

Abstract
In recent years there has been a growing interest among cancer biologists in cancer metabolism. This Review summarizes past and recent advances in our understanding of the reprogramming of glucose metabolism in cancer cells, which is mediated by oncogenic drivers and by the undifferentiated character of cancer cells. The reprogrammed glucose metabolism in cancer cells is required to fulfil anabolic demands. This Review discusses the possibility of exploiting the reprogrammed glucose metabolism for therapeutic approaches that selectively target cancer cells.
Until two decades ago, cellular glucose metabolism, and cancer metabolism in general, were not considered to be major branches of cancer biology. However, in the past 15 years, there has been a growing interest in cancer metabolism, particularly glucose metabolism in cancer cells; these topics have now become an integral part of cancer biology, similarly to signal transduction and transcription. Considering that accelerated aerobic glycolysis has been known to distinguish cancer cells from normal cells for many decades and that this distinction has been exploited to detect and image tumours in vivo, it is surprising that high cellular glucose metabolism has only recently been recognized as one of the hallmarks of cancer by cancer biologists. This renewed interest in glucose metabolism is coupled with the realization that one of the consequences of certain oncogenic drivers is increased cellular glucose metabolism and, indeed, metabolism in general. Perhaps one of the reasons for this renewed recognition is the realization that the PI3K–AKT–mTOR complex 1 (mTORC1) signalling pathway, which has an evolutionarily conserved function in metabolism, is also frequently activated in cancer cells.
The discovery of high rates of aerobic glycolysis in cancer cells by Otto Warburg in the late 1920s led him to assume that respiration, through the process of oxidative phosphorylation (OXPHO), is impaired or damaged in cancer cells1–3. In subsequent years, this idea that OXPHO in the mitochondria is suppressed in cancer cells was debated. Most notable is the debate between Warburg and Sidney Weinhouse4,5, in which Weinhouse disputed the assumption that tumour cells cannot oxidize glucose. Weinhouse and colleagues carried out isotope-tracing experiments to show that glucose can be oxidized to CO2. In other words, that OXPHO can occur in cancer cells at a rate similar to that in normal cells6–8. Retrospectively, it became clear that the data obtained by Warburg himself did not support the idea that respiration is diminished in cancer cells9. Instead, high rates of glucose metabolism and glycolysis are maintained in cancer cells despite the occurrence of OXPHO. Whereas most normal cells obey the Crabtree effect — which posits that if a high rate of glycolysis is maintained OXPHO is not — cancer cells do not. They maintain high rates of both glucose metabolism and OXPHO to fulfil the high demand for anabolic processes10. However, during tumour growth, the cells in the core of the tumour become hypoxic and only under these conditions are rates of OXPHO decreased while glycolysis is increased.
Glucose metabolism involves not only glycolysis but also other pathways that require glucose. These include the pentose phosphate pathway (PPP), which generates pentose phosphates for ribonucleotide synthesis and NADPH; the hexosamine pathway, which is required for glycosylation of proteins; glycogenesis, which generates glycogen for glucose storage; the serine biosynthesis pathway, which generates amino acids; which is followed by the one-carbon metabolism cycle, which generates NADPH, and which is required for purine and glutathione biosynthesis, and for methylation. This Review summarizes the mechanisms by which glucose metabolism is reprogrammed in cancer cells and how this reprogramming could be exploited to selectively target cancer cells.
Reprogramming of glucose metabolism
Committed reactions in glucose metabolism
The canonical glycolysis pathway comprises several reversible enzymatic reactions and three irreversible reactions, which are known as the committed steps. The first committed step is catalysed by hexokinases that phosphorylate glucose to produce glucose-6-phosphate (G6P). This reaction is perhaps the most important step in glucose metabolism — first, because it traps glucose inside cells, which could otherwise be exported by the glucose transporters, and second, because G6P is at the convergence point of not only glycolysis but also the PPP, the hexosamine pathway and glycogen synthesis (FIGS 1, 2).
Figure 1. Changes that occur in glucose metabolism of cancer cells.
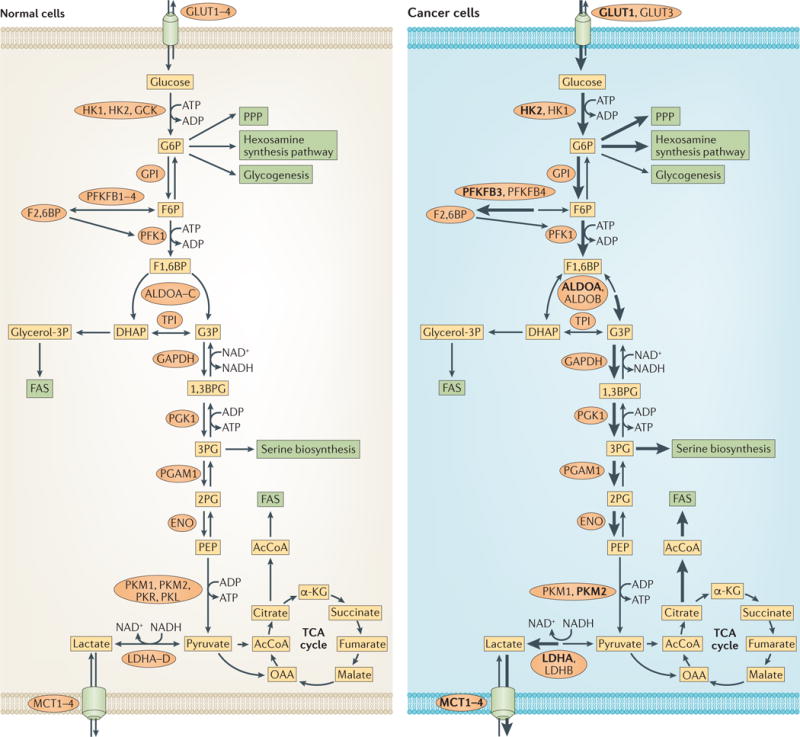
Compared with normal cells (left), the flux of glucose metabolism and glycolysis is accelerated in cancer cells (right) by preferential expression of transporters and enzyme isoforms that drive glucose flux forward and to adapt to the anabolic demands of cancer cells. Enzymes that catalyse the metabolic reactions are shown in ovals. Enzymes that are predominant in cancer cells are shown in bold. The thickness of the arrows indicates relative flux. 1,3BPG, 1,3-bisphosphoglycerate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; α-KG, α-ketoglutarate; AcCoA, acetyl-CoA; ALDO, aldolase; DHAP, dihydroxyacetone-phosphate; ENO, enolase; F1,6BP, fructose-1,6-bisphosphate; F2,6BP, fructose-2,6-bisphosphate; F6P, fructose-6-phosphate; FAS, fatty acid synthesis; G3P, glyceraldehyde-3-phosphate; G6P, glucose-6-phosphate; HK, hexokinase; LDH, lactate dehydrogenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GCK, glucokinase; GLUT, glucose transporter; glycerol-3P, glycerol-3-phosphate; GPI, glucose-6-phosphate isomerase; MCT, monocarboxylate transporter; OAA, oxaloacetate; PEP, phosphoenolpyruvate; PFK1, phosphofructokinase 1; PFKFB, 6-phosphofructo 2-kinase/fructose-2,6-bisphosphatase; PGAM1, phosphoglycerate mutase 1; PGK1, phosphoglycerate kinase 1; PK, pyruvate kinase; PPP, pentose phosphate pathway; TCA, tricarboxylic acid; TPI, triosephosphate isomerase.
Figure 2. Branching pathways from glucose-6-phosphate.
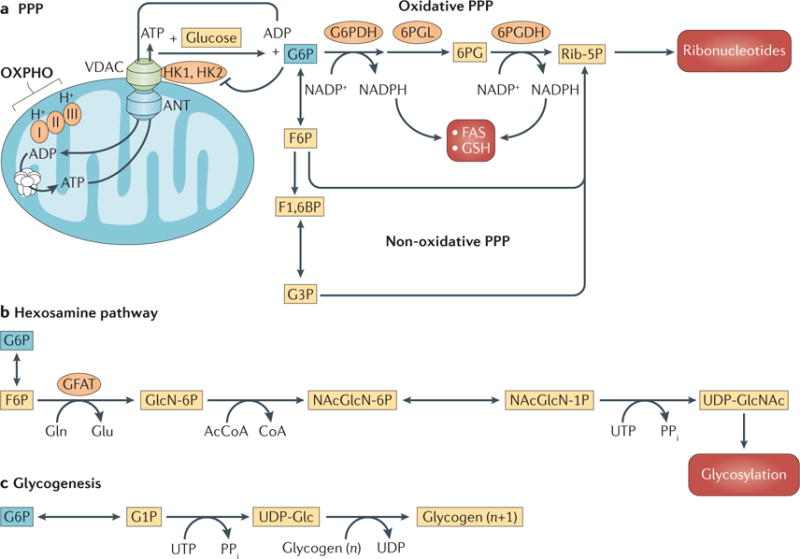
Mitochondrial hexokinase 1 (HK1) and HK2 phosphorylate glucose to glucose-6-phosphate (G6P) by preferentially using ATP derived from oxidative phosphorylation (OXPHO) in mitochondria. a | The pentose phosphate pathway (PPP), which generates NADPH and pentose phosphates. b | The hexosamine pathway that generates metabolites for glycosylation. c | Glycogenesis, which stores glycogen as an intracellular source of G6P. 6PG, 6-phosphogluconate; 6PGDH, 6-phosphogluconate dehydrogenase; 6PGL, 6-phosphogluconolactonase; AcCoA, acetyl-CoA; ANT, adenine nucleotide translocator; F1,6BP, fructose-1,6-bisphosphate; F6P, fructose-6-phosphate; FAS, fatty acid synthesis; G1P, glucose-1-phosphate; G3P, glyceraldehyde-3-phosphate; G6PDH, glucose-6-phosphate dehydrogenase; GFAT, glutamine:fructose-6-phosphate amidotransferase; GlcN-6P, glucosamine-6-phosphate; GSH, reduced glutathione; NAcGlcN-1P, N-acetyl D-glucosamine-1-phosphate; NAcGlcN-6P, N-acetyl D-glucosamine-6-phosphate; PPi, inorganic phosphate; Rib-5P, ribose-5-phosphate; UDP-Glc, UDP-glucose; UDP-GlcNAc, UDP-N-acetylglucosamine; VDAC, voltage-dependent anion channel.
