

Abstract
Like many other environments, Lake Mendota, WI, USA, is populated by many thousand microbial species. Only about 1,000 of these constitute between 80 and 99% of the total microbial community, depending on the season, whereas the remaining species are rare. The functioning and resilience of the lake ecosystem depend on these microorganisms, and it is therefore important to understand their dynamics throughout the year. We propose a two-layered set of dynamic mathematical models that capture and interpret the yearly abundance patterns of the species within the metapopulation. The first layer analyzes the interactions between 14 subcommunities (SCs) that peak at different times of the year and together contain all species whereas the second layer focuses on interactions between individual species and SCs. Each SC contains species from numerous families, genera, and phyla in strikingly different abundances. The dynamic models quantify the importance of environmental factors in shaping the dynamics of the lake’s metapopulation and reveal positive or negative interactions between species and SCs. Three environmental factors, namely temperature, ammonia/phosphorus, and nitrate+nitrite, positively affect almost all SCs, whereas by far the most interactions between SCs are inhibitory. As far as the interactions can be independently validated, they are supported by literature information. The models are quite robust and permit predictions of species abundances over many years both, under the assumption that conditions do not change drastically, or in response to environmental perturbations.
Introduction
Lake Mendota, WI, is home to over 18,000 microbial Operational Taxonomic Units (OTUs). The OTUs are defined at the 97% 16S rRNA gene identity level and serve as proxies for species.1 1,140 OTUs (5%) constitute between 80 and 99% of the total microbial community (Supplementary Figure S2), depending on the time of the year, whereas the remaining OTUs are rare. The OTU composition of the community changes markedly throughout the year, and the dynamics of these changes is an important determinant of the functionality of the lake. In particular, it has been shown that higher microbial species diversity is typically associated with more robust and resilient ecosystems.2 Thus, if the normal, healthy interaction dynamics could be quantified, then one could possibly develop tests, based on sentinels or early biomarkers, predicting ecosystem health or potential problems in the near future. The challenge is that the interaction dynamics of OTUs is difficult to assess owing to their sheer number, and because most of the microbes cannot be cultured in the laboratory.3 Simple algebra says that potentially over 300,000,000 pairwise interactions would have to be considered, because the interactions can easily be ‘asymmetric’ in a sense that the effect of OTU-A on OTU-B is different from the reverse effect.
Two approaches are currently used to infer the relationships among microbial species from 16S-rRNA amplicon data.4 The first establishes correlation networks that are based on the presence, absence, or abundance of the species across multiple locations or time points.5–9 The vertices represent species, whereas the edges represent either pairwise or complex relationships. Pairwise interactions are typically characterized with a similarity index or a modified Pearson Correlation Coefficient (PCC),5–9 while complex relationships are derived from regression or rule-based networks.4,10 Although static correlation networks can address large and complex communities of thousands of species across multiple environments,5,10 they do not capture potentially important dynamic trends and typically ignore the asymmetry of relationships between species.
The second approach utilizes differential equations to reconstruct dynamic networks.11–21 These equations often include terms that describe growth and decay, pairwise interactions between species, and the effects of nutrients or environment. Most of these approaches have been linear owing to the ease of parameter estimation. Among the nonlinear approaches, the Lotka–Volterra (LV) model has been used extensively,12–15,17,19–21 because it is easily interpreted and allows the incorporation of time-dependent external perturbation.22 The main challenge of this approach is the estimation of parameters.
Here we propose slightly modified LV models, which become manageable owing to a novel manner of parameter estimation based on linear regression.23 The models capture not only the metapopulation dynamics of the more than 1,000 highly abundant species in Lake Mendota, but also the pairwise interactions between individual OTUs and other SCs. To the best of our knowledge, this is the first time that LV models of the magnitude addressed here are applied to a real-world system.
Results
Yearly abundances of 14 subcommunities
The top 200 parametric instantiations of the SC model (see Materials and Methods) are able to capture the dynamic trends well (Figure 1). They also correlate well with the trends of the observed abundances. Although the figure only shows the abundances during 2 years, the models successfully run for at least 50 consecutive years, if the conditions do not change drastically (not shown).
