mL) was degassed by freeze pump thaw (4×) and then heated to 90 °C under Ar overnight. The reaction mixture was poured over 15 mL of H2O, the layers were separated, and the product was extracted from the aqueous layer with EtOAc (3 × 20 mL). The organic layers were combined, dried over MgSO4, and filtered, and the solvent was evaporated. The crude material was purified by silica gel chromatography (4:1 hexanes/EtOAc) to give 4 (0.29 g, 51%). 1H NMR (500 MHz, CDCl3) δ 8.75 (s, 1H), 7.91–7.88 (m, 1H), 7.76 (s, 1H), 7.57–7.52 (m, 3H), 7.34–7.30 (m, 1H), 6.88 (d, J = 5.2 Hz, 1H), 1.56 (s, 9H). ESI(+)MS (m/z) [M + H]+ calcd 333.1, found 333.1.
2-Chlorobenzo[c][1,7]naphthyridine (5)
To 0.291 g of 4 (0.877 mmol) were added 5 mL of CH2Cl2 and 0.5 mL of TFA. The solution was stirred overnight, and then 5 mL 5% Na2CO3 was added and the solution was stirred vigorously for 1.5 h. The layers were separated, and the product was extracted from the aqueous layer with EtOAc (2 × 20 mL). The organic layers were combined, washed with brine (1 × 25 mL), dried over MgSO4, and filtered, and the solvent was evaporated. The product was purified by column chromatography (4:1 hexanes/EtOAc) to give 5 (0.16 g, 87%). 1H NMR (500 MHz, CDCl3) δ 9.35 (s, 1H), 9.33 (s, 1H), 8.58 (d, J = 8.1 Hz, 1H), 8.39 (s, 1H), 8.15 (d, 7.9 Hz, 1H), 7.99 (ddd, J = 8.3 Hz, 7.2 Hz, 1.4 Hz, 1H), 7.92 (ddd, J = 8.1 Hz, 7.1 Hz, 1.1 Hz, 1H). ESI(+)MS (m/z) [M + H]+ calcd 215.0, 217.0; found 215.2, 217.0.
2-Bromobenzo[c][1,7]naphthyridine (6)
To a 5 mL microwave vial was added 0.010 g of 5 (0.047 mmol), 1 mL of HBr, and 2 mL of AcOH. The contents were heated at 130 °C for 2 h. The reaction mixture was then diluted with 50 mL of water, and the solution was neutralized to pH 7 with NaHCO3. The product was extracted from the aqueous mixture with CH2Cl2 (3 × 25 mL), and the organic layers were dried over MgSO4 and filtered to give 6 (0.0090 g, 75%). 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H), 9.32 (s, 1H), 8.61 (dq, J = 8.5, 1.0 Hz, 1H), 8.59 (s, 1H), 8.18 (ddd, J = 7.9 Hz, 1.4 Hz, 0.7 Hz, 1H), 8.03 (ddd, J = 8.3 Hz, 7.1 Hz, 1.4 Hz, 1H), 7.95 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H). ESI(+)MS (m/z) [M + H]+: calc. 259.0, 261.0, found 259.0, 261.0.
BNIQ Ligand (7)
To a flame-dried Schlenk flask under Ar were added 0.096 g of 1-iodoisoquinoline (0.37 mmol) and 0.016 g of LiCl (0.38 mmol). The flask was evacuated for 2 h, and anhydrous THF (1 mL) was added. The contents were stirred, and 190 μL of i-PrMgCl (2 M solution in THF) was added dropwise. The solution was stirred for 1 h, during which the reaction turned a dark purple. The contents were cooled to 0 °C, and 755 μL of ZnCl2 in THF (0.5 M) was added and stirred for 15 min at 0 °C. A solution of Pd2(dba)3 (0.009 g, 4 mol %) and PCy3 (0.011 g, 15 mol %) in THF (3 mL) under Ar was then added to the reaction solution, followed by a solution of 6 (0.065 g, 0.25 mmol) in THF (3 mL). The dark red-orange solution was stirred for 17 h at 65 °C. The solvent was removed in vacuo, and the contents were redissolved in EtOAc/H2O (25:20 mL). The layers were separated, and the EtOAc was washed with H2O (2 × 20 mL). The EtOAc was evaporated, and the crude material was purified by silica gel column chromatography with a solvent gradient (6:1 hexanes/EtOAc to 100% EtOAc) to obtain BNIQ ligand 7 (0.040 g, 52%). 1H NMR (500 MHz, CDCl3) δ 9.71 (s, 1H), 9.45 (s, 1H), 9.14 (s, 1H), 8.82–8.76 (m, 2H), 8.72 (d, J = 5.6 Hz, 1H), 8.18 (d, J = 7.9 Hz, 1H), 7.99 (ddd, J = 8.3 Hz, 7.1 Hz, 1.4 Hz, 1H), 7.95 (dt, J = 8.3 Hz, 1.0 Hz, 1H), 7.91 (ddd, J = 8.0 Hz, 7.1 Hz, 1.1 Hz, 1H), 7.80 (d, J = 5.7 Hz, 1H), 7.75 (ddd, J = 8.2 Hz, 6.8 Hz, 1.2 Hz, 1H), 7.66 (ddd, J = 8.3 Hz, 6.8 Hz, 1.3 Hz, 1H). ESI(+)MS (m/z) [M + H]+ calc. 308.1, found 308.4.