The PPP is composed of two branches: oxidative and non-oxidative. The oxidative branch generates NADPH and ribulose-5-phosphate in three irreversible reactions. The non-oxidative branch generates pentose phosphates for ribonucleotide synthesis in a series of reversible reactions that also recruit additional metabolites, such as fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphate (G3P) (reviewed in REFS 11,12) (FIG. 2a). Because the PPP produces NADPH, which is required to maintain intracellular redox homeostasis and fatty acid synthesis, as well as pentose phosphates, which are required for RNA and DNA synthesis (FIG. 2a), it has a pivotal role in cancer metabolism. The PPP is modulated by several metabolites, and in proliferating cancer cells both the oxidative and non-oxidative branches of the PPP are increased by several oncogenic drivers (reviewed in REFS 11,12). The extent to which the oxidative branch versus the non-oxidative branch is modulated depends on the redox and metabolic status of the cell.
The hexosamine pathway, which is initiated with G6P and F6P, is followed by four subsequent reactions that also use glutamine, acetyl-CoA and UTP to generate UDP-N-acetylglucosamine (UDP-GlcNAc) (FIG. 2b). UDP-GlcNAc is a sugar donor for the glycosylation of proteins13–15. In glycogenesis, glycogen, a branched polymer of glucose that functions as a repository of glucose units, is synthesized from G6P (FIG. 2c). Glycogen is synthesized mainly in the liver and skeletal muscles, but many cells have the capacity to synthesize glycogen, which can be a source of glucose for cancer cells in conditions in which glucose is limited (reviewed in REF. 16).
Cancer cells facilitate the first committed step in glucose metabolism by increasing glucose uptake and inducing high-level expression of hexokinase 2 (HK2) in addition to HK1, which is already expressed in normal cells17,18. There are four main hexokinases in mammalian cells — HK1–4, which are encoded by different genes19–21 — and a recently identified fifth hexokinase, hexokinase domain-containing protein 1 (HKDC1), which has not yet been fully characterized22,23. HK1–3 are all high-affinity hexokinases, but HK1 and HK2 have the unique ability to bind to mitochondria in a voltage-dependent anion channel (VDAC)-dependent manner (FIG. 2a). HK1–3 are inhibited by their own catalytic product, G6P, which also induces a conformational change that dissociates HK1 and HK2 from the mitochondria. By binding to the outer mitochondrial membrane and VDAC, HK1 and HK2 preferentially use ATP derived from mitochondria to phosphorylate glucose, thereby coupling OXPHO with glycolysis (reviewed in REF. 19) (FIG. 2a). HK3 has the highest affinity for glucose but is also inhibited by glucose at physiological levels, and its role in glucose metabolism in cancer cells is not clear. HK4 (also known as glucokinase) has a low affinity for glucose and is not inhibited by G6P, and its expression is restricted to the pancreas and liver19,21. HK1 is the most ubiquitous isoform and is found in most adult tissues, whereas HK2 is expressed at high levels only in skeletal muscle, heart and adipose tissues, although it is also highly expressed in embryonic tissues. However, HK2 expression is induced in cancer cells to increase the glucose flux into various metabolic pathways24,25. It is not completely clear why in most cancer cells only HK2 expression is induced without further increase in HK1 expression. One possibility is that the HK2 locus in the genome is more accessible to transcriptional manipulation. Interestingly, both HK1 and HK2 possess two tandem catalytic domains but only one of them is active in HK1, whereas both are active in HK2 (REF. 26).
The second committed step of glycolysis is catalysed by phosphofructokinase 1 (PFK1), and the third committed step is catalysed by pyruvate kinases. PFK1 catalyses the conversion of F6P into fructose-1,6-bisphosphate (F1,6BP), and pyruvate kinases convert phosphoenolpyruvate (PEP) into pyruvate (FIG. 1). The first two committed steps consume ATP whereas the third committed step generates ATP. Cancer cells use several mechanisms to increase the flux of glucose in glycolysis, with the exception of the third and last committed step (which generates ATP and pyruvate), which is attenuated in cancer cells (FIG. 1). The attenuation of the last committed step is achieved, in part, by predominantly using the low-affinity pyruvate kinase M2 (PKM2) isoform to catalyse this reaction27. There are four pyruvate kinase isoforms in mammalian cells — PKM1, PKM2, PKR and PKL — which are encoded by two separate genes28. The same gene, PKLR, encodes PKR and PKL through the use of alternative promoters. PKR expression is restricted to red blood cells, whereas PKL is expressed at high levels in the liver and to some extent in the kidney. PKM1 and PKM2 are encoded by another gene, PKM, through alternative splicing. The active pyruvate kinases are tetramers, but whereas PKM1 is a constitutively active tetramer, the tetrameric active form of PKM2 is allosterically regulated by various metabolites27. The activity of PKM2 can therefore be modulated to enable cancer cells to adapt to conditions that require different sets of metabolites for different anabolic processes. The expression of PKM2 varies between different types of cancer, and in a subset of breast cancer samples, its expression is hardly detected27. In a mouse model of breast cancer, the deletion of Pkm2 does not inhibit tumour development29, and similarly, PKM2 was found to be dispensable for the growth of colon cancer cells30. By contrast, PKM2 is required for the development of leukaemia31. The low level of PKM2 activity in cancer cells suggests that there is an alternative mechanism by which PEP is converted into pyruvate. One such mechanism is mediated by phosphoglycerate mutase 1 (PGAM1), which is known to catalyse the conversion of 3-phosphoglycerate (3PG) into 2-phosphoglycerate (2PG) in glycolysis. It was shown that PGAM1 converts PEP into pyruvate, possibly by transferring the PEP phosphate to the catalytic histidine of PGAM1 (REF. 32).
The attenuation of the last committed step of glycolysis diverts metabolites into the branching pathways, such as the PPP and the serine biosynthesis pathway, to generate sufficient metabolic intermediates and to augment the anabolic reactions required for cell growth and proliferation. Although the last committed step (the conversion of PEP into pyruvate) is attenuated, the subsequent conversion of pyruvate into lactate is markedly increased, and most of the lactate is secreted (FIG. 1). The increased conversion of pyruvate into lactate despite the inhibition of pyruvate generation might seem to be counterintuitive but this response has probably evolved to regenerate NAD+ and to maintain the flux to the branching pathways (see below). The enzymes that catalyse the reversible conversion of pyruvate into lactate are lactate dehydrogenases (LDHs) and are encoded by four separate genes (LDHA, LDHB, LDHC and LDHD). The two highly expressed isozymes, LDHA and LDHB, can form either homotetramers or heterotetramers. LDHA has a higher affinity for pyruvate, and LDHB has a higher affinity for lactate; thus, LDHA favours the forward reaction, and LDHB favours the reverse reaction. LDHA is the predominantly expressed LDH in cancer cells33,34. Because of the reversible activity of LDH, the secretion of lactate from the cell through monocarboxylate transporters (MCTs) is required to drive the reaction forward as well as to prevent a highly acidic intracellular environment. Also, because lactate inhibits the activity of PFK1 (REF. 35), it is important to prevent its intracellular accumulation to avoid inhibition of the second committed step in glycolysis. However, the secreted lactate could have supporting roles for the cancer cells. First, local acidification of the tumour microenvironment could potentially support tumour invasion36, in part through an increase in the level of extracellular vascular endothelial growth factor A (VEGFA)37,38 and proteases39,40. Second, the lactate can be taken up by adjacent stromal cells and used as an energy substrate to support growth or to generate pyruvate, which is then extruded by the stroma and taken up by the cancer cells41,42. Under conditions of low glucose availability, cancer cells can use the extracellular lactate or pyruvate to support the tricarboxylic acid cycle (TCA cycle) and to provide citrate and acetyl-CoA for fatty acid synthesis.
The increased pyruvate to lactate flux in cancer cells is catalysed by LDHA, in a reaction that generates NAD+ from NADH (FIG. 1). NAD+ is converted into NADH in glycolysis and is required to maintain a high flux of glucose metabolism. The replenished NAD+ is required as a coenzyme for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which catalyses the conversion of G3P into 1,3-bisphosphoglycerate (1,3BPG). Subsequently, 1,3BPG is converted into 3PG by phosphoglycerate kinase 1 (PGK1) in a reaction that generates ATP (FIG. 1). 3PG is not only used to generate PEP in the subsequent reaction of glycolysis but also participates in the serine biosynthesis pathway that branches from glycolysis to generate non-essential amino acids (FIGS 1, 3). The first step in the serine biosynthesis pathway, catalysed by phosphoglycerate dehydrogenase (PHGDH), also uses NAD+ to oxidize 3PG to the serine biosynthesis precursor, 3-phosphohydroxypyruvate. Thus, it would be interesting to determine whether inhibiting LDHA affects the serine biosynthesis pathway.
Figure 3. The serine biosynthesis pathway and extensions to the one-carbon metabolism, the methionine cycle, the purine biosynthesis pathway and the generation of glutathione.