Figure 1.
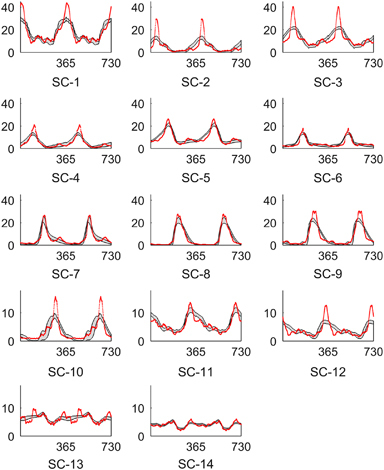
Predicted annual abundance of 14 subcommunities of bacteria. The x axis shows the days of a 2-year period, and the y axis shows the abundances of the subcommunities. The mean of the observed values measured between year 2000 and 2011 (red) and the maximum and minimum values of the ensemble of our top 200 parametric instantiations of the SC model (shaded gray) are shown. Note different scales of the y axes.
Twelve of the 14 SCs peak once per year, whereas SC 13 and SC 14 peaks twice (Figure 1 and Supplementary Figure S5). Supplementary Figure S5 shows that the fold-change profiles within SCs are very similar. It also reveals that the peaks are highly relevant, with an abundance that is 3- to 10-fold higher at the peak than the minimum abundance. Throughout the year, the total abundances of SC1–SC13 constitute 88.4–94.9% of the entire population (Supplementary Figure S6a).
Pairwise interactions between the 14 subcommunities
Using the best 200 model instantiations, we computed the means and s.d. of the parameter values (Figure 2). Among them, the estimated αij and βik values, when normalized (divided by − αii), are consistent with very small s.d. About two-thirds of all αij’s (62–67% in each model) are negative, which suggests strong competition between SCs (Supplementary Figure S7). Intriguingly, the terms βik×Xk are usually much higher than the corresponding terms αij×Xj: although Xj and Xk change over time, the median values of normalized αij×Xj and βik×Xk are 2.8 and 9.6, respectively. Expressed differently, the environmental conditions appear to have a greater effect (per unit of abundance) on the abundance of a SC than other SCs, at least qualitatively (Figures 2 and 3).
Figure 2.
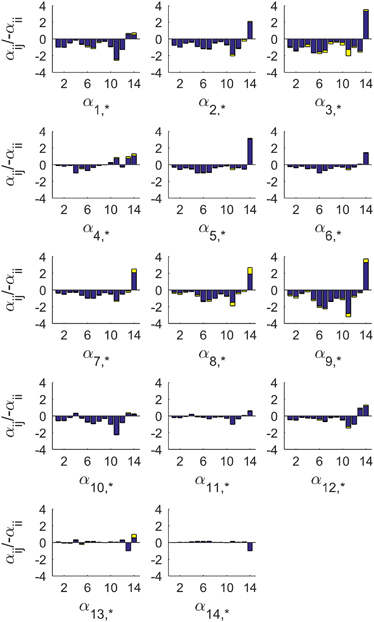
Estimated interactions (αij) between pairs of the 14 subcommunities (SCs). The y axis shows αij values, scaled by −1/αii, of SC 1 to 14. Each blue bar shows the mean value and each yellow extension represents the s.d. of the top 200 parametric instantiations of the SC model. It is evident that most interaction parameters are negative, at least for SC1 to SC12. SC14, and to a lesser degree SC13, are different in their effects and in how they are affected.
Figure 3.