[Ru(bpy)2(BNIQ)]X2 (8, X = PF6 or Cl)
BNIQ ligand (7) (0.015 g, 0.049 mmol) and Ru(bpy)2Cl2 (0.025 g, 0.048 mmol) were combined in 4 mL of ethylene glycol and heated at 130 °C for 17 h. The solution was cooled to room temperature, and diluted with 5 mL of H2O, and excess NH4PF6 was added to precipitate the product. The precipitate was collected via vacuum filtration, washed with H2O (2 × 5 mL), and dried (0.041 g, 82%). The complex was converted to the water-soluble Cl salt by anion exchange chromatography (Sephadex QAE) and further purified by preparative HPLC using a gradient of H2O (with 0.1% TFA) to CH3CN over 1 h. 1H NMR (500 MHz, DMSO-d6) δ 9.99 (s, 1H), 9.59 (s, 1H), 9.20–9.15 (m, 2H), 8.97 (d, J = 8.2 Hz, 1H), 8.94–8.86 (m, 3H), 8.45 (s, 1H), 8.44 (d, J = 8.2 Hz, 1H), 8.30–8.20 (m, 4H), 8.15–8.08 (m, 4H), 8.03 (td, J = 7.0, 1.1 Hz, 2H), 7.95 (ddd, J = 5.7, 1.4, 0.7 Hz, 1H), 7.85 (dddd, J = 5.4, 4.6, 1.5, 0.7 Hz, 2H), 7.79 (ddd, J = 5.6, 1.5, 0.7 Hz), 7.69 (d, J = 6.2 Hz, 1H), 7.64 (ddd, J = 7.6, 5.6, 1.3 Hz, 1H), 7.59 (ddd, J = 7.6, 5.6, 1.3 Hz, 1H), 7.45 (ddd, 7.2, 5.6, 1.3 Hz, 1H), 7.40 (ddd, J = 7.3, 5.7, 1.3 Hz, 1H). ESI(+)MS (m/z) [M-2Cl]2+ calcd 360.6, found 360.6. UV–vis in H2O, λ/nm (ε x 104/M–1 cm–1): 287 (8.0), 366 (3.0), 431 (1.7).
Luminescence Measurements
Steady-State Luminescence
Luminescence spectra were recorded on an ISS-K2 spectrofluorometer at 25 °C. The ruthenium complex was excited at 440 nm, and emission spectra were integrated from 590 to 850 nm. The chloride salt of the complex was used for all DNA experiments. In appropriate figures, [DNA] is defined as the concentration of the full sequence oligonucleotide. The concentration of the ruthenium complex for steady-state measurements was 4 μM.
Time-Resolved Luminescence
Time-resolved spectroscopic measurements were carried out at the Beckman Institute Laser Resource Center and were conducted using instrumentation that has been described.11 Briefly, a 460 nm light produced by OPO pumped with a 10 Hz, Qswitched Nd:YAG laser (Spectra-Physics Quanta-Ray PRO-Series) was used as an excitation source (pump pulse duration ≈ 8 ns). The emitted light was detected at 700 nm with a photomultiplier tube (Hamamatsu R928) following wavelength selection by a double monochromator (Instruments SA DH-10). Scattered laser light was removed from the detectors using suitable filters. The samples were held in 1 cm path length quartz cuvettes (Starna) equipped with stir bars and irradiated at 460 nm with 500–1000 laser pulses at 3 mJ/pulse. Kinetic traces were fit to exponential equations of the form I(t) = a0 + Σan exp(−t/τn), where I(t) is the signal intensity as a function of time, a0 is the intensity at long time, an is a pre-exponential factor that represents the relative contribution from the nth component to the trace, and τn is the lifetime of the nth component, convoluted with a Gaussian function to take into account the Instrument Response Function (fwmh = 8 ns). The errors are evaluated to be equal to 5–10% for mono- and biexponential decays, respectively.
Results and Discussion
BNIQ Ligand Design and Synthesis
In this work, we designed a new sterically expansive polypyridyl ligand that possesses a similar width to the mismatch-specific chrysi ligand but lacks imine protons characteristic of chrysi. We have proposed that exchangeable imine protons are responsible for quenching any ambient temperature luminescence of compounds like [Ru(bpy)2(chrysi)]2+.8a As such, BNIQ was designed to contain heterocyclic nitrogen atoms that coordinate directly to the ruthenium core, analogously to bpy or phen, and an additional nonchelating nitrogen atom that could enhance the sensitivity of the complex to its environment. BNIQ also possesses similar structural features to the tpqp ligand, which has been shown to exhibit some degree of mismatch specificity in binding.8c
BNIQ was synthesized in seven steps (Scheme 1) from the starting material 2-chloro-5-nitropyridine. A key step in the ligand synthesis is the conversion of the chlorine intermediate 5 into its bromine analogue 6, a transformation that was executed in order to generate a more reactive substrate in the subsequent Negishi coupling. Using an adapted literature procedure for a related 2-chloropyridine derivative,125 was heated in a 2:1 (v/v) mixture of acetic acid/aqueous HBr at 130 °C in a microwave reactor to yield 6. Negishi coupling conditions were adapted from the literature13,14 to synthesize the BNIQ ligand (7) from 6 and 1-iodoisoquinoline.