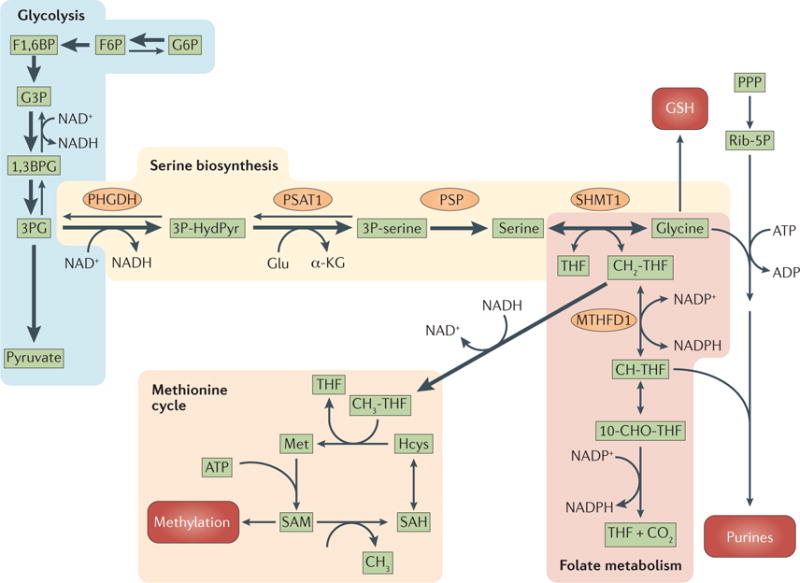
3-phosphoglycerate (3PG) generated by glycolysis provides the initial substrate for serine biosynthesis. In the first step in the serine biosynthesis pathway, 3PG is oxidized by phosphoglycerate dehydrogenase (PHGDH) in a reaction that consumes NAD+. The second step is catalysed by phosphoserine aminotransferase (PSAT1) in a reaction that is coupled to deamination of glutamate (Glu) to α-ketoglutarate (α-KG). The last step is catalysed by phosphoserine phosphatase (PSP). The conversion of serine to glycine generates 5,10-methylenetetrahydrofolate (CH2-THF), which is then used in folate metabolism and in the methionine cycle. Glycine is used to generate glutathione (GSH), and together with ribose-5-phosphate (Rib-5P) to generate purines. The folate pathway can generate NADPH, and 10-formyl-THF (10-CHO-THF). 10-CHO-THF together with Rib-5P and glycine participates in the generation of purines. Demethylation of 5-methyl-THF (CH3-THF) contributes one carbon to the methionine cycle by the methylation of homocysteine (Hcys) to generate methionine (Met). Methionine is converted into S-adenosylmethionine (SAM) and is used by methyltransferases. Demethylation of SAM generates S-adenosylhomocysteine (SAH), which is converted back into Hcys by deadenylation. The thickness of the arrows indicatesrelative flux. 1,3BPG, 1,3-bisphosphoglycerate; 3P-HydPyr, 3-phosphohydroxypyruvate; 3P-serine, 3-phosphoserine; CH-THF, 5,10 methenyl-THF; F1,6BP, fructose-1,6-bisphosphate; F6P, fructose-6-phosphate; G3P, glyceraldehyde-3-phosphate; G6P, glucose-6-phosphate; MTHFD1, methylenetetrahydrofolate dehydrogenase 1; PPP, pentose phosphate pathway; SHMT1, serine hydroxymethyltransferase 1.
If the slow conversion of PEP into pyruvate, catalysed by PKM2, increases channelling to the serine biosynthesis pathway in cancer cells, it would require more NAD+, which in turn would increase NADH levels. This may explain, at least in part, why there is increased conversion of pyruvate into lactate (which generates NAD+) when the PEP to pyruvate flux is low43. Notably, despite the slow conversion of PEP into pyruvate, cancer cells seem to have sufficient pyruvate to import into the mitochondria for the TCA cycle, which is further facilitated by exogenous glutamine44.
Interestingly, in many cancer cells, the first steps in the serine biosynthesis pathway are augmented by either overexpression or gene amplification of PHGDH45–49. It is not clear why cancer cells accelerate the first steps of the serine biosynthesis pathway, as PHGDH seems to be required for cancer cell proliferation regardless of the presence or absence of exogenous serine46,50. One possible explanation is that the subsequent reaction in the serine biosynthesis pathway, catalysed by phosphoserine aminotransferase (PSAT1), converts 3-phosphohydroxypyruvate into serine, but also generates α-ketoglutarate (α-KG) from glutamine by transamination (FIG. 3). It was reported that in cells with overexpression of PHGDH, this transamination reaction supplies a significant portion of α-KG to the TCA cycle46. Another potential explanation for the requirement of PHGDH despite exogenous serine, is due to reduced purine biosynthesis51 (see below). Notably, although PHGDH is required for cancer cell proliferation and tumour initiation of breast cancer cells, it was reported that it is dispensable for the tumour maintenance of oestrogen receptor (ER)− breast cancer cells overexpressing PHGDH43.
The subsequent conversion of serine into glycine by serine hydroxymethyltransferase 1 (SHMT1) in the one-carbon metabolism cycle in the cytosol is coupled to the conversion of tetrahydrofolate (THF) into 5,10-methylene-THF (CH2-THF). Then, CH2-THF is converted into 5,10-methenyl-THF (CH-THF), which enters the folate cycle (FIG. 3). CH2-THF is also converted into 5-methyl-THF (CH3-THF), which provides the methyl group to generate S-adenosylmethionine in the methionine cycle (FIG. 3). S-Adenosylmethionine is a substrate for methyltransferases, which methylate metabolites, nucleic acids and proteins. The conversion of CH2-THF into CH-THF is catalysed by methylenetetra hydrofolate dehydrogenases (MTHFD1 and MTHFD2) in both the cytoplasm and mitochondria in a reaction that generates NADPH. CH-THF can also be converted into 10-formyl-THF (10-CHO-THF), which participates in purine biosynthesis, or to THF in a reaction that generates NADPH (FIG. 3). Recently, it was shown that folate metabolism is an important source of NADPH in cancer cells in addition to the PPP52,53. However, unlike the PPP, the reaction that generates NADPH in the folate cycle, which is catalysed by MTHFD1 in the cytoplasm and MTHFD2 in the mitochondria, is reversible (FIG. 3). Notably, if the therapeutic inhibition of the serine biosynthesis pathway increases the intracellular levels of 3PG it might also inhibit NADPH production by the PPP, as 3PG was shown to inhibit 6-phosphogluconate dehydrogenase (6PGDH), which catalyses the second step in the oxidative PPP54. Finally, glycine, together with 10-CHO-THF, contributes to the generation of purine nucleotides in a series of reactions that use ribose-5-phosphate generated from the PPP as the initial substrate. Glycine is also used together with glutamate and cysteine to generate glutathione (FIG. 3). It was recently reported that when PHGDH is inhibited glycine is converted back into serine by the cytoplasmic SHMT1, and this reversed reaction occurs even when glycine is derived from exogenous serine. The reversed reaction also consumes CH2-THF51 (FIG. 3). Therefore, less glycine and 10-CHO-THF are available for the synthesis of purines51. This could explain, at least in part, why PHGDH is required despite the availability of exogenous serine. It is not completely clear, however, why SHMT1 continues to regenerate glycine from serine when both serine and glycine are available from exogenous sources. It also remains to be determined whether PHGDH inhibition reduces the flux of glycine into glutathione and the generation of NADPH by the cytoplasmic folate pathway (FIG. 3).
In summary, glycolysis is accelerated in cancer cells to feed the branching pathways that generate nucleotides, amino acids and fatty acids for the anabolic processes that generate nucleic acids, proteins and membranes. Notably, the activities of many of the glycolytic enzymes are positively and negatively regulated to maintain homeostasis. Cancer cells often exploit these regulatory pathways to fulfil their anabolic needs and for adaptation to various microenvironments.
Regulation of glycolytic enzymes
The enzymes that catalyse the committed steps of glycolysis are tightly controlled and allosterically regulated, both positively and negatively. As indicated above, the mitochondria-associated high-affinity HK1 and HK2 enzymes that catalyse the first committed step are allosterically inhibited by their own catalytic product, G6P, which causes them to dissociate from mitochondria. However, it is likely that this feedback inhibition is minimal in rapidly dividing cancer cells, because G6P is rapidly consumed by the increased flux of downstream pathways.
The tetrameric PFK1 that catalyses the conversion of F6P into F1,6BP (the second committed step in glycolysis) is controlled by multiple positive and negative regulators. The PFK1 subunits are encoded by different genes — PFKM in muscle, PFKL in liver and PFKP in platelets55. PFK1 is allosterically activated by fructose-2,6-bisphosphate (F2,6BP), which is generated from F6P by 6-phosphofructo 2-kinase/fructose-2,6-bisphosphatase (PFK2/F2,6BPase or PFKFB). PFKFB contains two tandem domains with opposing activities. One domain has a kinase activity that phosphorylates F6P to F2,6BP, and the other domain has a phosphatase activity that dephosphorylates F2,6BP back to F6P. Because F2,6BP is an allosteric activator of PFK1, the relative kinase to phosphatase activities of PFKFB can determine PFK1 activity, and the relative intracellular levels of F6P and F1,6BP. There are four major isoforms of PFKFB (PFKFB1–4), which are encoded by separate genes. The relative kinase to phosphatase activity of each isoform is different, and their relative expression levels vary in different mammalian tissues and in different types of cancer (reviewed in REF. 56). In general, the PFKFB3 isoform, which has greater kinase activity than phosphatase activity, is increased in cancer57, thereby increasing the intracellular level of F2,6BP and the allosteric activation of PFK1.
PFK1 is inhibited by PEP, lactate, citrate, palmitoyl-CoA and ATP, which are downstream products of glycolysis (FIG. 4). It is possible that the inhibition of PFK1 by these products, in particular citrate and palmitoyl-CoA, has evolved to redirect glucose into the PPP to generate sufficient NADPH levels for lipogenesis and to combat oxidative stress, as well as sufficient ribose-5-phosphate for ribonucleotide biosynthesis. Interestingly, PFK1 activity is inhibited by glycosylation at serine 529, the same amino acid that is required to bind to its allosteric activator F2,6BP58. Therefore, glycosylation prohibits the allosteric activation of PFK1 by F2,6BP.
Figure 4. Positive and negative regulation of enzymes in glucose metabolism.
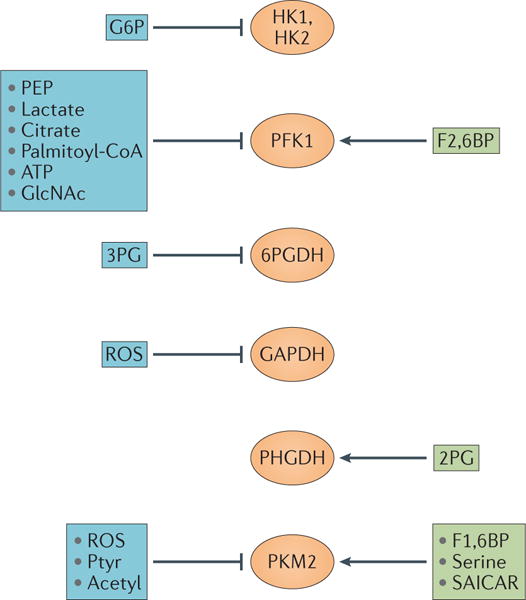
Metabolites can either positively or negatively regulate the activities of enzymes in glucose metabolism. Reactive oxygen species (ROS) are known to oxidize and inhibit glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and pyruvate kinase M2 (PKM2) activities. PKM2 is also inhibited by tyrosine phosphorylation (Ptyr) or by acetylation (Acetyl). Positive regulators are in green boxes and negative regulators are in blue boxes. 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; 6PGDH, 6-phosphogluconate dehydrogenase; F1,6BP, fructose-1,6-bisphosphate; F2,6BP, fructose-2,6-bisphosphate; G6P, glucose-6-phosphate; GlcNAc, N-acetylglucosamine; HK, hexokinase; PEP, phosphoenolpyruvate; PFK1, phosphofructokinase 1; PHGDH, phosphoglycerate dehydrogenase; SAICAR, phosphoribosylaminoimidazolesuccinocarboxamide.