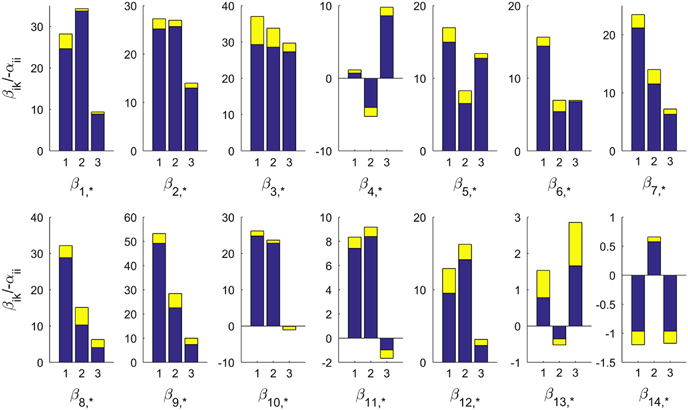
Estimated environmental effects (βik) on the 14 subcommunities. The plot shows βik values, scaled by −1/αii of SC 1 to 14. Each bar shows the mean value and each yellow extension represents the s.d. of the top 200 parametric instantiations of the SC model. The three environmental conditions are ordered as water temperature, ammonia, and nitrate–nitrite. Most environmental effects are positive.
Interestingly, the means of the αii values for SC 7, 8, and 9 are the smallest in magnitude (Supplementary Figure S7). This result may reflect that these SCs, which peak in July through September when the water temperature is highest and biomass is higher, have the lowest death rates due to ‘crowding.’
The αij matrix is asymmetrical, because interaction effects are not necessarily reciprocal. Among the pairs αij and αji, about 75%, 4%, and 21% are −/−, +/− or +/+, respectively. The number of positive αji values is smaller than in studies of communities growing in human or mouse gut or on spoiling pork,13,14,21 suggesting that the availability of food sources may affect the types of relationships differently within each habitat.
Except for SC 4, 11, 13 and 14, all SCs are positively affected by environmental conditions (Figures 3 and 4). Ammonia/phosphorus, which rapidly declines in April, negatively affects SC 4, which peaks in April. Nitrate+nitrite, which is low in November, negatively affects SC 11, which peaks in November. In SC 11, two of the five top OTUs belong to the family Oxalobacteraceae and one to the family ACK-M1. These families are either responsive to ammonia24 or have members that fix nitrogen25 (Supplementary Figure S10, Supplementary Table S5). Interestingly, SC 13 and 14 have very small (β ik) values, indicating relative tolerance to variations in environmental conditions.
Figure 4.
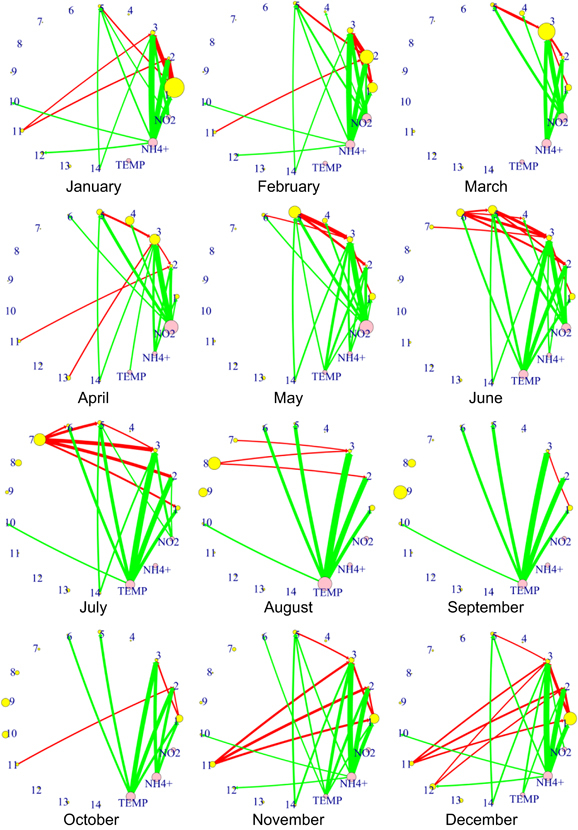
Networks of strongest interactions among the 14 subcommunities, as well as environmental conditions, by month. The interaction network for each month was computed from the best subcommunity model and weighted by the monthly average abundance of the subcommunities. The mean and s.d. of all values were computed, and only those interactions were retained that are at least one s.d. away from the mean. This cutoff corresponds to 31.73% of all interactions. Networks for other cutoffs are shown in the Supplementary Information. Each vertex size is proportional to the size of the subcommunity (yellow) or the abundance of environmental conditions (pink). The thickness of each edge is proportional to the strength of a positive (green) or negative (red) interaction.