Synthesis and Characterization of the [Ru(bpy)2(BNIQ)]2+ Complex
Coordination of BNIQ to ruthenium (Scheme 2) is a facile synthesis, achieved by heating the ligand in a 1:1 ratio with Ru(bpy)2Cl2 in ethylene glycol at 130 °C. [Ru(bpy)2(BNIQ)]2+ was isolated from the reaction mixture as its PF6– salt before conversion to its water-soluble chloride salt by anion exchange chromatography and further purification by preparative HPLC.
Scheme 2. Synthesis of [Ru(bpy)2(BNIQ)]2+.

The UV–visible spectrum of [Ru(bpy)2(BNIQ)]2+ shows a characteristic MLCT transition in the visible region at 430–440 nm (Figure S1). Upon excitation at 440 nm in aqueous solution, [Ru(bpy)2(BNIQ)]2+ exhibits a broad emission centered at 700 nm that is nearly insensitive to the presence of oxygen in solution (Figure S2).
Steady-State Luminescence of [Ru(bpy)2(BNIQ)]2+ in the Presence of DNA
We investigated the steady-state emission of [Ru(bpy)2(BNIQ)]2+ in the presence of three different 27-mer DNA duplexes: one that is completely well-matched, one that contains a single CC mismatch, and one containing an abasic site (sequences provided in Figure 2). Titrations (Figure 2) reveal that the emission intensity of the complex increases upon the addition of each duplex; however, the emission intensity is brighter for the samples containing the CC and abasic sites compared to the well-matched DNA. The maximum ruthenium emission intensities reached for the CC mismatch and abasic DNA samples are approximately 1.7-fold larger than the emission intensity of the free ruthenium complex, and 1.5-fold greater than the well-matched DNA sample. The binding affinities evaluated by a global analysis of the titration curves (Table 1, Figure S3) reveal that [Ru(bpy)2(BNIQ)]2+ has an ∼500-fold higher affinity for oligomers containing CC mismatch or abasic sites (3.5 × 106 and 3.8 × 106 M–1, respectively) compared to oligomers with fully well-matched sites (7.3 × 103 M–1). Thus, the titrations demonstrate that this new ruthenium complex is very specific for the thermodynamically destabilized sites in DNA. Interestingly, analysis of the titrations indicates that the differences in emission intensities observed in Figure 2 are mainly related to the higher affinity of the complex toward mismatched and abasic sites. From the analysis of the steady-state titration curves (Figure S3), we evaluated that [Ru(bpy)2(BNIQ)]2+ has similar intrinsic emissivity when bound to a well-matched or mismatched site (1.36, 1.42, and 1.46 for well-matched, CC, and abasic sequences, respectively) relative to free complex. Thus, the inherent brightness of a complex bound to a mismatch or a well-matched site is rather similar, and the dramatic differences in steady-state emission intensities observed are correlated with the higher affinity of the compound toward the destabilized base pairs.
Figure 2.

Steady-state emission titrations of [Ru(bpy)2(BNIQ)]2+ with well-matched, mismatched, and abasic DNA duplexes at 25 °C. λex = 440 nm, [Ru] = 4 μM. [DNA] reflects the concentration of full sequence. Samples were prepared in 5 mM Tris, 200 mM NaCl, pH 7.5. DNA sequences are provided at the top of the figure; the underscore represents the absence of a base in the abasic (AB) sequence. Emission spectra were integrated from 590 to 850 nm.
Table 1. Binding Affinities of [Ru(bpy)2(BNIQ)]2+ with Well-Matched, Mismatched, and Abasic DNA Duplexes.
| well-matched | mismatched | abasic | |
|---|---|---|---|
| Ka (M–1)a | 7.3 × 103 | 3.5 × 106 | 3.8 × 106 |
Time-Resolved Luminescence of [Ru(bpy)2(BNIQ)]2+ with and without DNA
In order to confirm the steady-state behavior, we studied the luminescence lifetimes of [Ru(bpy)2(BNIQ)]2+ in the presence of the three DNA duplexes (Table 2). The 700 nm emission of the free complex decays as a monoexponential function with a lifetime of 215 ns in water. In the presence of 2 equiv of the well-matched sequence, this emission decays as a biexponential with short (235 ns) and long (487 ns) components. The shorter component, which contributes approximately 75% to the overall decay, we assign to the free complex in solution. The longer component, responsible for the other 25% of the decay, is thus attributed to the complex bound to DNA. In the presence of 2 equiv of the mismatched sequence, the emission decays monoexponentially with a lifetime of 416 ns. A comparable single lifetime of 421 ns is found in the presence of 2 equiv of the abasic duplex.