The two major activators of PKM2, which catalyses the third committed step of glycolysis, are F1,6BP (the catalytic product of PFK1) and serine (FIG. 4). Both F1,6BP and serine increase the abundance of the tetrameric active form of PKM2 by direct binding (reviewed in REF. 27). The metabolic advantage of this activation is not obvious. However, high levels of F1,6BP and serine could indicate that there is no need to shunt F6P back to the PPP or to shunt other metabolites to the serine biosynthesis pathway, and therefore that glycolysis can move forward to generate ATP. The activation of PKM2 by F1,6BP could, however, be balanced by pro-proliferation signals that increase tyrosine kinase activity. Phosphorylation of PKM2 on tyrosine 105 or the binding of PKM2 to phosphotyrosine peptides inhibits its binding and activation by F1,6BP. Importantly, PKM2 activity is inhibited by reactive oxygen species (ROS) through the oxidation of cysteine 358 and thus is regulated by the redox state of the cell59. This inhibition of PKM2 could potentially channel metabolites to the serine biosynthesis and one-carbon metabolism pathways, and possibly increase the flux of glucose to the oxidative PPP59 to combat oxidative stress. Under oxidative stress conditions, the channelling of metabolites to the oxidative PPP is further exacerbated by the oxidative inhibition of GAPDH activity60. Other mechanisms that activate or inhibit PKM2 are described elsewhere27.
Mechanisms of reprogramming
There are no universal mechanisms known to be used by all types of cancer cells to reprogramme glucose metabolism, but there are several common mechanisms by which cancer cells modulate and hijack the three committed steps in glycolysis, as well as glucose uptake, to fulfil their anabolic demands. Individual oncoproteins and tumour suppressors can affect glucose metabolism in cancer cells by multiple mechanisms. Some of these mechanisms overlap with the mechanisms mediated by hypoxia and the transcription factor hypoxia inducible factor 1 (HIF1) to accelerate glucose metabolism61. HIF1 increases the expression of the glucose transporter GLUT1 (also known as SLC2A1) and of HK2 to increase glucose uptake and glucose phosphorylation (the first committed step of glycolysis), respectively25,62. Similarly, oncogenic KRAS, oncogenic BRAF and activated AKT increase the expression and translocation to the plasma membrane of GLUT1 and other glucose transporters63–66. HK2 expression is markedly induced in cancer cells by multiple mechanisms and oncogenic drivers and is transcriptionally upregulated by MYC18,67–69. AKT promotes the association of HK1 and HK2 with the mitochondria70, and HK2 is phosphorylated by AKT, which in turn increases the association of HK2 with mitochondria71 and possibly increases its intracellular activity.
The expression of PFKFB3, which generates F2,6BP, the allosteric activator of PFK1, is induced by HIF1, and PFKFB3 is overexpressed in cancer cells even in normoxic conditions72–75. AKT activates PFK1 by the phosphorylation and activation of PFKFB2 (REF. 56). However, constitutive activation of PFK1 may not be beneficial for cancer cells in all conditions. For example, under oxidative stress conditions, it may be important to attenuate the activity of PFK1 to divert glucose into the PPP and to generate NADPH to combat oxidative stress. Similarly, activated PFK1 in cancer cells should still be susceptible to inhibition by citrate and palmitoyl-CoA when they are present at excess levels to shunt F6P back into the PPP and to provide NADPH for lipogenesis.
HIF1 also increases the expression of LDHA and MCT4 to increase the conversion of pyruvate into lactate and its secretion from the cell, respectively61. Similarly, expression levels of LDHA, MCT1 and MCT4 are increased in cancer (FIG. 1). MYC, which is frequently deregulated in various types of cancer, transcriptionally increases the expression of LDHA and MCT1 (FIG. 5). The roles of MYC in cancer metabolism are reviewed elsewhere76–78. Finally, both the oxidative and non-oxidative branches of the PPP are regulated by several oncoproteins and tumour suppressors (reviewed in REF. 11) (FIG. 5).
Figure 5. Regulation of glucose metabolism by oncoproteins and tumour suppressors.
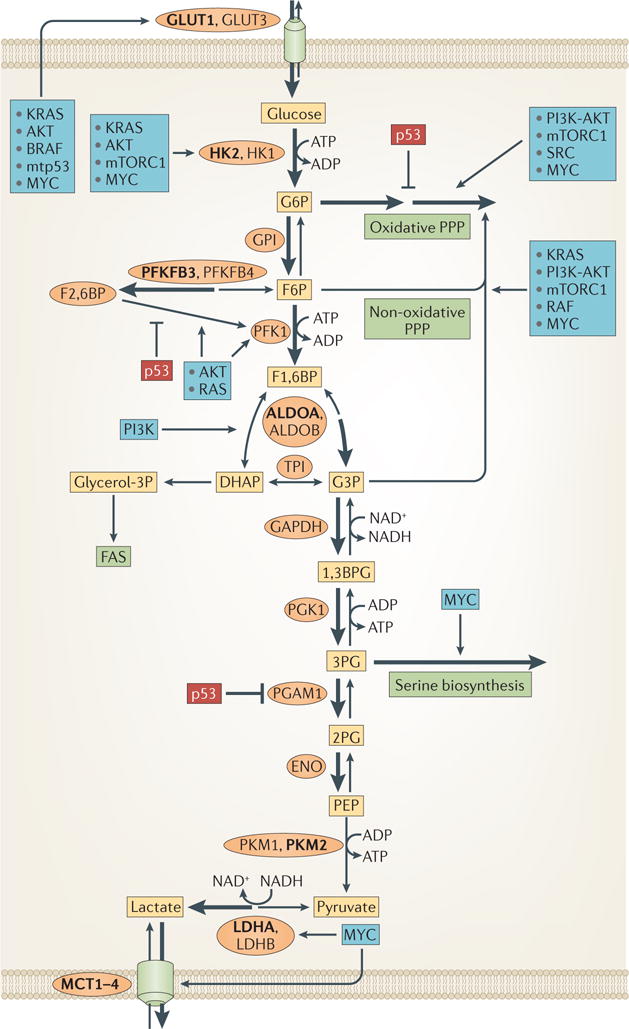
Several oncoproteins (in blue boxes) are known to either elevate the expression or induce the activity of enzymes and transporters that facilitate a high rate of glucose metabolism in cancer cells. The tumour suppressor p53 (in dark red boxes) is known to inhibit certain glucose metabolism pathways. Enzymes that are predominant in cancer cells are shown in bold. The thickness of the arrows indicates relative flux. 1,3BPG, 1,3-bisphosphoglycerate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; ALDO, aldolase; DHAP, dihydroxyacetone phosphate; ENO, enolase; F1,6BP, fructose-1,6-bisphosphate; F2,6BP, fructose-2,6-bisphosphate; F6P, fructose-6-phosphate; FAS, fatty acid synthesis; G3P, glyceraldehyde-3-phosphate; G6P, glucose-6-phosphate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GLUT, glucose transporter; glycerol-3P, glycerol-3-phosphate; GPI, glucose-6-phosphate isomerase; HK, hexokinase; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; mTORC1, mTOR complex 1; mtp53, mutant p53; PEP, phosphoenolpyruvate; PFK1, phosphofructokinase 1; PFKFB, 6-phosphofructo 2-kinase/fructose-2,6-bisphosphatase; PGAM1, phosphoglycerate mutase 1; PGK1, phosphoglycerate kinase 1; PK, pyruvate kinase; PPP, pentose phosphate pathway; TPI, triosephosphate isomerase.
Both wild-type and gain-of-function mutants of the tumour suppressor p53 have been implicated in the control of glucose metabolism (reviewed in REFS 79,80) (FIG. 5). For example, p53 induces the expression of TP53-induced glycolysis and apoptosis regulator (TIGAR), which, similarly to PFKFB, can decrease the level of F2,6BP, the allosteric activator of PFK1. Consequently, F6P levels are increased as a result of decreased PFK1 activity and F6P is redirected to the PPP to generate NADPH, maintain the redox state of the cell and increase the level of ribose-5-phosphate for ribonucleotide synthesis. Because the conversion of G6P into F6P is reversible, the accumulation of F6P could also increase the steady-state level of G6P and therefore, the inhibition and detachment from the mitochondria of hexokinases. However, TIGAR binds to HK2 and promotes its activity at the mitochondria through a PFKFB-independent mechanism, especially under hypoxic conditions81. A gain-of-function mutant p53 was shown to increase the plasma membrane translocation of GLUT1 by affecting RHOA–Rho-associated protein kinase (ROCK) signalling82.
Perhaps the best and most comprehensive example of the reprogramming of glucose metabolism in cancer cells is found in hepatocellular carcinoma (HCC) (FIG. 6). The primary function of normal differentiated hepatocytes is to regulate the circulating levels of glucose. Therefore, their intracellular glucose metabolism is adapted to this function. Normal hepatocytes not only consume glucose but also export it; their predominant glucose transporter is GLUT2, which is characterized by high levels of reversible flux. The phosphorylation of glucose is mediated by the low-affinity hexokinase, HK4. Normal hepatocytes store glucose as glycogen or break it down depending on systemic needs. Hepatocytes are also distinguished from most other cells in the body by their ability to carry out gluconeogenesis, which runs in the opposite direction to glycolysis and is catalysed by enzymes that override the committed steps in glycolysis. These enzymes are glucose 6-phosphatase (G6Pase; also known as G6PC), fructose-1,6-bisphosphatase 1 (FBP1) and phosphoenolpyruvate carboxykinase (PEPCK; also known as PCK), which override the first, second and third committed steps of glycolysis, respectively (FIG. 6). In HCC, however, extensive reprogramming of these metabolic pathways occurs. First, the main glucose transporter in HCC is GLUT1, not GLUT2, which has a relatively high reversed glucose flux83. Second, and perhaps the most profound change, is the suppression of HK4 expression and the induction of the high-affinity hexokinase HK2 (REF. 84). Third, there is an isoform switch of aldolase, the enzyme that reversibly cleaves F1,6BP to dihydroxyacetone phosphate (DHAP) and G3P. There are three aldolase isoforms, which are encoded by three different genes (ALDOA, ALDOB and ALDOC). Aldolase B is the major isoform in normal differentiated hepatocytes, but its expression is suppressed in HCC, whereas aldolase A expression is induced85,86. Aldolase B catalyses the condensation of DHAP and G3P to F1,6BP more efficiently than aldolase A, whereas aldolase A is more efficient at cleaving F1,6BP compared with aldolase B87. Thus, aldolase B is better suited to gluconeogenesis in differentiated hepatocytes, whereas aldolase A is better suited to the glycolytic flux in HCC. Aldolase A is also the predominant isoform expressed in other types of cancer88. It is associated with F-actin, and recently, it was reported that PI3K activation mediates the dissociation of aldolase A from the actin cytoskeleton in an AKT-independent manner. The dissociation from the actin cytoskeleton increases the abundance and activity of aldolase A in the cytoplasm89. Fourth, the expression of gluconeogenic enzymes is markedly suppressed in HCC cells90. In addition, whereas the pyruvate kinase isoform, PKL, is the predominant isoform in normal hepatocytes, PKM2, which is more suited to cancer cells, is the main isoform in HCC cells91,92. Finally, to increase the flux of pyruvate to lactate the expression level of LDHA is increased in HCC cells compared with normal hepatocytes91. Similarly to HCC cells, clear cell renal cell carcinoma (ccRCC) cells undergo extensive reprogramming of glucose metabolism compared with their cells of origin93. Differentiated kidney cells, like differentiated hepatocytes, express gluconeogenic genes that are suppressed in ccRCC93. Most notable is the ubiquitous loss of FBP1 in ccRCC, which was attributed not only to the loss of its catalytic activity but specifically to the loss of its non-catalytic activity in the nucleus94.