To summarize, the pairwise interactions between SCs are mostly negative, whereas the environmental effects on SCs are mostly positive.
Bacterial distribution within subcommunities
Almost all (18,642) of the identified OTUs were classified into 63 phyla; only 12 OTUs do not have a phylum classification. For each OTU, we computed the average abundance over all data points, and for each phylum, we summed the abundances for all OTUs, then ranked them based on the total abundance. The top seven phyla, accounting for 92.6–99.9% of the population are: Actinobacteria, dominant in SC1, 3, 4, and 13; Proteobacteria in SC2, 7–11 and 14; and Bacteroidetes in SC5 and 12 (Table 1). Notwithstanding the dominance of particular phyla, each bacterial SC contains bacterial OTUs from a broad range of taxonomic groups. This result is not surprising, because each SC has to execute a wide array of tasks. It also reveals why clustering by taxonomy is not an effective strategy for characterizing the interaction dynamics in the lake.
Table 1. Distribution of the top 7 phyla within the 14 bacterial subcommunities (ordered from top to bottom) examined in our model.
| Peak Time | Actino. | Proteo. | Bactero. | Cyano. | Verruco. | Chlorobi. | Plancto. |
|---|---|---|---|---|---|---|---|
| January | 54.86 | 42.85 | 0.80 | 1.17 | 0.32 | 0.01 | 0.00 |
| February | 4.77 | 47.34 | 21.60 | 26.20 | 0.07 | 0.01 | 0.01 |
| March | 66.51 | 23.67 | 4.29 | 0.17 | 4.38 | 0.00 | 0.97 |
| April | 50.54 | 22.72 | 25.17 | 1.19 | 0.12 | 0.00 | 0.26 |
| May | 31.75 | 16.82 | 47.31 | 4.03 | 0.03 | 0.01 | 0.04 |
| June | 34.90 | 22.25 | 33.02 | 2.31 | 7.46 | 0.00 | 0.05 |
| July | 3.33 | 34.16 | 21.80 | 26.24 | 13.80 | 0.02 | 0.66 |
| August | 8.20 | 28.13 | 11.21 | 19.85 | 21.76 | 8.04 | 2.80 |
| September | 21.74 | 23.01 | 13.88 | 17.90 | 16.63 | 3.40 | 3.44 |
| October | 8.20 | 31.48 | 24.68 | 18.14 | 10.05 | 0.64 | 6.81 |
| November | 19.87 | 59.15 | 13.50 | 0.24 | 4.93 | 2.16 | 0.16 |
| December | 22.34 | 24.79 | 46.40 | 0.07 | 6.40 | 0.00 | 0.00 |
| Twice | 99.99 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Twice | 32.62 | 37.61 | 29.21 | 0.05 | 0.50 | 0.01 | 0.01 |
The abundance of each phylum, scaled by the abundance of all phyla in the same subcommunity, is shown. The phyla in columns 2–8 are Actinobacteria, Proteobacteria, Cyanobacteria, Verrucomicrobia, Chlorobi, and Planctomycetes, respectively.
Using the software PICRUSt26 and the Greengenes Database,27 we assigned KEGG functions to the the OTUs presented in 14 SCs. In total, 42.2% of the total community were mapped to KEGG pathways. We observed specific enrichment of certain pathways in SCs, as shown in Supplementary Figure S13. Data are available at http://www.bst.bme.gatech.edu/research.php.
Abundances of individual OTUs
We assessed the abundances of individual OTUs using three models, as described in the Methods Section. Among the top 1,140 OTUs, 89.3% can be predicted successfully when the individual OTU is implemented as a new group and the parameters are reoptimized (Model #3; Supplementary Figure S14). Interestingly, the αij×Xj and βik×Tk terms of OTUs belonging to the same phylum, class, or order cluster together and are significantly different from random clusters (Supplementary Table S4). For the top 1,140 OTUs, we extracted 922 OTUs whose abundances are predicted best by Model #3. We found that the pairs αi,sc/αi,sc are often positively correlated, whereas the pairs βik/αi,sc are often negatively correlated (data not shown). This result suggests that the change in the abundance of an OTU is driven either by competition with other bacteria in the community or by positive influences from the environment. Examples of the dynamics of individual OTUs are given in Figure 5. The Supplement Information provides further details.