Table 2. [Ru(bpy)2(BNIQ)]2+ Emission Lifetimes in Various Solvents and in the Presence of Well-Matched, Mismatched, and Abasic DNA Duplexesa.
| lifetime (ns) | |
|---|---|
| Milli-Q H2O | 215 |
| bufferb | 217 |
| D2O | 389 |
| CH3CN (anhydrous) | 265 |
| well-matched | 235 (75%) |
| 487 (25%) | |
| mismatched | 416 |
| abasic | 421 |
Two important conclusions can be drawn from these measurements. First, the absence of a short lifetime component with the mismatched or abasic sequences indicates that the complex is fully bound to the DNA duplex, which is in agreement with the evaluated binding affinities, based on which only 3% of the complex remains free in solution. Second, the lifetimes seen with the mismatched and abasic sequence are close to the long component observed with the well-matched sequence. As expected from the analysis of the steady-state titrations, the lifetimes associated with the complex bound to DNA for well-matched, mismatched, and abasic sites are similar. Thus, the monoexponential decay in the 416 and 421 ns lifetimes should correspond to an average de-excitation process of complexes bound to DNA (well-matched and mismatched/abasic) and the small population of free complex in solution. These data support the conclusion that the brighter steady-state emission spectra observed with the mismatched and abasic duplexes (Figure 2) are due primarily to a higher binding affinity toward the destabilized sites compared to well-matched base pairs.
To elucidate the mechanism that gives rise to the enhanced luminescence observed with the duplexes, the emission lifetimes of [Ru(bpy)2(BNIQ)]2+ were further characterized in several solvent systems (Table 2). Interestingly, in anhydrous acetonitrile, the emission lifetime (265 ns) is only 50 ns longer than the lifetime in water. Thus, water itself does not dramatically quench the luminescence of [Ru(bpy)2(BNIQ)]2+. This lack of quenching contrasts to what is observed with phenazine and acridine-based complexes like [Ru(bpy)2(dppz)]2+, which are extremely sensitive to quenching by water via hydrogen bonding to nonchelated ligand nitrogen atoms.6 A similar effect is not observed for the BNIQ complex. Nevertheless, upon interacting with DNA, the solvation sphere around the complex is greatly altered; in the restricted environment bound to DNA, there is reduced dissipation of energy from the excited complex to solvent through vibrational modes. The importance of the nonradiative decay via vibrational relaxation can be probed using deuterated solvent. The effect of solvent deuteration on the emission of [Ru(bpy)3]2+ was first investigated by Watts and Van Houten, and it was found that deuteration of H2O leads to a doubling of the [Ru(bpy)3]2+ emission lifetime (0.58–1.02 μs at 25 °C).15,16 It was proposed that the ability of the solvent vibrational modes to deactivate the ruthenium excited state was attenuated upon solvent deuteration. For [Ru(bpy)2(BNIQ)]2+, a similar phenomenon is occurring, since a significantly longer lifetime for the complex in D2O (389 ns, Table 2) compared to water is detected. This longer component is more consistent with the longer lifetime detected for complexes bound to DNA. One could also argue that the increase in luminescence lifetime for [Ru(bpy)2(BNIQ)]2+ when bound to DNA is due to greater rigidity and lower frequency of collisions compared to the free complex in solution.17 A combination of these factors could give rise to the enhanced luminescence observed upon DNA binding.
[Ru(bpy)2(BNIQ)]2+ Luminescence with Different Base Mismatches
Owing to the large size of the BNIQ ligand and the observation that [Ru(bpy)2(BNIQ)]2+ preferentially targets mismatched and abasic sites in DNA, we hypothesize that the complex binds to these defects by metalloinsertion. To test this hypothesis, we investigated whether [Ru(bpy)2(BNIQ)]2+ is capable of targeting other types of mismatches in addition to the CC mismatch. An important characteristic of metalloinsertors is that the extent of mismatch binding correlates with the thermodynamic destabilization associated with the mismatch; the more destabilized the mismatch, the easier it is to displace the mismatched bases by the inserted ligand.2c,18 Thus, for luminescent metalloinsertors, we anticipate that the more destabilized the mismatch, the tighter the binding of the complex and the larger the observed emission enhancement.5,7,18
Luminescence titrations were performed with [Ru(bpy)2(BNIQ)]2+ and hairpin oligonucleotides containing the variable base pair XY (Figure 3). Indeed, we detect the greatest emission in the presence of the most destabilized mismatch, CC. Additionally, little emission enhancements are seen for the well-matched GC and GG mismatched hairpins. This small change is to be expected, given that G-containing mismatches are similar in stability to well-matched base pairs.19,20 However, we note a few variations with respect to the predicted trend of mismatch instability and luminescence enhancement. [Ru(bpy)2(BNIQ)]2+ exhibits a brighter emission in the presence of the “like-with-like” base mismatches AA and TT relative to CT and CA, even though we expect CT and CA to be more destabilized compared to AA and TT.19 Nonetheless, the emission of [Ru(bpy)2(BNIQ)]2+ is clearly sensitive to the identity of the single base mismatch, consistent with binding through metalloinsertion. We should note that binding to the bulged hairpin sites in these sequences is also possible; however, we do see comparable emission enhancements with the 27-mer and hairpin GC sequences. Binding to single base bulges has been reported for the metalloinsertor [Rh(bpy)2(chrysi)]3+, albeit with lower affinity than mismatch sites.21
Figure 3.

Steady-state emission titrations of [Ru(bpy)2(BNIQ)]2+ with DNA hairpins containing a variable XY base pair at 25 °C. λex = 440 nm, [Ru] = 4 μM. [DNA] reflects concentration of full sequence. Samples were prepared in 5 mM Tris, 50 mM NaCl, pH 7.5. Emission spectra were integrated from 590 to 850 nm.