Figure 6. Reprogramming of glucose metabolism in hepatocellular carcinoma.
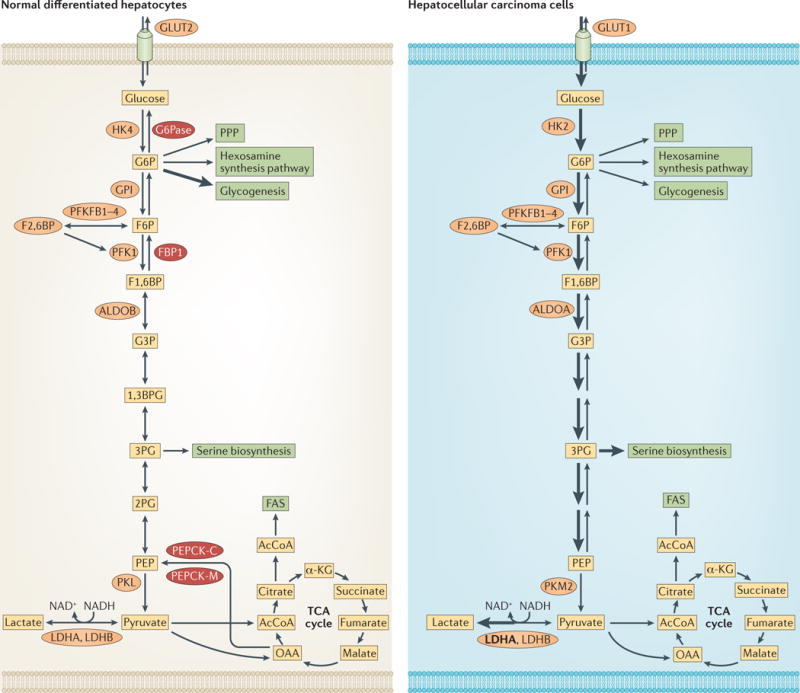
Differentiated hepatocytes use the reversal glucose transporter, GLUT2, for the uptake and export of glucose. The first committed step in glucose metabolism is attenuated because it is catalysed by the low-affinity hexokinase, HK4 (also known as glucokinase). The three committed steps in glucose metabolism could be reversed by the gluconeogenic enzymes (dark red). Glucose-6-phosphatase (G6Pase) dephosphorylates glucose-6-phosphate (G6P) back to glucose. Fructose-1,6-bisphosphatase (FBP1) dephosphorylates fructose-1,6-bisphosphate (F1,6BP) back to fructose-6-phosphate (F6P) and phosphoenolpyruvate carboxykinase (PEPCK) reverses the last committed step in glycolysis by converting oxaloacetate (OAA) to phosphoenolpyruvate (PEP), both in the mitochondria by the mitochondrial enzyme, PEPCK-M, and in the cytoplasm by PEPCK-C. In hepatocellular carcinoma the suppression of HK4 expression and the induction of the high-affinity hexokinase HK2 and by suppressing the expression of gluconeogenic (HCC), glucose metabolism is accelerated by the expression of GLUT1, enzymes. Unlike differentiated hepatocytes, HCC cells express relatively high levels of aldolase A (ALDOA), they express pyruvate kinase M2 (PKM2) instead of PKL, and they elevate the expression of lactate dehydrogenase A (LDHA; shown in bold). The thickness of the arrows indicates relative flux. 1,3BPG, 1,3-bisphosphoglycerate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; α-KG, α-ketoglutarate; AcCoA, acetyl-CoA; F2,6BP, fructose-2,6-bisphosphate; F6P, fructose-6-phosphate; FAS, fatty acid synthesis; G3P, glyceraldehyde-3-phosphate; GPI, glucose-6-phosphate isomerase; PFK1, phosphofructokinase 1; PFKFB, 6-phosphofructo 2-kinase/fructose-2,6-bisphosphatase; PPP, pentose phosphate pathway; TCA, tricarboxylic acid.
Interestingly, under certain conditions when glucose and nutrients are limited, cancer cells may adapt by using gluconeogenic enzymes to generate glycolytic intermediates. For example, the mitochondrial isoform of PEPCK, PEPCK-M (also known as PCK2), which is expressed in breast and lung cancer cells, as well as in several other cancer cell lines, was reported to be increased and activated under limited glucose conditions95–97. It was also reported that the cytosolic form of PEPCK, PEPCK-C (also known as PCK1) is overexpressed in colon cancer, and it accelerates the generation of glycolytic intermediates98. Surprisingly, cytoplasmic PEPCK-C also increased the use of glucose by a mechanism that may involve the activation of mTORC1 that is not fully understood.
Glucose metabolism and cancer cell survival
Glucose deprivation can lead to energetic stress and to the selective death of cancer cells compared with normal cells. However, the energetic stress per se may not be the main cause of this selective cell death. Glucose deprivation reduces the intracellular redox power of cancer cells because it decreases the production of NADPH from the PPP and from glucose-derived one-carbon metabolism. Thus, glucose deprivation markedly increases the intracellular level of ROS. Because, in general, highly metabolic cancer cells have higher levels of ROS than normal cells, they may be more vulnerable to ROS-induced cell death99. Interestingly, during extracellular matrix detachment, cells markedly decrease glucose uptake. Therefore, during solid tumour formation when cancer cells detach from the extracellular matrix or when breast cancer cells migrate to the lumen of the mammary gland (FIG. 7), they cease to consume glucose and undergo energetic stress and decreased ATP production100. In addition, NADPH levels decline, and thus, intracellular ROS levels are increased. Under these conditions, cancer cells may be more susceptible to cell death than other cells. However, as a consequence of the energetic stress and reduced intracellular ATP levels, AMP-activated protein kinase (AMPK), the sensor of ATP levels, is activated. In turn, AMPK inhibits acetyl-CoA carboxylase 1 (ACC1; also known as ACACA) and ACC2 (also known as ACACB), which are required for fatty acid synthesis (FAS) and for the inhibition of fatty acid oxidation (FAO). Thus, AMPK activation inhibits FAS while promoting FAO. By inhibiting FAS, AMPK reduces NADPH consumption. By promoting FAO, AMPK increases the flux from malate to pyruvate, which generates NADPH, and promotes the conversion of isocitrate into α-KG by isocitrate dehydrogenase 1 (IDH1), which also generates NADPH101 (FIG. 7). Thus, AMPK activation, following reduced glucose consumption, prevents a sharp increase in intracellular ROS that may kill the cancer cells.
Figure 7. Energetic and oxidative stress during solid tumour formation.
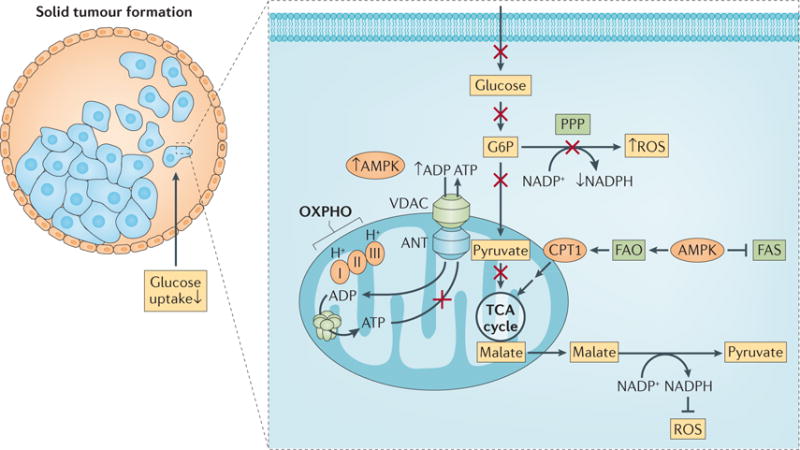
During migration to the lumen, tumour cells suppress glucose uptake. Consequently NADPH and ATP levels decline. The decline in NADPH level increases the intracellular level of reactive oxygen species (ROS), which can cause cell death. The decline in ATP level induces the activation of AMP-activated protein kinase (AMPK), which in turn inhibits fatty acid synthesis (FAS) and induces fatty acid oxidation (FAO). The inhibition of FAS reduces NADPH consumption, and the induction of FAO could generate malate independently of pyruvate. The conversion of malate into pyruvate regenerates NADPH. Therefore NADPH homeostasis is maintained to reduce the elevated ROS. A similar scenario may occur in circulating metastatic cells. ANT, adenine nucleotide translocator; CPT1, carnitine O-palmitoyltransferase; G6P, glucose-6-phosphate; OXPHO, oxidative phosphorylation; PPP, pentose phosphate pathway; TCA, tricarboxylic acid; VDAC, voltage-dependent anion channel.