Figure 5.
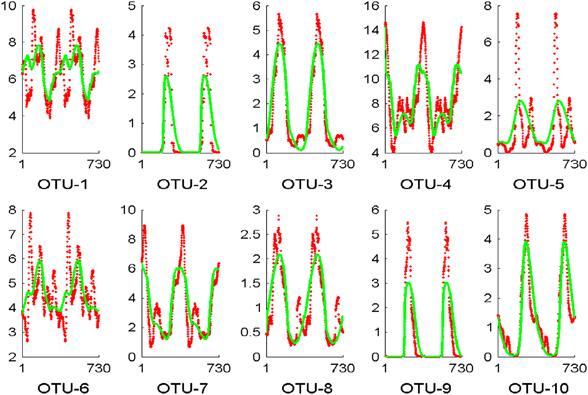
Predictions of abundances of individual OTUs plotted over a period of 2 years. Each subplot shows the mean of observed abundances (red dots) and the annual predicted values of Model #3 (green). The x axis shows the day within a 2-year period, and the y axis represents the abundances as percentages of the overall population.
The individual OTU–SC interaction network adds a second layer to our investigation. The first layer (SC model) captures pairwise interactions between SCs that reflect average effects contributed by all OTUs in each SC. At the second layer, OTU–SC interactions describe the effects of each SC and of the environmental conditions on an individual OTU. As an example, OTU#141903 (a member of the family Nitrosomonadaceae) has a large positive βi,2 value, which indicates that it is strongly, positively affected by ammonia. Although we cannot assign this OTU to a more specific taxonomic group, previous studies suggest that all cultivated representatives of this group are able to oxidize ammonia,28 which reflects our result. OTU#517152 (a member of the genus Roseomonas) has a small negative αii value and a large positive βi,2 value, suggesting that this species has a relatively low death rate and is strongly affected by temperature. Various members of this genus are well-studied aquatic organisms. They were described as slow growing29 and growing better at 25–28 °C,29 than in colder water, in some cases thriving up to 42 °C.30 The Supplementary Information offers further discussions (Supplementary Table S6).
Among these results, we identified 33 OTUs with outliers in βik or αi,sc values and good abundance prediction results and searched the literature for evidence to support or reject our predictions. We found indirect evidence to support the prediction of 15 OTUs and evidence for one, suggesting that further investigation is needed (Supplementary Tables S7a,b). For the remaining OTUs, little is known about their characteristics. These results are summarized in the Supplementary Information. A table with notable interactions among SC-OTUs is available at http://www.bst.bme.gatech.edu/research.php.
Discussion
Naturally occurring microbial consortia in lakes, and elsewhere, follow annual cycles, where species abundances are correlated with seasonal changes in environmental conditions.31–34 It is important to understand this dynamics because it is, without doubt, associated with the health of the ecosystem.
Recent metagenomic sequencing technologies have revolutionized this line of investigation. However, while OTU abundances are informative, they do not by themselves convey the dynamics within a metapopulation, but require computational analysis. We perform such an analysis here with LV models (Supplementary Figures S8–S12 and S15, Supplementary Tables S1–S3 and S5). Our models suggest that the dynamics of OTUs can be described in terms of the parameters αij and βik, and that these parameters are biologically relevant, as they signify the strength and nature of interactions between OTU groups as competitive, parasitic, commensal, or neutral (Supplementary Figure S8). The interaction models of individual OTUs furthermore generate hypotheses about the importance of environmental factors and other bacterial groups on the growth of individual OTUs. The models could in principle be used to predict consequences of changes in OTU distribution, but it is unclear how to validate such predictions. For example, we used the SC model to test the effect of environmental conditions on the abundances of SCs (Supplementary Figure S11). Seven SCs (1, 3, 5, 10, 11, 13, and 14) were predicted to return to their normal abundance patterns when the disturbances ended. Other SCs were strongly affected by the environment and their abundance profiles did not recover even several years after the disturbances stopped.