Luminescence Quenching with Cu(phen)22+
To further elucidate the binding mode of [Ru(bpy)2(BNIQ)]2+ at the mismatch site, we employed the quencher Cu(phen)22+. Cu(phen)22+ binds in the DNA minor groove22−24 and has been used to selectively quench the luminescence of ruthenium complexes bound to a mismatch in the minor groove.5,25 We applied the Cu(phen)22+ quencher to samples containing [Ru(bpy)2(BNIQ)]2+ with the well-matched and mismatched duplexes (Figure 4). For the mismatched sample, as the concentration of Cu(phen)22+ is increased, we observe quenching of the luminescence of the ruthenium complex. This quenching suggests that the complex binds to the mismatch in the minor groove, consistent with metalloinsertion. Interestingly, the enhanced emission associated with binding to the well-matched duplex also decreases as Cu(phen)22+ is added. In fact, one can see that, at the highest [Cu]/[Ru] for the well-matched DNA sample, the emission spectrum overlays precisely with the spectrum corresponding to free ruthenium (Figure 4). Given the very low binding affinity of [Ru(bpy)2(BNIQ)]2+ toward well-matched base pairs (7.3 × 103 M–1) compared to the CC mismatch (3.5 × 106 M–1), the observed decrease in emission intensity with the well-matched sample is likely a reflection of excess Cu(phen)22+ displacing the weakly associated ruthenium complex from well-matched sites in the duplex to yield the free ruthenium complex in solution. This result suggests that [Ru(bpy)2(BNIQ)]2+ interacts with well-matched sites from the minor groove.
Figure 4.

Steady-state emission spectra of [Ru(bpy)2(BNIQ)]2+ (4 μM) with well-matched (blue) and mismatched (red) DNA duplexes (12 μM) at 25 °C. Cu(phen)22+ was added to the samples such that [Cu]/[Ru] = 6, 12, 24, and 36 (indicated in light blue for well-matched and orange for mismatched samples). Black lines represent samples of Ru in the absence of DNA or Cu. λex = 440 nm. Samples were prepared in 5 mM Tris, 200 mM NaCl, pH 7.5. DNA sequences shown in Figure 2 were used.
Luminescence Quenching with [Fe(CN)6]3–
We have proposed that [Ru(bpy)2(BNIQ)]2+ binds to the mismatched site via metalloinsertion. Therefore, we predict that, at the mismatch, the complex is bound deeper and more tightly compared to well-matched sites. We used [Fe(CN)6]3– to quench the emission of [Ru(bpy)2(BNIQ)]2+ when bound to the well-matched and mismatched duplexes (Table 3, Figure S4). [Fe(CN)6]3–, an anionic quencher, is repelled by the negatively charged phosphate backbone of the DNA.26 As such, its ability to quench [Ru(bpy)2(BNIQ)]2+ will be dictated by how well the ruthenium complex is protected by the DNA duplex.
Table 3. [Ru(bpy)2(BNIQ)]2+ Emission Lifetimes in the Presence and Absence of [Fe(CN)6]3– Quenchera.
| [Fe] = 0 mM | [Fe] = 8 mM | |
|---|---|---|
| free Ru | 215 | 9 |
| well-matched | 235 (75%), 487 (25%) | 24 (40%), 215 (60%) |
| mismatched | 416 | 335 |
Samples containing 6 μM Ru and 12 μM DNA were prepared in 5 mM Tris, 200 mM NaCl, pH 7.5 using DNA sequences shown in Figure 2. λex = 460 nm, λem = 700 nm. Percentages reflect the relative contributions of each lifetime to the overall decay. K3[Fe(CN)6](aq) was added to a final concentration of 8 mM.
As expected, [Fe(CN)6]3– dramatically quenches the emission lifetime of free [Ru(bpy)2(BNIQ)]2+ in solution (Table 3); we also observe quenching in the steady-state spectra (Figure S4). Some static quenching is likely also occurring. In the presence of the well-matched duplex, the shorter lifetime component is nearly eliminated, consistent with quenching of free ruthenium. The complexes weakly bound to well matched sites are also quenched to some extent dynamically by [Fe(CN)6]3– as we also see that the steady-state emission intensity for the well-matched sample is significantly quenched (Figure S4). Conversely, the longer component is quenched by over 50%. This differential quenching of the two lifetime components reveals that binding to well-matched sites does in fact protect the complex from quenching and likely occurs through intercalation.
Importantly, we observe that the emission lifetime associated with binding to the mismatched site is quenched to a much lesser extent compared to well-matched binding, which is also evident from the steady-state [Fe(CN)6]3– quenching. This observation illustrates that the complex is bound deeply at the mismatched site and is less accessible to the quencher. This result supports the notion that compared to well-matched sites, the complex binds to destabilized sites through an alternate binding mode, ostensibly metalloinsertion.