Interestingly, similarly reduced glucose consumption and enhanced energetic stress may occur during metastasis, when cancer cells migrate from the primary tumour site to the metastatic site. Indeed, it was recently shown that during metastasis, ROS levels increased in circulating melanoma cells102. The reason for the increased ROS was not clearly elucidated, but it may be due to a decrease in glucose consumption. It was reported that to combat ROS and to maintain cell survival, the circulating melanoma cells increased NADPH levels by promoting the folate pathway in one-carbon metabolism102. However, it is also possible that AMPK is activated in the circulating melanoma cells and thus reduces NADPH consumption by FAS and regenerates NADPH through malate-to-pyruvate and isocitrate-to-α-KG reactions by promoting FAO.
Exploiting glucose metabolism for therapy
As described in this Review, it is clear that the reprogramming of glucose metabolism in cancer cells distinguishes them from their cells of origin and from most other normal cells in the body. This distinction is exploited for visualizing tumours in vivo by imaging their uptake of the radiolabelled glucose analogue [18F] fluoro-2-deoxyglucose (FDG) using positron emission tomography (PET). The question then arises, if elevated glucose metabolism in cancer cells is exploited to selectively detect them, can it be exploited to selectively eradicate them? The simplest answer to that question is that although glucose metabolism is increased in cancer cells, they mostly use the same glycolytic enzymes as normal cells. Thus, the inhibition of glycolytic enzymes as a cancer treatment may increase the risk of adverse and undesirable consequences. Nevertheless, it might be possible to target glucose transporters and glycolytic enzymes that are preferentially used by cancer cells compared with normal cells.
Glucose transporters
There are 14 glucose transporter isoforms, with different affinities for glucose and other hexoses, encoded by different genes in humans103–105. The four main classical glucose transporters are GLUT1, GLUT2 (also known as SLC2A2), GLUT3 (also known as SLC2A3) and GLUT4 (also known as SLC2A4), which have different affinities for glucose. In principle, glucose transporters are reversible, but of the four main glucose transporters, only GLUT2 is highly reversible. The relative expression levels of the different isoforms vary in different mammalian tissues and are often suited to the function of the tissue in the context of the whole body. Although GLUT1, which has a high affinity for glucose, is overexpressed in many cancers, cancer cells also express GLUT2 and GLUT3 (REF. 104). Interestingly, GLUT4 is a predominantly expressed isoform, which is required for glucose uptake in multiple myeloma106. As indicated earlier, oncogenic KRAS was reported to induce the expression of GLUT1, and cancer cells expressing oncogenic KRAS or oncogenic BRAF require GLUT1 (REF. 63). There are several small molecules that inhibit GLUT1, and they have been shown to selectively kill cancer cells in vitro107,108. However, the widespread expression of GLUT1 in different types of normal mammalian cells may preclude the clinical use of these inhibitors. Germline deletion of Glut1 in mice causes embryonic lethality, and mice with heterozygous deletion of Glut1 survive but have seizures, hypoglycorrhachia and impaired motor activity109. However, the consequences of systemic deletion of Glut1 in adult mice have yet to be determined.
GLUT1, together with other glucose transporters, is also a transporter for the oxidized form of vitamin C (ascorbic acid), dehydroascorbic acid (DHA)110,111. Inside cells, DHA is reduced to vitamin C and thus increases the consumption of reduced glutathione (GSH) and NADPH. It was recently reported that high doses of DHA deplete intracellular GSH levels in colorectal cancer (CRC) cells that carry activating KRAS or BRAF mutations and express high levels of GLUT1 (REF. 112). Consequently, intracellular levels of ROS were increased, which in turn oxidized and inhibited GAPDH112. The inhibition of GAPDH suppressed the subsequent steps in glycolysis and the LDHA-mediated conversion of NADH to NAD+ that is required for GAPDH activity. Further depletion of NAD+ was possibly due to the consumption of NAD+ in the activation of poly-(ADP-ribose) polymerase 1 (PARP1)112,113. It was therefore concluded that DHA selectively kills CRC cells that carry activating KRAS and BRAF mutations by inducing an energy crisis. These results suggest the possibility of selectively targeting cancer cells that express high levels of glucose transporters on their plasma membranes. High doses of vitamin C have been reported to benefit patients with cancer, and there have been several preclinical and clinical studies assessing the use of vitamin C114,115 (see also ClinicalTrials.gov), but the exact mechanism by which it exerts its effect is unknown. The recently proposed mechanism could raise several issues. First, the inhibition of GAPDH could divert glucose into the PPP to generate higher levels of NADPH, which would then reduce the oxidized glutathione to GSH and reactivate GAPDH. Second, oxidation by ROS would inhibit PKM2, and metabolites could then be diverted from glycolysis to the one-carbon metabolism cycle and the folate pathway that generates NADPH. Third, the depletion of ATP would activate AMPK, which increases NADPH levels by reducing FAS, which consumes NADPH, and inducing FAO, which generates ATP and NADPH.
Because cancer cells have high intracellular ROS levels, DHA could selectively kill cancer cells by increasing ROS above critical levels. Indeed, it was documented that cancer cells carrying activated RAS, BCR–ABL or activated PI3K–AKT signalling have high levels of ROS, and they can be selectively eradicated by exposure to β-phenethyl isothiocyanate (PEITC)116,117. PEITC is present in relatively high quantities in cruciferous vegetables, and it increases intracellular ROS levels by conjugation with GSH, which is then extruded from the cells, thereby rapidly depleting both cytoplasmic and mitochondrial GSH116,118.
Hexokinases
The ability of the FDG–PET scan to selectively detect cancer cells is not dependent solely on the high level of glucose transporters in cancer cells, because the reversible glucose transporters could export FDG unless it is phosphorylated and trapped inside the tumour cells. The phosphorylation of FDG is catalysed by hexokinases. As cancer cells have higher hexokinase activity than most normal cells, the FDG–PET scan could be an indirect readout for the high hexokinase activity of cancer cells.
The high hexokinase activity in cancer cells is due largely to the induction of HK2 expression. As HK2 is not expressed in most normal adult cells, its systemic ablation could selectively target cancer cells. Indeed, although germline deletion of Hk2 causes embryonic lethality, its systemic deletion in adult mice is well tolerated18,119. Moreover, systemic deletion of Hk2 after tumour onset inhibited tumour development in mouse models of cancer18. Importantly, no compensatory induction of HK1 expression was observed. These results have provided genetic proof of concept that HK2 can be systemically inhibited to treat cancer without adverse physiological consequences. However, because of the structural similarities between HK1 and HK2 (REFS 19–21), developing small-molecule inhibitors that preferentially inhibit HK2 could be challenging.
Nevertheless, the allosteric inhibition of HK1 and HK2 by their own product, G6P, could be exploited to preferentially target HK2. Although G6P inhibits both HK1 and HK2, its inhibitory effect on HK2 increases in the presence of orthophosphate, whereas the inhibition of HK1 is decreased21. Thus, it might be possible to develop G6P mimetics that preferentially inhibit HK2. Importantly, both HK1 and HK2 are high-affinity hexokinases, and normal cells may not need their full activation, whereas cancer cells do. As indicated above, HCC cells are distinct from normal hepatocytes by the suppression of HK4 expression and the induction of HK2 expression. Thus, systemic delivery of an HK2 inhibitor, even if it also inhibits HK1, could lead to accumulation of the drug at relatively high levels in the liver and could selectively target HCC cells and not normal hepatocytes, which do not express HK2. This strategy may also apply to other glycolytic enzymes that are expressed only in HCC cells and not in mature hepatocytes.
In addition to their roles in glucose metabolism, HK1 and HK2 promote cell survival by binding to mitochondria70,120–122. The mitochondrial binding is inhibited by G6P, and thus, a G6P mimetic or other small molecules that detach HK2 from mitochondria could increase the sensitivity of cancer cells to chemotherapeutic drugs.
The second committed step
Targeting the PFK1 isoforms that catalyse the second committed step in glycolysis might not be feasible because of their essential role in this process. However, it might be possible to preferentially inhibit the second committed step in cancer cells by an indirect mechanism. F2,6BP, which is an allosteric activator of PFK1 (FIG. 1), is generated by PFKFBs that possess both kinase and phosphatase activities. F2,6BP levels are dependent on the relative kinase and phosphatase activities of the different PFKFB isoforms. Thus, inhibiting their kinase activity while retaining their phosphatase activity will inhibit PFK1 activity by decreasing levels of F2,6BP. Cancer cells express relatively high levels of PFKFB3, which has higher kinase activity than phosphatase activity. Thus, small-molecule inhibitors that inhibit the kinase activity of PFKFB3 could preferentially inhibit PFK1 in cancer cells. Indeed, a selective inhibitor of PFKFB3 kinase was developed, and it selectively inhibited the proliferation of cancer cells123,124. PFKFB4 is also expressed in cancer cells, and its expression might be increased to compensate for inhibition of PFKFB3. Recently, PFKFB4-specific inhibitors were developed125, and therefore, both PFKFB3 and PFKFB4 could be inhibited to effectively inhibit PFK1 in cancer cells. As the PFKFB3 inhibitor was also shown to inhibit angiogenesis of endothelial cells and vessel sprouting126,127, systemic inhibition of PFKFB3 could also inhibit the supply of nutrients and oxygen to the tumour cells. The inhibition of PFK1 by PFKFB inhibitors could increase the flux to the PPP but also the intracellular level of G6P, and therefore could also inhibit hexokinase activity.
The last committed step
On the basis of recent results showing that a low level of PKM2 activity is beneficial for cancer progression, it is not clear whether inhibitors or activators of PKM2 should be developed for cancer therapy. Finally, inhibitors of lactate metabolism are being developed. LDHA inhibitors deplete NAD+ and therefore inhibit glycolysis. MCT1 and MCT4 inhibitors inhibit the export of intracellular lactate. These inhibitors could force the intracellular re-conversion of lactate into pyruvate and therefore deplete NAD+ levels, but they could also be cytotoxic by decreasing the intracellular pH below a crucial level (reviewed in REF. 31).
Serine biosynthesis and one-carbon metabolism
The inhibition of PHGDH in some cancer cells that carry amplification or overexpression of PHGDH could selectively target these cells and, recently, small-molecule inhibitors of PHGDH have been identified51,128. It was shown that these inhibitors preferentially inhibit the proliferation and tumorigenesis of cancer cells that have high levels of de novo serine biosynthesis. Finally, there are several inhibitors of the enzymes involved in one-carbon metabolism. Some of these inhibitors have been approved for cancer therapy and some are in clinical trials (reviewed in REF. 129).