In most other network models, the grouping of OTUs has been based on taxonomy,12–14,35 resulting in very large networks with millions of pairwise interactions that are difficult to manage. In contrast, our model captures the bacterial dynamics in the lake at the levels of SCs and individual OTUs. This approach succeeded due to the grouping of OTUs into SCs based on their abundance peak times and to our novel estimation strategy. Notably, the OTUs in each SC are taxonomically very diverse at the species and genus levels, suggesting that taxonomically related OTUs are distributed over SCs throughout the year, such that each SC contains representatives of all functionally important taxonomic genera. Horizontal gene transfer, which is frequent in the microbial world and often accounts for the functional redundancy among phyla,36 is likely to contribute to the widely distributed abundances.
Although the paper focuses on an aquatic metapopulation, it is easy to imagine that similar types of analyses could be applied to other microbial consortia that display periodic annual or daily patterns.
Materials and methods
Data
The data were collected at 91 time points, from March 2000 to June 2011,31,32,37 and made publicly available at www.lter.limnology.wisc.edu.37,38 The dataset consists of abundance measurements, which were interpreted through 16S-sequences. Using the software Qiime,39 with 97% identity as a cutoff, and the Greengenes database27 (greengenes.lbl.gov), 18,696 OTUs were identified (see Supplementary Information for details).
Also measured were nineteen physical and chemical conditions of the lake, collected from 1995 to 2013;38 see references 6,40 and Supplementary Figure S1. Fourteen of these remain fairly constant, while water temperature, nitrate+nitrite, ammonia, total phosphorus unfiltered, dissolved reactive phosphorus, and dissolved reactive silica vary substantially over time.
Data processing
In order to manage the large number of OTUs, we first followed conventional wisdom and clustered the OTUs by taxonomy (cf. references 12–14,35). Specifically, we identified the top seven phyla, but found that their abundance profiles varied widely among OTUs within the phyla (Supplementary Figures S3 and S6b). Grouping by order, class, or genus yielded similar results. In spite of extensive efforts, none of these taxonomic clustering modalities led to new insights or interesting results.
We therefore decided to cluster differently, based on the annual peak time for each OTU. For each OTU, the abundances throughout the years 2000–2011 were superimposed, which resulted in a single, ‘collective 1-year period.’ The results were smoothed by computing the mean value of each 30-day window (Supplementary Figure S4). These smoothed profiles reflect the seasonal changes in abundances well. We omitted from clustering OTUs with only one observed data point and OTUs whose abundances were indicated by the smoothed curves to be zero.
For each OTU profile, we identified the positions of the top one or two abundance peaks and then clustered OTUs based on these peak profiles. This analysis resulted in 13 groups plus one additional group for all remaining OTUs. We refer to these 14 groups as subcommunities (SCs).
We chose water temperature and two chemical conditions (ammonia and total nitrate+nitrite) that follow distinct annual pattern. The patterns of other chemical conditions were omitted because they were highly related to the chosen patterns (Supplementary Figure S1a). The data were processed similarly to the abundance data. Their variability over the years fell within ranges of the mean±s.d. of the observed data, superimposed onto one ‘typical year’ (Supplementary Figure S1b).
Model
In our modeling format, Xi is the abundance of an OTU or SC i. The interactions between Xi and other Xj’s and with environmental conditions Tk are described through product terms, which have their origin in mass action kinetics.41 The model takes the form:
Ẋi is the rate of change of variable i, and the indexed parameters α and β indicate the type and strength of an interaction between pairs of OTUs or between OTUs and the environment, respectively. We use this structure to represent interactions among the 14 bacterial SCs and among individual OTUs and SCs. The quality of results is assessed with two similarity scores (see Supplementary Information).
Ignoring the less interesting situation that Xi=0, Equation (1) can be rewritten as
If abundances and slopes can be determined from the time courses of all SCs, this equation becomes an algebraic system of linear equations.23,42–45 Thus, even though the system is highly nonlinear, linear regression can be used to solve for all parameter values (for details, see Supplementary Information).