Model for [Ru(bpy)2(BNIQ)]2+ Binding to the Destabilized DNA Mismatch
Figure 5 illustrates our model for binding by [Ru(bpy)2(BNIQ)]2+ to a destabilized mismatch site. We propose that the complex binds by metalloinsertion. On the basis of the increase in excited state lifetime, the BNIQ ligand is deeply inserted into the helix and the Cu titrations suggest binding occurs from the minor groove side. Based upon the relative thermodynamics in binding different mismatches, we propose binding of the complex is by metalloinsertion with extrusion of the destabilized mismatched bases. Remarkably, the binding is highly specific for the mismatched site given the 500× increase in binding affinity versus well-matched duplex DNA. This work illustrates that the design of novel sterically demanding ligands is a valid approach in the development of mismatch-specific coordination complexes.
Figure 5.
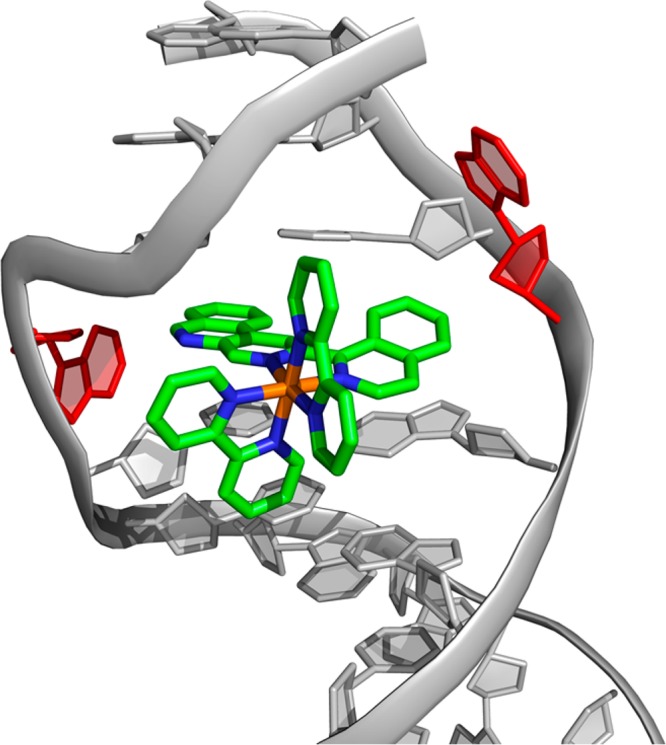
Model of [Ru(bpy)2(BNIQ)]2+ bound to a mismatch via metalloinsertion. Consistent with other metalloinsertors, we propose that the large BNIQ ligand is capable of extruding the destabilized bases (red) from the DNA π-stack and inserting deeply into the helix. The [Ru(bpy)2(BNIQ)]2+ structure was generated in Spartan 14 (Wavefunction, Inc.) and modeled into the X-ray crystal structure of [Rh(bpy)2(chrysi)]2+ bound to an AA mismatch (PDB: 3GSK)2a using PyMOL.
Acknowledgments
We thank the NIH (GM033309) for its financial support. L.M. thanks the Belgian American Educational Foundation for the Cabeaux–Jacobs Fellowship. We are grateful to the Beckman Institute Laser Resource Center (BILRC) at Caltech for facilities. We also thank Scott Virgil of the Caltech Center for Catalysis and Chemical Synthesis for his assistance in the BNIQ ligand synthesis and Shuo Shi for preliminary work on the ligand synthesis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.7b01037.
Figure S1: UV–visible spectrum of [Ru(bpy)2(BNIQ)]2+. Figure S2: Steady-state emission spectra of [Ru(bpy)2(BNIQ)]2+ in aerated and deoxygenated water. Figure S3: Steady-state emission titrations of [Ru(bpy)2(BNIQ)]2+ with 27-mer DNA sequences and the corresponding fits from the global fitting process. Figure S4: Steady-state luminescence quenching by ferricyanide (PDF)
Author Present Address
† L.M.: Engineering of Molecular Nanosystems, Université libre de Bruxelles, 50 Av F.D. Roosevelt, CP165/64 1050, Brussels, Belgium.
Author Present Address
‡ A.J.M.: Otto Diels Institute of Organic Chemistry, University of Kiel, Otto-Hahn-Platz 4, Kiel 24098, Germany.
The authors declare no competing financial interest.