Conclusions and future directions
The exploitation of the reprogrammed glucose metabolism to selectively target cancer cells may provide attractive and effective therapeutic approaches. However, one concern with this approach is the existence of multiple isoforms of the glycolytic enzymes and the fact that small-molecule inhibitors may not distinguish between the predominant isoform expressed by cancer cells and the isoforms expressed by normal cells. Even if relative specificity of the inhibitor is achieved, expression of the other isoforms might be induced in response to inhibition of the cancer-specific isoform.
Another concern is that the adaptation of metabolic pathways in conjunction with the use of alternative nutrients could overcome the inhibition of glucose metabolism in cancer cells. It is likely that in many cases in which glycolysis is inhibited, cells will respond by increasing OXPHO. One way to circumvent this compensation is to combine the glycolytic inhibitor with an inhibitor of OXPHO. Perhaps the most effective combination would be with metformin, which inhibits mitochondrial complex I, and which is being used for the treatment of diabetes.
To avoid the adaptation and resistance of cancer cells to glycolytic inhibitors, it might be better to use these inhibitors as an adjuvant therapy to an already-approved therapy to increase their efficacy. One attractive strategy is the combination of glycolytic inhibitors with immunotherapy. Recent reports have shown that there is competition between tumour-infiltrating T cells and tumour cells, whereby the highly glycolytic tumour cells deprive the microenvironment of glucose and other nutrients130,131. Because tumour-infiltrating activated T cells also have high levels of aerobic glycolysis, the glucose-depleted tumour microenvironment inhibits their activity, thereby dampening the ability of infiltrating T cells to respond to the tumour cells. Another known mechanism by which tumour cells evade T cell surveillance is through tumour cell surface expression of the ligand, programmed cell death 1 ligand 1 (PDL1; also known as CD274) for the programmed cell death 1 (PD1; also known as PDCD1) receptor on the surface of T cells132. Thus, the combination of a therapy that targets reprogrammed glucose metabolism in cancer cells with a recently approved immunotherapy, such as PD1- or PDL1-targeted antibodies, could further improve cancer therapy.
In sum, the growing interest in reprogrammed glucose metabolism in cancer cells will probably lead to innovative approaches that exploit the high glucose flux in cancer cells for cancer therapy. These approaches could be challenging because they will be required to overcome the compensatory and adaptive responses mediated by the tangled and sophisticated nature of glucose metabolism.
Acknowledgments
The work is supported by grants CA090764, and AG016927 from the National Institutes of Health and by a Veteran Affairs Merit Award BX000733 to N.H.
Glossary
- Aerobic glycolysis
Conversion of glucose into lactate that takes place in the presence of oxygen
- Oxidative phosphorylation
(OXPHO). A metabolic process of nutrient oxidation that generates ATP in mitochondria
- Pentose phosphate pathway
(PPP). A metabolic process in which glucose is used to generate NADPH and ribose-5-phosphate for nucleotide biosynthesis
- Hexosamine pathway
A metabolic process in which an amine group is added to hexoses to generate a sugar donor for the glycosylation of proteins
- One-carbon metabolism
Biochemical reactions catalysed by a set of enzymes and coenzymes in which the transfer of one-carbon groups occurs to provide precursors for purine synthesis and the methionine cycle
- Tricarboxylic acid cycle
(TCA cycle). A series of chemical reactions that start with oxidation of acetyl-CoA to generate precursors for certain amino acids and a reducing agent for oxidative phosphorylation
- Folate cycle
A metabolic pathway, included in one-carbon metabolism, that uses tetrahydrofolates as cofactors and precursors for purine synthesis and the methionine cycle
- Methionine cycle
Part of one-carbon metabolism, the methionine cycle generates S-adenosylmethionine, which is a substrate for methyltransferases
- Hypoglycorrhachia
An abnormally low glucose level in the cerebrospinal fluid
Footnotes
Competing interests statement
The author declares no competing interests.
References
- 1.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warburg O. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology, Berlin-Dahlen. Constable; 1930. [Google Scholar]
- 3.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 4.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 5.Weinhouse S. On respiratory impairment in cancer cells. Science. 1956;124:267–269. doi: 10.1126/science.124.3215.267. [DOI] [PubMed] [Google Scholar]
- 6.Weinhouse S. Studies on the fate of isotopically labeled metabolites in the oxidative metabolism of tumors. Cancer Res. 1951;11:585–591. References 3–6 include the original Warburg theory that respiration is impaired in cancer cells followed by the debate between Warburg and Weinhouse on whether respiration is impaired in cancer cells, and a paper by Weinhouse describing the use of isotope tracing to determine oxidative metabolism in tumours. [PubMed] [Google Scholar]
- 7.Weinhouse S. Oxidative metabolism of neoplastic tissues. Adv Cancer Res. 1955;3:269–325. doi: 10.1016/s0065-230x(08)60922-7. [DOI] [PubMed] [Google Scholar]
- 8.Weinhouse S. Glycolysis, respiration, and anomalous gene expression in experimental hepatomas: G.H.A Clowes memorial lecture. Cancer Res. 1972;32:2007–2016. [PubMed] [Google Scholar]
- 9.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 10.Crabtree HG. The carbohydrate metabolism of certain pathological overgrowths. Biochem J. 1928;22:1289–1298. doi: 10.1042/bj0221289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39:347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Timm KN, et al. Hyperpolarized [U-2H, U-13C] Glucose reports on glycolytic and pentose phosphate pathway activity in EL4 tumors and glycolytic activity in yeast cells. Magn Reson Med. 2015;74:1543–1547. doi: 10.1002/mrm.25561. [DOI] [PubMed] [Google Scholar]
- 13.Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE. 2005;312:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- 14.Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta. 2010;1800:80–95. doi: 10.1016/j.bbagen.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jozwiak P, Forma E, Brys M, Krzeslak A. O-GlcNAcylation and metabolic reprogramming in cancer. Front Endocrinol (Lausanne) 2014;5:145. doi: 10.3389/fendo.2014.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zois CE, Favaro E, Harris AL. Glycogen metabolism in cancer. Biochem Pharmacol. 2014;92:3–11. doi: 10.1016/j.bcp.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Patra KC, Hay N. Hexokinase 2 as oncotarget. Oncotarget. 2013;4:1862–1863. doi: 10.18632/oncotarget.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patra KC, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213–228. doi: 10.1016/j.ccr.2013.06.014. A demonstration that it is feasible to systemically delete a major glycolytic enzyme for cancer therapy in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–4696. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- 20.Sui D, Wilson JE. Structural determinants for the intracellular localization of the isozymes of mammalian hexokinase: intracellular localization of fusion constructs incorporating structural elements from the hexokinase isozymes and the green fluorescent protein. Arch Biochem Biophys. 1997;345:111–125. doi: 10.1006/abbi.1997.0241. [DOI] [PubMed] [Google Scholar]
- 21.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 22.Irwin DM, Tan H. Molecular evolution of the vertebrate hexokinase gene family: identification of a conserved fifth vertebrate hexokinase gene. Comp Biochem Physiol Part D Genom Proteom. 2008;3:96–107. doi: 10.1016/j.cbd.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Guo C, et al. Coordinated regulatory variation associated with gestational hyperglycaemia regulates expression of the novel hexokinase HKDC1. Nat Commun. 2015;6:6069. doi: 10.1038/ncomms7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinohara Y, Yamamoto K, Kogure K, Ichihara J, Terada H. Steady state transcript levels of the type II hexokinase and type 1 glucose transporter in human tumor cell lines. Cancer Lett. 1994;82:27–32. doi: 10.1016/0304-3835(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 25.Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem. 2001;276:43407–43412. doi: 10.1074/jbc.M108181200. [DOI] [PubMed] [Google Scholar]
- 26.Tsai HJ, Wilson JE. Functional organization of mammalian hexokinases: both N- and C-terminal halves of the rat type II isozyme possess catalytic sites. Arch Biochem Biophys. 1996;329:17–23. doi: 10.1006/abbi.1996.0186. [DOI] [PubMed] [Google Scholar]
- 27.Israelsen WJ, Vander Heiden MG. Pyruvate kinase: function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51. doi: 10.1016/j.semcdb.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamada K, Noguchi T. Nutrient and hormonal regulation of pyruvate kinase gene expression. Biochem J. 1999;337:1–11. [PMC free article] [PubMed] [Google Scholar]
- 29.Israelsen WJ, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409. doi: 10.1016/j.cell.2013.09.025. This paper showed that PKM2 deficiency accelerates mammary tumour formation in a mouse model of breast cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cortes-Cros M, et al. M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proc Natl Acad Sci USA. 2013;110:489–494. doi: 10.1073/pnas.1212780110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang YH, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–1323. doi: 10.1016/j.cell.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vander Heiden MG, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valvona CJ, Fillmore HL, Nunn PB, Pilkington GJ. The regulation and function of lactate dehydrogenase a: therapeutic potential in brain tumor. Brain Pathol. 2016;26:3–17. doi: 10.1111/bpa.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123:3685–3692. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costa Leite T, Da Silva D, Guimaraes Coelho R, Zancan P, Sola-Penna M. Lactate favours the dissociation of skeletal muscle 6-phosphofructo-1-kinase tetramers down-regulating the enzyme and muscle glycolysis. Biochem J. 2007;408:123–130. doi: 10.1042/BJ20070687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res. 2006;66:5216–5223. doi: 10.1158/0008-5472.CAN-05-4193. [DOI] [PubMed] [Google Scholar]
- 37.Fukumura D, et al. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001;61:6020–6024. [PubMed] [Google Scholar]
- 38.Shi Q, et al. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene. 2001;20:3751–3756. doi: 10.1038/sj.onc.1204500. [DOI] [PubMed] [Google Scholar]
- 39.Baumann F, et al. Lactate promotes glioma migration by TGF-β2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009;11:368–380. doi: 10.1215/15228517-2008-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rozhin J, Sameni M, Ziegler G, Sloane BF. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res. 1994;54:6517–6525. [PubMed] [Google Scholar]
- 41.Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. 2009;92:329–333. doi: 10.1016/j.radonc.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 42.Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71:6921–6925. doi: 10.1158/0008-5472.CAN-11-1457. [DOI] [PubMed] [Google Scholar]
- 43.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 44.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Locasale JW, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. References 45 and 46 showed overexpression of PHGDH in cancer cells, which diverts metabolism into the serine biosynthesis pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeNicola GM, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gromova I, et al. High level PHGDH expression in breast is predominantly associated with keratin 5-positive cell lineage independently of malignancy. Mol Oncol. 2015;9:1636–1654. doi: 10.1016/j.molonc.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun L, et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015;25:429–444. doi: 10.1038/cr.2015.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen J, et al. Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget. 2013;4:2502–2511. doi: 10.18632/oncotarget.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pacold ME, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452–458. doi: 10.1038/nchembio.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fan J, et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis CA, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hitosugi T, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22:585–600. doi: 10.1016/j.ccr.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dunaway GA, Kasten TP, Sebo T, Trapp R. Analysis of the phosphofructokinase subunits and isoenzymes in human tissues. Biochem J. 1988;251:677–683. doi: 10.1042/bj2510677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ros S, Schulze A. Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013;1:8. doi: 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chesney J. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase and tumor cell glycolysis. Curr Opin Clin Nutr Metab Care. 2006;9:535–539. doi: 10.1097/01.mco.0000241661.15514.fb. [DOI] [PubMed] [Google Scholar]