Predicting the abundances of individual OTUs
We also used the model to predict the abundances of individual OTUs. Formally, the model has exactly the same format as in Equation (1). However, to test whether environmental conditions alone could model the data (Model #1), all αij parameters were set to zero, except for αii, and the βik values were re-estimated. For Model #2, αij and βik values were chosen from the filtered parameter values described in the Supplementary Information. For Model #3, we removed the OTU of interest from its SC and considered it as a new group. The αij and βik values were then re-estimated for individual OTUs, based on 100 values for αii selected from the range [−1, 0]. The goodness of fit was evaluated with similarity scores (see Supplementary Information).
Acknowledgments
This work was funded by grant DEB-1241046 of the National Science Foundation, USA. The authors are grateful to Luis-Miguel Rodriguez Rojas and Despina Tsementzi for their help with the OTU identification, beneficial discussions, and valuable feedback.
Footnotes
Supplementary Information accompanies the paper on the Systems Biology and Applications website (http://www.nature.com/npjsba)
The authors declare no conflict of interest.
References
- Konstantinidis, K. T. & Tiedje, J. M. Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr. Opin. Microbiol. 10, 504–509 (2007). [DOI] [PubMed] [Google Scholar]
- Shade, A. et al. Fundamentals of microbial community resistance and resilience. Front. Microbiol. 3, 417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann, R. I. , Ludwig, W. & Schleifer, K. H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust, K. & Raes, J. Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550 (2012). [DOI] [PubMed] [Google Scholar]
- Barberan, A. , Bates, S. T. , Casamayor, E. O. & Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, J. A. et al. Defining seasonal marine microbial community dynamics. ISME J. 6, 298–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust, K. et al. Microbial co-occurrence relationships in the human microbiome. PLoS Computat. Biol. 8, e1002606 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, J. & Alm, E. J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8, e1002687 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, Q. S. et al. Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics 22, 2532–2538 (2006). [DOI] [PubMed] [Google Scholar]
- Chaffron, S. , Rehrauer, H. , Pernthaler, J. & von Mering, C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 20, 947–959 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner, D. E. & Blaser, M. J. The dynamics of Helicobacter pylori infection of the human stomach. J. Theor. Biol. 176, 281–290 (1995). [DOI] [PubMed] [Google Scholar]
- Marino, S. , Baxter, N. T. , Huffnagle, G. B. , Petrosino, J. F. & Schloss, P. D. Mathematical modeling of primary succession of murine intestinal microbiota. Proc. Natl Acad. Sci. USA 111, 439–444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, C. K. & Mehta, P. Identifying keystone species in the human gut microbiome from metagenomic timeseries using sparse linear regression. PLoS ONE 9, e102451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, R. R. et al. Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Comput. Biol. 9, e1003388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier, J. et al. Microbial interactions within a cheese microbial community. Appl. Environ. Microbiol. 74, 172–181 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanly, T. J. , Urello, M. & Henson, M. A. Dynamic flux balance modeling of S. cerevisiae and E. coli co-cultures for efficient consumption of glucose/xylose mixtures. Appl. Microbiol. Biotechnol. 93, 2529–2541 (2012). [DOI] [PubMed] [Google Scholar]
- Balagadde, F. K. et al. A synthetic Escherichia coli predator-prey ecosystem. Mol. Syst. Biol. 4, 187 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith, J. J. , McNulty, N. P. , Rey, F. E. & Gordon, J. I. Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science 333, 101–104 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa, H. & Sakha, M. Z. Prediction of competitive microbial growth in mixed culture at dynamic temperature patterns. Biocontrol Sci. 19, 121–127 (2014). [DOI] [PubMed] [Google Scholar]
- Berry, D. & Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 5, 219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F. , Guo, Y. Z. & Li, Y. F. Interactions of microorganisms during natural spoilage of pork at 5 degrees C. J. Food Eng. 72, 24–29 (2006). [Google Scholar]
- Bucci, V. & Xavier, J. B. Towards predictive models of the human gut microbiome. J. Mol. Biol. 426, 3907–3916 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voit, E. O. & Chou, I.-C. Parameter estimation in canonical biological systems models. Int. J. Syst. Synth. Biol. 1, 1–19 (2010). [Google Scholar]
- Dennis, P. G. , Seymour, J. , Kumbun, K. & Tyson, G. W. Diverse populations of lake water bacteria exhibit chemotaxis towards inorganic nutrients. Isme Journal 7, 1661–1664 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldani J. I. et al. in The Prokaryotes—Alphaproteobacteria and Betaproteobacteria (eds Rosenberg E., Delong E. F., Lory S., Stackebrandt E. & thompson F.) 919–974 (Springer-Verlag Berlin Heidelberg, 2014).
- Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser J. , Head I. , Stein L. in The Prokaryotes (eds Rosenberg E., DeLong E., Lory S., Stackebrandt E. & Thompson F.) 901–918 (Springer Berlin Heidelberg, 2014).
- Gallego, V. , Sanchez-Porro, C. , Garcia, M. T. & Ventosa, A. Roseomonas aquatica sp nov., isolated from drinking water. Int. J. Syst. Evol. Microbiol., 56, 2291–2295 (2006). [DOI] [PubMed] [Google Scholar]
- Rihs, J. D. et al. Roseomonas, a new genus associated with bacteremia and other human infections. J. Clin. Microbiol. 31, 3275–3283 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara, E. L. , Hanson, P. C. , Hu, Y. H. , Winslow, L. & McMahon, K. D. A decade of seasonal dynamics and co-occurrences within freshwater bacterioplankton communities from eutrophic Lake Mendota, WI, USA. ISME J. 7, 680–684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shade, A. et al. Interannual dynamics and phenology of bacterial communities in a eutrophic lake. Limnol. Oceanogr. 52, 487–494 (2007). [Google Scholar]
- Fuhrman, J. A. & Steele, J. A. Community structure of marine bacterioplankton: patterns, networks, and relationships to function. Aquat. Microb. Ecol. 53, 69–81 (2008). [Google Scholar]
- Fuhrman, J. A. et al. A latitudinal diversity gradient in planktonic marine bacteria. Proc. Natl Acad. Sci. USA 105, 7774–7778 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiler, A. , Heinrich, F. & Bertilsson, S. Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J. 6, 330–342 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Quintero, A. & Konstantinidis, K. T. Inter-phylum HGT has shaped the metabolism of many mesophilic and anaerobic bacteria. ISME J. 9, 958–967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- NTL-LTER. Time series of bacterial community dynamics in Lake Mendota. North Temperate Lakes Long-Term Ecological Research (NTL-LTER) program, NSF, Katherine Trina McMahon, Center for Limnology, University of Wisconsin-Madison. http://lter.limnology.wisc.edu (2014).
- NTL-LTER. Chemical Limnology of North Temperate Lakes LTER Primary Study Lakes: Nutrients, pH and Carbon. North Temperate Lakes Long-Term Ecological Research (NTL-LTER) program, NSF, Center for Limnology, University of Wisconsin-Madison. http://lter.limnology.wisc.edu (2012).
- Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- North Temperatue Lakes LTER. https://lter.limnology.wisc.edu/about/overview (15 October 2014).
- Voit, E. O. , Martens, H. A. & Omholt, S. W. 150 years of the mass action law. PLoS Comput. Biol. 11, e1004012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voit, E. O. & Savageau, M. A. Power-law approach to modeling biological systems; III. Methods of analysis. J. Ferment. Technol. 60, 233–241 (1982). [Google Scholar]
- Varah, J. M. A spline least-squares method for numerical parameter-estimation in differential-equations. Siam J. Sci. Stat. Comput. 3, 28–46 (1982). [Google Scholar]
- Voit, E. O. & Almeida, J. Decoupling dynamical systems for pathway identification from metabolic profiles. Bioinformatics 20, 1670–1681 (2004). [DOI] [PubMed] [Google Scholar]
- Chou, I. C. & Voit, E. O. Recent developments in parameter estimation and structure identification of biochemical and genomic systems. Math. Biosci. 219, 57–83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.