Supplementary Material
References
- a Boyle K. M.; Barton J. K. Targeting DNA mismatches with rhodium metalloinsertors. Inorg. Chim. Acta 2016, 452, 3–11. 10.1016/j.ica.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Granzhan A.; Kotera N.; Teulade-Fichou M.-P. Finding needles in a basestack: recognition of mismatched base pairs in DNA by small molecules. Chem. Soc. Rev. 2014, 43, 3630–3665. 10.1039/c3cs60455a. [DOI] [PubMed] [Google Scholar]
- a Zeglis B. M.; Pierre V. C.; Kaiser J. T.; Barton J. K. A Bulky Rhodium Complex Bound to an Adenosine-Adenosine DNA Mismatch: General Architecture of the Metalloinsertion Binding Mode. Biochemistry 2009, 48, 4247–4253. 10.1021/bi900194e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pierre V. C.; Kaiser J. T.; Barton J. K. Insights into finding a mismatch through the structure of a mispaired DNA bound by a rhodium intercalator. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 429–434. 10.1073/pnas.0610170104. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jackson B. A.; Barton J. K. Recognition of Base Mismatches in DNA by 5,6-Chrysenequinone Diimine Complexes of Rhodium(III): A Proposed Mechanism of Preferential Binding in Destabilized Regions of the Double Helix. Biochemistry 2000, 39, 6176–6182. 10.1021/bi9927033. [DOI] [PubMed] [Google Scholar]
- a Komor A. C.; Barton J. K. An Unusual Ligand Coordination Gives Rise to a New Family of Rhodium Metalloinsertors with Improved Selectivity and Potency. J. Am. Chem. Soc. 2014, 136, 14160–14172. 10.1021/ja5072064. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Komor A. C.; Schneider C. J.; Weidmann A. G.; Barton J. K. Cell-Selective Biological Activity of Rhodium Metalloinsertors Correlates with Subcellular Localization. J. Am. Chem. Soc. 2012, 134, 19223–19233. 10.1021/ja3090687. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ernst R. J.; Komor A. C.; Barton J. K. Selective Cytotoxicity of Rhodium Metalloinsertors in Mismatch Repair-Deficient Cells. Biochemistry 2011, 50, 10919–10928. 10.1021/bi2015822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gill M. R.; Thomas J. A. Ruthenium(II) polypyridyl complexes and DNA – from structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012, 41, 3179–3192. 10.1039/c2cs15299a. [DOI] [PubMed] [Google Scholar]; b Baggaley E.; Weinstein J. A.; Williams J. A. Lighting the way to see inside the live cell with luminescent transition metal complexes. Coord. Chem. Rev. 2012, 256, 1762–1785. 10.1016/j.ccr.2012.03.018. [DOI] [Google Scholar]; c Lo K. Luminescent Rhenium(I) and Iridium(III) Polypyridine Complexes as Biological Probes, Imaging Reagents, and Photocytotoxic Agents. Acc. Chem. Res. 2015, 48, 2985–2995. 10.1021/acs.accounts.5b00211. [DOI] [PubMed] [Google Scholar]; d Ji L.-N.; Zou X.-H.; Liu J.-G. Shape- and enantioselective interaction of Ru(II)/Co(III) polypyridyl complexes with DNA. Coord. Chem. Rev. 2001, 216–217, 513–536. 10.1016/S0010-8545(01)00338-1. [DOI] [Google Scholar]; e Li G.; Sun L.; Ji L.; Chao H. Ruthenium(II) Complexes with dppz: from molecular photoswitch to biological applications. Dalton Trans. 2016, 45, 13261–13276. 10.1039/C6DT01624C. [DOI] [PubMed] [Google Scholar]; f McKinley A. W.; Lincoln P.; Tuite E. M. Environmental effects on the photophysics of transition metal complexes with dipyrido[2,3-a:3′,2′-c]phenazine (dppz) and related ligands. Coord. Chem. Rev. 2011, 255, 2676–2692. 10.1016/j.ccr.2011.06.012. [DOI] [Google Scholar]; g Lo L.; Choi A.; Law W. Applications of luminescent inorganic and organometallic transition metal complexes as biomolecular and cellular probes. Dalton Trans. 2012, 41, 6021–6047. 10.1039/c2dt11892k. [DOI] [PubMed] [Google Scholar]
- Boynton A. N.; Marcelis L.; Barton J. K. [Ru(Me4phen)2(dppz)]2+, a Light Switch for DNA Mismatches. J. Am. Chem. Soc. 2016, 138, 5020–5023. 10.1021/jacs.6b02022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Friedman A. E.; Chambron J. C.; Sauvage J. P.; Turro N. J.; Barton J. K. Molecular “Light Switch” for DNA: Ru(bpy)2(dppz)2+. J. Am. Chem. Soc. 1990, 112, 4960–4962. 10.1021/ja00168a052. [DOI] [Google Scholar]; b Jenkins Y.; Friedman A. E.; Turro N. J.; Barton J. K. Characterization of Dipyridophenazine Complexes of Ruthenium(II): The Light Switch Effect as a Function of Nucleic Acid Sequence and Conformation. Biochemistry 1992, 31, 10809–10816. 10.1021/bi00159a023. [DOI] [PubMed] [Google Scholar]; c Hartshorn R. M.; Barton J. K. Novel Dipyridophenazine Complexes of Ruthenium(II): Exploring Luminescent Reporters of DNA. J. Am. Chem. Soc. 1992, 114, 5919–5925. 10.1021/ja00041a002. [DOI] [Google Scholar]