- 58.Yi W, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anastasiou D, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seidler NW. Basic biology of GAPDH. Adv Exp Med Biol. 2013;985:1–36. doi: 10.1007/978-94-007-4716-6_1. [DOI] [PubMed] [Google Scholar]
- 61.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519–9525. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- 63.Yun J, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. This paper showed that the oncogenic activity of KRAS or BRAF requires GLUT1 and that glucose deprivation can induce oncogenic KRAS or BRAF mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barthel A, et al. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- 65.Foran PG, et al. Protein kinase B stimulates the translocation of GLUT4 but not GLUT1 or transferrin receptors in 3T3-L1 adipocytes by a pathway involving SNAP-23, synaptobrevin-2, and/or cellubrevin. J Biol Chem. 1999;274:28087–28095. doi: 10.1074/jbc.274.40.28087. [DOI] [PubMed] [Google Scholar]
- 66.Von der Crone S, et al. Glucose deprivation induces Akt-dependent synthesis and incorporation of GLUT1, but not of GLUT4, into the plasma membrane of 3T3-L1 adipocytes. Eur J Cell Biol. 2000;79:943–949. doi: 10.1078/0171-9335-00118. [DOI] [PubMed] [Google Scholar]
- 67.Katabi MM, Chan HL, Karp SE, Batist G. Hexokinase type II: a novel tumor-specific promoter for gene-targeted therapy differentially expressed and regulated in human cancer cells. Hum Gene Ther. 1999;10:155–164. doi: 10.1089/10430349950018959. [DOI] [PubMed] [Google Scholar]
- 68.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta. 2002;1555:14–20. doi: 10.1016/s0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- 69.Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gottlob K, et al. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 72.Minchenko A, et al. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem. 2002;277:6183–6187. doi: 10.1074/jbc.M110978200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Atsumi T, et al. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002;62:5881–5887. [PubMed] [Google Scholar]
- 74.Minchenko OH, et al. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie. 2005;87:1005–1010. doi: 10.1016/j.biochi.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 75.Kessler R, Bleichert F, Warnke JP, Eschrich K. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) is up-regulated in high-grade astrocytomas. J Neurooncol. 2008;86:257–264. doi: 10.1007/s11060-007-9471-7. [DOI] [PubMed] [Google Scholar]
- 76.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18:5546–5553. doi: 10.1158/1078-0432.CCR-12-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hsieh AL, Walton ZE, Altman BJ, Stine ZE, Dang CV. MYC and metabolism on the path to cancer. Semin Cell Dev Biol. 2015;43:11–21. doi: 10.1016/j.semcdb.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC metabolism, and cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. 2013;18:617–633. doi: 10.1016/j.cmet.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356:197–203. doi: 10.1016/j.canlet.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci USA. 2012;109:20491–20496. doi: 10.1073/pnas.1206530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang C, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karim S, Adams DH, Lalor PF. Hepatic expression and cellular distribution of the glucose transporter family. World J Gastroenterol. 2012;18:6771–6781. doi: 10.3748/wjg.v18.i46.6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guzman G, et al. Evidence for heightened hexokinase II immunoexpression in hepatocyte dysplasia and hepatocellular carcinoma. Dig Dis Sci. 2015;60:420–426. doi: 10.1007/s10620-014-3364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Castaldo G, et al. Quantitative analysis of aldolase A mRNA in liver discriminates between hepatocellular carcinoma and cirrhosis. Clin Chem. 2000;46:901–906. [PubMed] [Google Scholar]
- 86.Wang Y, et al. Identification of four isoforms of aldolase B down-regulated in hepatocellular carcinoma tissues by means of two-dimensional western blotting. In Vivo. 2011;25:881–886. [PubMed] [Google Scholar]
- 87.Penhoet EE, Rutter WJ. Catalytic and immunochemical properties of homomeric and heteromeric combinations of aldolase subunits. J Biol Chem. 1971;246:318–323. [PubMed] [Google Scholar]
- 88.Asaka M, et al. Alteration of aldolase isozymes in serum and tissues of patients with cancer and other diseases. J Clin Lab Anal. 1994;8:144–148. doi: 10.1002/jcla.1860080306. [DOI] [PubMed] [Google Scholar]
- 89.Hu H, et al. Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell. 2016;164:433–446. doi: 10.1016/j.cell.2015.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang B, Hsu SH, Frankel W, Ghoshal K, Jacob ST. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor γ, coactivator 1α. Hepatology. 2012;56:186–197. doi: 10.1002/hep.25632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wong CC, et al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS ONE. 2014;9:e115036. doi: 10.1371/journal.pone.0115036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pan D, Mao C, Wang YX. Suppression of gluconeogenic gene expression by LSD1-mediated histone demethylation. PLoS ONE. 2013;8:e66294. doi: 10.1371/journal.pone.0066294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Massari F, et al. Metabolic alterations in renal cell carcinoma. Cancer Treat Rev. 2015;41:767–776. doi: 10.1016/j.ctrv.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 94.Li B, et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251–255. doi: 10.1038/nature13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mendez-Lucas A, Hyrossova P, Novellasdemunt L, Vinals F, Perales JC. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability. J Biol Chem. 2014;289:22090–22102. doi: 10.1074/jbc.M114.566927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Leithner K, et al. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene. 2015;34:1044–1050. doi: 10.1038/onc.2014.47. [DOI] [PubMed] [Google Scholar]
- 97.Vincent EE, et al. Mitochondrial phosphoenolpyruvate carboxykinase regulates metabolic adaptation and enables glucose-independent tumor growth. Mol Cell. 2015;60:195–207. doi: 10.1016/j.molcel.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 98.Montal ED, et al. PPEPCK coordinates the regulation of central carbon metabolism to promote cancer cell growth. Mol Cell. 2015;60:571–583. doi: 10.1016/j.molcel.2015.09.025. References 95–98 showed that the gluconeogenic enzymes, PEPCK-M and PEPCK-C, are expressed in some cancer cells to promote tumour growth when glucose is limited. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nogueira V, Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res. 2013;19:4309–4314. doi: 10.1158/1078-0432.CCR-12-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schafer ZT, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. References 100 and 101 showed that extracellular matrix detachment induces energetic and oxidative stress, and AMPK activation. AMPK activation is required to maintain NADPH homeostasis to promote cell survival during extracellular matrix detachment and solid tumour formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Piskounova E, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650–654. doi: 10.1097/CCO.0b013e328356da72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Szablewski L. Expression of glucose transporters in cancers. Biochim Biophys Acta. 2013;1835:164–169. doi: 10.1016/j.bbcan.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 105.Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34:121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McBrayer SK, et al. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood. 2012;119:4686–4697. doi: 10.1182/blood-2011-09-377846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chan DA, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3:94ra70. doi: 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu Y, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012;11:1672–1682. doi: 10.1158/1535-7163.MCT-12-0131. [DOI] [PubMed] [Google Scholar]
- 109.Wang D, et al. A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet. 2006;15:1169–1179. doi: 10.1093/hmg/ddl032. [DOI] [PubMed] [Google Scholar]
- 110.Rumsey SC, et al. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J Biol Chem. 1997;272:18982–18989. doi: 10.1074/jbc.272.30.18982. [DOI] [PubMed] [Google Scholar]
- 111.Corpe CP, Eck P, Wang J, Al-Hasani H, Levine M. Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J Biol Chem. 2013;288:9092–9101. doi: 10.1074/jbc.M112.436790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yun J, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Uetaki M, Tabata S, Nakasuka F, Soga T, Tomita M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci Rep. 2015;5:13896. doi: 10.1038/srep13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen Q, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci USA. 2008;105:11105–11109. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mikirova N, Casciari J, Rogers A, Taylor P. Effect of high-dose intravenous vitamin C on inflammation in cancer patients. J Transl Med. 2012;10:189. doi: 10.1186/1479-5876-10-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Trachootham D, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 117.Nogueira V, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xu K, Thornalley PJ. Involvement of glutathione metabolism in the cytotoxicity of the phenethyl isothiocyanate and its cysteine conjugate to human leukaemia cells in vitro. Biochem Pharmacol. 2001;61:165–177. doi: 10.1016/s0006-2952(00)00526-8. [DOI] [PubMed] [Google Scholar]
- 119.Heikkinen S, et al. Hexokinase II-deficient mice Prenatal death of homozygotes without disturbances in glucose tolerance in heterozygotes. J Biol Chem. 1999;274:22517–22523. doi: 10.1074/jbc.274.32.22517. [DOI] [PubMed] [Google Scholar]
- 120.Majewski N, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 121.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004;24:730–740. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Robey RB, Hay N. Mitochondrial hexokinases: guardians of the mitochondria. Cell Cycle. 2005;4:654–658. doi: 10.4161/cc.4.5.1678. [DOI] [PubMed] [Google Scholar]
- 123.Clem B, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008;7:110–120. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 124.Clem BF, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. 2013;12:1461–1470. doi: 10.1158/1535-7163.MCT-13-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chesney J, Clark J, Lanceta L, Trent JO, Telang S. Targeting the sugar metabolism of tumors with a first-in-class 6-phosphofructo-2-kinase (PFKFB4) inhibitor. Oncotarget. 2015;6:18001–18011. doi: 10.18632/oncotarget.4534. References 124 and 125 describe inhibitors of PFKFB3 and PFKFB4 that can be used for cancer therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.De Bock K, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 127.Schoors S, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 128.Mullarky E, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci USA. 2016;113:1778–1783. doi: 10.1073/pnas.1521548113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chang CH, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ho PC, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. References 130 and 131 showed that there is metabolic competition in the microenvironment between tumour-infiltrating T cells and the tumour cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]