- Fung S. K.; Zou T.; Cao B.; Chen T.; To W.-P.; Yang C.; Lok C.-N.; Che C.-M. Luminescent platinum(II) complexes with functionalized N-heterocyclic carbine or diphosphine selectively probe mismatched and abasic DNA. Nat. Commun. 2016, 7, 10655. 10.1038/ncomms10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a McConnell A. J.; Lim M. H.; Olmon E. D.; Song H.; Dervan E. E.; Barton J. K. Luminescent Properties of Ruthenium(II) Complexes with Sterically Expansive Ligands Bound to DNA Defects. Inorg. Chem. 2012, 51, 12511–12520. 10.1021/ic3019524. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zeglis B. M.; Barton J. K. Binding of Ru(bpy)2(eilatin)2+ to Matched and Mismatched DNA. Inorg. Chem. 2008, 47, 6452–6457. 10.1021/ic8006537. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rüba E.; Hart J. R.; Barton J. K. [Ru(bpy)2(L)]Cl2: Luminescent Metal Complexes that Bind DNA Base Mismatches. Inorg. Chem. 2004, 43, 4570–4578. 10.1021/ic0499291. [DOI] [PubMed] [Google Scholar]
- Sullivan B. P.; Salmon D. J.; Meyer T. J. Mixed Phosphine 2,2′-Bipyridine Complexes of Ruthenium. Inorg. Chem. 1978, 17, 3334. 10.1021/ic50190a006. [DOI] [Google Scholar]
- Hughes R. O.; et al. Design, Synthesis, and Biological Evaluation of 3-[4-(2-Hydroxyethyl)piperazin-1-yl]-7-(6-methoxypyridin-3-yl)-1-(2-propoxyethyl)pyrido[3,4-b]pyrazin-2(1H)-one, A Potent, Orally Active, Brain Penetrant Inhibitor of Phosphodiesterase 5 (PDE5). J. Med. Chem. 2010, 53, 2656–2600. 10.1021/jm901781q. [DOI] [PubMed] [Google Scholar]
- Dempsey J. L.; Winkler J. R.; Gray H. B. Kinetics of Electron Transfer Reactions of H2-Evolving Cobalt Diglyoxime Catalysts. J. Am. Chem. Soc. 2010, 132, 1060–1065. 10.1021/ja9080259. [DOI] [PubMed] [Google Scholar]
- Qiujiang D. Preparation of 2-bromopyridine from 2-chloropyridine by halogen exchange. Chem. Ind. Times 2005, 2, 39–40. [Google Scholar]
- Luzung M. R.; Patel J. S.; Yin J. A Mild Negishi Cross-Coupling of 2-Heterocyclic Organozinc Reagents and Aryl Chlorides. J. Org. Chem. 2010, 75, 8330–8332. 10.1021/jo1018798. [DOI] [PubMed] [Google Scholar]
- Milne J. E.; Buchwald S. L. An Extremely Active Catalyst for the Negishi Cross-Coupling Reaction. J. Am. Chem. Soc. 2004, 126, 13028–13032. 10.1021/ja0474493. [DOI] [PubMed] [Google Scholar]
- Van Houten J.; Watts R. J. The Effect of Ligand and Solvent Deuteration on the Excited State Properties of the Tris(2,2′-bipyridyl)ruthenium(II) Ion in Aqueous Solution. Evidence for the Electron Transfer to Solvent. J. Am. Chem. Soc. 1975, 97, 3843–3844. 10.1021/ja00846a062. [DOI] [Google Scholar]
- Van Houten J.; Watts R. J. Temperature Dependence of the Photophysical and Photochemical Properties of the Tris(2,2′-bipyridyl)ruthenium(II) Ion in Aqueous Solution. J. Am. Chem. Soc. 1976, 98, 4853–4858. 10.1021/ja00432a028. [DOI] [Google Scholar]
- Barton J. K.; Danishefsky A. T.; Goldberg J. M. Tris(phenanthroline)ruthenium(II): Stereoselectivity in Binding to DNA. J. Am. Chem. Soc. 1984, 106, 2172–2176. 10.1021/ja00319a043. [DOI] [Google Scholar]
- Lim M. H.; Song H.; Olmon E. D.; Dervan E. E.; Barton J. K. Sensitivity of Ru(bpy)2dppz2+ Luminescence to DNA Defects. Inorg. Chem. 2009, 48, 5392–5397. 10.1021/ic900407n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyret N.; Seneviratne A.; Allawi H. T.; SantaLucia J. Nearest-Neighbor Thermodynamics and NMR of DNA Sequences with Internal A·A, C·C, G·G, and T·T Mismatches. Biochemistry 1999, 38, 3468–3477. 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- SantaLucia J.; Hicks D. The Thermodynamics of DNA Structural Motifs. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 415–440. 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- Zeglis B. M.; Boland J. A.; Barton J. K. Recognition of Abasic Sites and Base Bulges in DNA by a Metalloinsertor. Biochemistry 2009, 48, 839–849. 10.1021/bi801885w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigman D. S.; Chen C.-H. B. Chemical Nucleases: New Reagents in Molecular Biology. Annu. Rev. Biochem. 1990, 59, 207–236. 10.1146/annurev.bi.59.070190.001231. [DOI] [PubMed] [Google Scholar]
- Sigman D. S.; Mazumder A.; Perrin D. M. Chemical Nucleases. Chem. Rev. 1993, 93, 2295–2316. 10.1021/cr00022a011. [DOI] [Google Scholar]
- Lim M. H.; Lau I. H.; Barton J. K. DNA Strand Cleavage Near a CC Mismatch Directed by a Metalloinsertor. Inorg. Chem. 2007, 46, 9528–9530. 10.1021/ic701598k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.; Kaiser J. T.; Barton J. K. Crystal Structure of Δ-[Ru(bpy)2dppz]2+ bound to mismatched DNA reveals side-by-side metalloinsertion and intercalation. Nat. Chem. 2012, 4, 615–620. 10.1038/nchem.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar C. V.; Barton J. K.; Turro N. J. Photophysics of Ruthenium Complexes Bound to Double Helical DNA. J. Am. Chem. Soc. 1985, 107, 5518–5523. 10.1021/ja00305a032. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.